

Abstract
We describe a new class of subunit-selective antagonists of N-methyl D-Aspartate (NMDA)-selective ionotropic glutamate receptors that contain the (E)-3-phenyl-2-styrylquinazolin-4(3H)-one backbone. The inhibition of recombinant NMDA receptor function induced by these quinazolin-4-one derivatives is non-competitive and voltage-independent, suggesting that this family of compounds does not exert action on the agonist binding site of the receptor or block the channel pore. The compounds described here resemble CP-465,022 ((S)-3-(2-chlorophenyl)-2-[2-(6-diethylaminomethyl-pyridin-2-yl)-vinyl]-6-fluoro-3H-quinazolin-4-one), a non-competitive antagonist of AMPA-selective glutamate receptors. However, modification of ring substituents resulted in analogues with greater than 100-fold selectivity for recombinant NMDA receptors over AMPA and kainate receptors. Furthermore, within this series of compounds, analogues were identified with 50-fold selectivity for recombinant NR2C/D-containing receptors over NR2A/B containing receptors. These compounds represent a new class of non-competitive subunit-selective NMDA receptor antagonists.
INTRODUCTION
N-methyl-D-aspartate (NMDA) receptors are ligand-gated cation-selective channels that mediate glutamatergic excitatory synaptic transmission in the central nervous system.1 A wide range of roles have been proposed for NMDA receptors, including neuronal development2-5 and synaptic plasticity underlying learning.6-7 In addition, aberrant activation of NMDA receptors has been suggested to participate in neuropathological conditions such as stroke, epilepsy, schizophrenia, depression, Alzheimer's disease, Huntington's disease, and Parkinson's disease.8-15
NMDA receptors are tetrameric assemblies of two glycine-binding NR1 subunits and two glutamate-binding NR2 subunits, of which there are four subtypes (NR2A, NR2B, NR2C, NR2D). The NR2 subunit appears to control pharmacological properties, response time course, and channel properties.16-22 In particular, receptors containing NR2C or NR2D subunits are activated by glycine and glutamate with higher potency than receptors containing NR2A or NR2B. NMDA receptors containing NR2C or NR2D also show lower maximal open probability than receptors containing NR2A or NR2B. NR2D-containing NMDA receptors have been hypothesized to show exceptionally slow deactivation following removal of glutamate. NR2C and NR2D subunits also show localization patterns distinct from NR2A and NR2B, with prominent expression in cerebellum, discrete nuclei within the basal ganglia, and select populations of interneurons.23-37 The distinct anatomical localization of the NR2 subunits raises the possibility that subunit-selective antagonists and allosteric modulators might provide well-tolerated therapeutic treatments for a wide range of indications.1
Ifenprodil was the first subunit-selective NMDA receptor antagonist identified, showing over 200-fold selectivity at NR2B over NMDA receptors containing other NR2 subunits.38-41 Despite intense interest in NMDA receptors and their hypothesized roles in numerous neurological disorders, no antagonists that are more than 10-fold selective for NR2A, NR2C, or NR2D have yet been identified.42-46 The inability to identify subunit-selective competitive antagonists may reflect the highly conserved glutamate-binding pocket within the NR2 subunit.47-48 Channel blockers are similarly poorly selective for NMDA receptors comprised of different NR2 subunits,49 presumably due to structural conservation of the permeation pore. The lack of subunit-selective pharmacological tools for this receptor class has been an impediment to understanding the functional roles of the NR2A, NR2C, and NR2D subunits in neurons. Because NMDA receptor architecture appears to reflect the assembly of multiple extracellular semi-autonomous domains,50-52 we hypothesized that the multiple protein-protein interfaces within the NMDA receptor may provide new targets for modulating NMDA receptor function. To search for modulators that might interact at these interfaces, we executed a multi-well fluorescence-based assay to identify novel non-competitive inhibitors of recombinant NMDA receptors.53 Screening a diversity library of compounds with this assay allowed us to identify a new class of recombinant NMDA receptor antagonists that contains the quinazolin-4-one backbone, which is shared with the previously reported α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor-selective noncompetitive antagonist CP-465,022 (1, Figure 1).54-56 NMDA receptor inhibition was voltage-independent and non-competitive (Figure S1). Synthetic efforts towards optimizing backbone substituents led to the development of a structure-activity relationship (SAR), ultimately yielding novel compounds with between 50-100 fold selectivity for recombinant NR2C/D- over NR2A/B-containing receptors. The half-maximal inhibiting concentrations of members of this class are in the low micromolar range (IC50 = 0.6-6 μM) at recombinant NMDA receptors, with no detectable activity at recombinant kainate receptors and variable activity at recombinant AMPA receptors.
Figure 1.

Structures for CP-465,022 (1), (E)-4-(2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (2), and (E)-3-(2-methoxyphenyl)-2-(2-nitrostyryl)quinazolin-4(3H)-one (3).
RESULTS
Chemistry
We evaluated both commercially available analogues and a number of synthetic styryl quinazolin-4-one analogues that were generated via a three step sequence which combines commercially available fragments: anthranilic acids, anilines, and aldehydes (Scheme 1). Briefly, a substituted anthranilic acid (4) was converted to the benzoxazin-4-one (5) by refluxing in acetic anhydride. The quinazolin-4-one core (6) was generated by a ring opening-ring closure reaction under acidic conditions in the presence of a substituted aniline. Finally, acid-catalyzed condensation reaction of 6 with a substituted aldehyde yielded the target (E)-3-phenyl-2-styrylquinazolin-4(3H)-one.
Scheme 1.

Synthesis of (E)-3-phenyl-2-styrylquinazolin-4(3H)-ones. (a) Ac2O, reflux. (b) AcOH, reflux. (c) Ac2O, AcOH, NaOAc, reflux.
An alternative methodology was utilized to synthesize analogues for which the starting material anthranilic acids were not commercially available (Scheme 2). Protection of the carboxylic acid of 78 with tert-butyldimethylsilyl chloride (TBDMSCl) and N-methylmorpholine gave 10.57 Suzuki coupling between 10 and the appropriately substituted boronic acid afforded C ring R8-substituted quinazolinones 55a and 56a. The TBDMS protecting group was removed with the addition of 1.0 N hydrochloric acid (HCl) during work-up. A condensation reaction of 55a or 56a with meta-nitrobenzaldehyde gave 55 and 56. Reduction of the styryl linker present in 80 with hydrogenolysis yielded the fully saturated linker compound 81 (Scheme 3).
Scheme 2.

Suzuki route for synthesis of (E)-3-phenyl-2-styrylquinazolin-4(3H)-ones. (a) TBDMSCl, NMM, THF. (b) RB(OH)2, Pd(PPh3)4, aq. NaHCO3, 1,2-DME, reflux. (c) Ac2O, AcOH, NaOAc, reflux.
Scheme 3.

Synthesis of 81. (a) H2, Pd/C, DMF.
Quniazolin-4-ones inhibit NMDA receptors
Completion of a fluorescence-based58 screen of 83,880 compounds from ChemDiv and Asinex libraries of small molecules on NMDA receptor responses in both NR1/NR2D and NR1/NR2C cell lines yielded a 0.16% hit rate; all hits were confirmed by two-electrode voltage-clamp recording. A number of active compounds in this assay shared a quinazolin-4-one backbone, typified by (E)-4-(2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (2) and (E)-3-(2-methoxyphenyl)-2-(2-nitrostyryl)quinazolin-4(3H)-one (3) (see Figure 1). Quinazolin-4-one derivatives have previously been shown to be potent and relatively selective AMPA receptor antagonists.54-56 Compounds 2 and 3 inhibited NR1/NR2C and NR1/NR2D responses completely with fitted IC50 values in the micromolar range (IC50 values 9 and 5 μM, respectively, at NR1/NR2D). Inhibition by 30 μM of compound 2 was non-competitive at NR1/NR2D receptors, in that it could not be surmounted by increasing the concentration of glutamate and glycine from 100/30 μM to 1000/300 μM (n=4; Figure S1 in Supporting Information). Moreover, inhibition of NR1/NR2D responses by 10 μM of compound 2 was voltage-independent, with no significant difference in inhibition at −40 mV compared to +40 mV (p>0.05; t-test; n=5; Figure S1 in Supporting Information). These data suggest that quinazolin-4-ones are noncompetitive antagonists of NR1/NR2D NMDA receptors that likely act at a site independent of the channel pore. Inhibition of NR1/NR2C receptors by compound 2 was similarly complete at saturating concentrations (n=8), non-competitive (n=6), and voltage-independent (n=6).
Positional Isomer Manipulation of Screening Hits 2 and 3
The screening hits, compounds 2 and 3, were inactive at kainate receptors, and 4-5-fold more potent (i.e. lower fitted IC50) at NMDA receptors than AMPA receptors (Table 1). Analogues lacking substitutions on all aryl rings were inactive at NMDA and AMPA receptors (Figure S2 in Supporting Information), suggesting substitutions of aryl rings A and B are essential for activity. To evaluate whether we could improve the selectivity for NMDA receptors over AMPA receptors, positional isomer combinations of both 2 (A ring R2 = NO2, B ring R4 = COOH) and 3 (A ring R3 = NO2, B ring R6 = OMe) were evaluated (Table 1). All positional isomer modifications of A and B ring substitution led to significantly decreased potency (increased IC50) compared to the screening hit 3, and thus no further work was conducted within this series. Two positional isomeric analogues of compound 2 appeared to inhibit NR1/NR2D responses with IC50 values of 15 μM (7) and 6 μM (13). Notably, 2,7 and 13 all contained a para-carboxylic acid B ring substituent despite differential ortho-, meta-, or para-substitution of the A ring nitro group. This suggested optimal placement of the para-carboxylic acid B ring substituent would likely yield the most active analogues within this structural series. Moreover, a number of these compounds showed improved selectivity for NMDA receptors over the AMPA receptor GluR1, as well as improved selectivity for NR2D-containing NMDA receptors over receptors containing the NR2A subunit, although the substitution patterns controlling these effects were unclear. The IC50 values for inhibition of NR2C- and NR2D-containing receptors were more similar than those for NR2A- and NR2B-containing receptors. These initial experiments suggested that it might be possible to identify derivatives within this class that selectively inhibit NR2C/D-containing recombinant NMDA receptors compared to AMPA, kainate, or NR2A/B-containing NMDA receptors.
Table 1.
Optimization of subunit selectivity through evaluation of Ring A and Ring B substituent positions.
 |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 values in μM were determined by fitting the Hill equation to average composite concentration-effect | ||||||||||||||
| Number | R1 | R2 | R3 | R4 | R5 | R6 | NR2A IC50 |
NR2B IC50 |
NR2C IC50 |
NR2D IC50 |
GluR1 IC50 |
GluR6 IC50 |
NR2Aa NR2D |
GluR1b NR2D |
| 7 | NO2 | COOH | 64 | 93 | 15 | 15 | >300 | NE | 4 | >20 | ||||
| 8 | NO2 | COOH | 135 | 177 | 33 | 27 | >300 | NE | 5 | >10 | ||||
| 9 | NO2 | COOH | >300 | 197 | >300 | 278 | >300 | NE | 1 | 1 | ||||
| 2 | NO2 | COOH | 21 | 30 | 12 | 9 | 32 | NE | 2 | 4 | ||||
| 11 | NO2 | COOH | 100 | 69 | 19 | 15 | 32 | NE | 7 | 2 | ||||
| 12 | NO2 | COOH | >300 | 235 | 258 | 148 | >300 | NE | 2 | 2 | ||||
| 13 | NO2 | COOH | 11 | 17 | 8 | 6 | 6 | NE | 2 | 1 | ||||
| 14 | NO2 | COOH | 17 | 20 | 8 | 7 | 5 | NE | 2 | 1 | ||||
| 15 * | NO2 | OCH3 | >300 | 98 | 181 | 221 | >300 | NE | 1 | 1 | ||||
| 16 * | NO2 | OCH3 | >300 | >300 | >300 | >300 | >300 | NE | 1 | 1 | ||||
| 17 * | NO2 | OCH3 | >300 | >300 | >300 | 290 | >300 | NE | 1 | 1 | ||||
| 18 * | NO2 | OCH3 | >300 | >300 | 152 | 168 | 178 | NE | 2 | 1 | ||||
| 19 * | NO2 | OCH3 | >300 | 262 | 205 | 186 | >300 | NE | 2 | 2 | ||||
| 20 * | NO2 | OCH3 | >300 | >300 | >300 | >300 | >300 | NE | 1 | 1 | ||||
| 21 * | NO2 | OCH3 | >300 | >300 | 143 | 125 | 223 | NE | 2 | 2 | ||||
| 22 * | NO2 | OCH3 | >300 | >300 | >300 | >300 | >300 | NE | 1 | 1 | ||||
| 3 * | NO2 | OCH3 | 4 | 7 | 4 | 5 | 14 | NE | 1 | 3 | ||||
curves from 3-17 oocytes injected with NR1/NR2A, NR1/NR2B, NR1/NR2C, NR1/NR2D, GluR1, or GluR6 cRNA. Oocytes were obtained from 1-3 frogs; NE indicates no effect. IC50 values greater than 300 μM were determined as described in the Experimental Methods.
(IC50 NR2A)/(IC50 NR2D).
(IC50 GluR1)/(IC50 NR2D).
1-10 mM 2-hydroxypropyl-β-cyclodextrin was included for 30 and 100 μM concentrations of test compound. See Figure S2 (Supporting Information) for full general structure.
The effect of substituent position and identity on rings A,B,C
To further evaluate the effects of different aryl ring substituents, we measured the fitted IC50 values for inhibition at recombinant NR1/NR2A, NR1/NR2B, NR1/NR2C, NR1/NR2D, and GluR1 receptor current responses under voltage clamp for each test compound. The position of a variety of A ring substituents was tested while holding the B ring substituent constant (R4 = COOH) (Table 2). Given the lack of any detectable activity at recombinant kainate receptors (Table 1), responses at GluR6 were not studied further. A ring substitution at the meta position (R2) was either equipotent or modestly improved potency compared to ortho (R3) or para (R1) A ring substitutions at NR2D-containing receptors among all analogues tested. For example, analogues containing meta methoxy (25) and trifluoromethyl (28) were most potent among positional isomers, whereas meta and ortho hydroxyl (31, 32) and nitro (2, 13) A ring substituents had nearly equivalent potencies.
Table 2.
Optimization of subunit selectivity through evaluation of Ring A substituents.
 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number | R1 | R2 | R3 | NR2A IC50 |
NR2B IC50 |
NR2C IC50 |
NR2D IC50 |
GluR1 IC50 |
NR2Aa NR2D |
GluR1b NR2D |
| 23 | 84 | 85 | 39 | 33 | 25 | 3 | 1 | |||
| 24 | OCH3 | 100 | 98 | 42 | 31 | 250 | 3 | 8 | ||
| 25 | OCH3 | 24 | 36 | 17 | 9 | 27 | 3 | 3 | ||
| 26 | OCH3 | 88 | 51 | 32 | 16 | 38 | 6 | 2 | ||
| 27 | CF3 | 61 | 55 | 23 | 15 | 233 | 4 | 16 | ||
| 28 | CF3 | 13 | 26 | 8 | 12 | 22 | 1 | 2 | ||
| 29 | CF3 | >300 | >300 | 88 | 28 | >300 | >10 | >10 | ||
| 30 | OH | 38 | 35 | 50 | 36 | 110 | 1 | 3 | ||
| 31 | OH | 52 | 30 | 24 | 17 | 24 | 3 | 1 | ||
| 32 | OH | 20 | 18 | 20 | 18 | 36 | 1 | 2 | ||
| 7 | NO2 | 64 | 93 | 15 | 15 | >300 | 4 | >20 | ||
| 2 | NO2 | 21 | 30 | 12 | 9 | 32 | 2 | 4 | ||
| 13 | NO2 | 11 | 17 | 8 | 6 | 6 | 2 | 1 | ||
| 33 | CH3 | 41 | 138 | 28 | 17 | >300 | 2 | >17 | ||
| 34 | CH3 | 21 | 34 | 28 | 18 | >300 | 1 | >16 | ||
| 35 | CH3 | 86 | 93 | 80 | 81 | 62 | 1 | 1 | ||
IC50 values in μM were determined by fitting the Hill equation to average composite concentration-effect curves from 4-17 oocytes injected with NR1/NR2A, NR1/NR2B, NR1/NR2C, NR1/NR2D, or GluR1 cRNA. Oocytes were obtained from 1-3 frogs. Compounds 2 and 7, previously shown in Table 1, are included here to facilitate comparison. IC50 values greater than 300 μM were determined as described in the Experimental Methods.
(IC50 NR2A)/(IC50 NR2D)
(IC50 GluR1)/(IC50 NR2D). See Figure S2 (Supporting Information) for full general structure.
The positional preference for C ring substitutions was explored utilizing chlorinated derivatives while retaining optimal A ring (R2 = NO2) and B ring (R4 = COOH) substitutions (Table 3; Fig. S2 in Supporting Information). Substitution at position R8, specifically compound 37, gave both improved potency (IC50 1 μM) and selectivity (IC50 NR2A/ IC50 NR2D = 16) for mono-substituted compounds. Interestingly, the R8,R10-dichloro compound 40 showed enhanced NR2D selectivity (IC50 NR2A/ IC50 NR2D = 22) with decreased on-target potency (IC50 10 μM).
Table 3.
Optimization of subunit selectivity through evaluation of Ring C substituents.
 |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number | R1 | R2 | R7 | R8 | R9 | R10 | NR2A IC50 |
NR2B IC50 |
NR2C IC50 |
NR2D IC50 |
GluR1 IC50 |
NR2Aa NR2D |
GluR1b NR2D |
| 36 | NO2 | Cl | >300 | 110 | 22 | 14 | >300 | >20 | >20 | ||||
| 37 | NO2 | Cl | 16 | 13 | 2 | 1 | 7 | 16 | 7 | ||||
| 38 | NO2 | Cl | 32 | 52 | 16 | 15 | 41 | 2 | 3 | ||||
| 39 * | NO2 | Cl | 56 | 243 | 12 | 9 | 87 | 6 | 10 | ||||
| 40 | NO2 | Cl | Cl | 224 | 109 | 16 | 10 | >300 | 22 | 30 | |||
| 41 | NO2 | Cl | Cl | >300 | >300 | 9 | 6 | >300 | >50 | >50 | |||
| 2 | NO2 | H | 21 | 30 | 12 | 9 | 32 | 2 | 4 | ||||
| 42 | NO2 | F | 36 | 33 | 14 | 7 | 7 | 5 | 1 | ||||
| 37 | NO2 | Cl | 16 | 13 | 2 | 1 | 7 | 16 | 7 | ||||
| 43 | NO2 | Br | 17 | 13 | 2 | 1 | 8 | 17 | 8 | ||||
| 44 | NO2 | I | 18 | 6 | 1 | 0.6 | 31 | 18 d | 52 | ||||
| 45 | NO2 | CH3 | 12 | 15 | 5 | 2 | 27 | 6 | 14 | ||||
| 46 * | NO2 | OCH3 | 229 | >300 | 6 | 3 | >300 | 76 | >100 | ||||
| 47 * | NO2 | NO2 | NE | NE | 204 | 90 | >300 | - | - | ||||
| 48 | NO2 | OH | 237 | >300 | 43 | 10 | 96 | 24 | 10 | ||||
| 49 | NO2 | C6H5 | 11 | 3 | 2 c | 2 c | 5 | 6 | 3 | ||||
| 50 | NO2 | R8-C4H4-R9 | 11 | 5 | 2 c | 2 c | 4 | 6 | 2 | ||||
| 51 | NO2 | R8-OCH2CH2O-R9 | >300 | >300 | 281 | 213 | >300 | 1 | 1 | ||||
| 52 | NO2 | R8-OCH2O-R9 | 112 | 162 | 40 | 36 | >300 | 3 | >8 | ||||
| 53 * | NO2 | CH(CH3)2 | 101 | 98 | 6 | 7 | 200 | 14 | 29 | ||||
| 54 | NO2 | CH2CH2CH3 | 28 | 18 | 3 | 4 | 50 | 7 | 13 | ||||
| 55 | NO2 | 2-thiophene | 157 | 129 | 5 | 3 | 280 | 52 | 93 | ||||
| 56 | NO2 | CHCHC6H5 | 19 | 30 | 4 | 4 | 131 | 5 | 33 | ||||
IC50 values in μM were determined by fitting the Hill equation to average composite concentration-effect curves from 7-26 oocytes injected with NR1/NR2A, NR1/NR2B, NR1/NR2C, NR1/NR2D, GluR1 cRNA. Oocytes were obtained from 2-5 frogs. Compound 2, previously shown in Table 1, is included to facilitate comparison. IC50 values greater than 300 μM were determined as described in Experimental Methods. NE indicates no effect.
(IC50 NR2A)/(IC50 NR2D)
(IC50 GluR1)/(IC50 NR2D)
data were fitted with a variable minimum (see text)
selectivity was calculated from IC50 values rounded to nearest value in μM.
1-10 mM 2-hydroxypropyl-β-cyclodextrin was included for 30 and/or 100 μM concentrations of test compound. See Figure S2 (Supporting Information) for full general structure.
Evaluation of a series of R8 C ring substituents revealed a correlation between van der Waals radii of the series H, F, Cl, Br, I and both the IC50 value at NR1/NR2D (r = −0.95; p<0.01; Figure S3 in Supporting Information) and the selectivity for NR2D over NR2A (r = 0.97; p<0.01). In particular, iodinated derivative 44 inhibited NR1/NR2D receptor responses with an IC50 value of 600 nM and is 18-and 52-fold selective over recombinant NR2A and AMPA GluR1 receptors, respectively (Table 3). It is unclear whether the increased potency and selectivity of 44 can be attributed to an electropositive effect or a steric effect. Consistent with a steric effect, we observed that other analogues containing large R8 substituents also improve NR2D apparent selectivity such as R8 = Ph (49) and R8,R9-naphthyl (50) when it is assumed that inhibition is complete at saturating concentrations. However, the inhibition curves with these larger hydrophobic substituents revealed incomplete inhibition. Fitting the concentration-effect data for these compounds with a variable minimum (see Experimental) resulted in little difference in potency between the various NR2 subunits (see IC50 values in Table 3). Nevertheless, the fitted curves reveal striking differences in the degree of inhibition. For example, a saturating concentration of compound 50 is predicted to reduce responses at receptors containing NR2A, NR2B, NR2C, or NR2D to 68, 50, 26, or 20% of control respectively. Similarly, a saturating concentration of compound 49 is predicted to reduce responses at saturating concentrations for receptors containing NR2A, NR2B, NR2C, or NR2D to 68, 51, 22, or 14% of control, respectively. These classes of compounds were not studied further.
Robust combined selectivity for NR2C and NR2D over NR2A, NR2B and GluR1 was achieved with methoxy at the R8 position (46, IC50 3 μM), which was greater than 100-fold selective for NR1/NR2D over GluR1. This compound series has a maximum solubility of 70 μM (n=2), necessitating the use of 1 mM β-cyclodextrin for concentrations above 70 μM. IC50 values for 46 at NR2C were similar to those at NR2D. Likewise, IC50 values at NR2A were typically similar to NR2B, and were >50 fold higher than at NR2C/D containing receptors (see Table 3).
We subsequently tested the effect of altering the nitro and carboxylic acid substituents on the A and B rings, respectively, for compounds where R8 was methoxy (C ring) (Table 4). Replacement of the p-carboxylic acid functionality on the B ring with hydrogen (57), cyano (58), amide (59), or methyl ester (60) moieties led to compounds that were either substantially less potent or inactive (Table 4). These results suggest an anionic component such as the carboxylate is crucial for binding. Substitutions of the A ring nitro group that maintained some activity included R2 = OMe (61, NR2D IC50 9 μM) and R2 = COOCH3 (63, NR2D IC50 6 μM). Only modest activity was maintained with para substituents on the A ring (Table 4). These data suggest the binding pocket prefers the electron rich nitro and carboxylic acid groups on rings A and B, respectively.
Table 4.
Substitutions for Ring B carboxylic acid and Ring A nitro groups.
 |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Number | R1 | R2 | R4 | R8 | NR2A IC50 |
NR2B IC50 |
NR2C IC50 |
NR2D IC50 |
GluR1 IC50 |
NR2Aa NR2D |
GluR1b NR2D |
| 57 * | NO2 | >300 | >300 | >300 | >300 | >300 | 1 | 1 | |||
| 46 * | NO2 | COOH | OCH3 | 229 | >300 | 6 | 3 | >300 | 76 | >100 | |
| 58 | NO2 | CN | OCH3 | >300 | >300 | >300 | >300 | >300 | 1 | 1 | |
| 59 | NO2 | CONH2 | OCH3 | >300 | 82 | 58 | >300 | >300 | 1 | 1 | |
| 60 | NO2 | COOCH3 | OCH3 | 218 | 46 | 39 | 38 | 138 | 6 | 4 | |
| 61 | OCH3 | COOH | OCH3 | 73 | 109 | 18 | 9 | 220 | 8 | 24 | |
| 62 | COOH | COOH | OCH3 | >300 | >300 | >300 | 131 | >300 | >2 | >2 | |
| 63 | COOCH3 | COOH | OCH3 | 80 | 39 | 15 | 6 | 41 | 13 | 7 | |
| 64 | COOCH3 | COOCH3 | COOH | OCH3 | >300 | >300 | 124 | 52 | >300 | >6 | >6 |
| 7 | NO2 | COOH | 64 | 93 | 15 | 15 | >300 | 4 | >20 | ||
| 65 | SCH3 | COOH | 211 | 79 | 42 | 22 | 244 | 10 | 11 | ||
| 66 | CN | COOH | 94 | 106 | 28 | 34 | >300 | 3 | >9 | ||
| 67 | N(CH3)2 | COOH | >300 | >300 | 63 | 53 | >300 | >5 | >5 | ||
| 68 | OCHF2 | COOH | 73 | 89 | 33 | 22 | 208 | 3 | 9 | ||
| 69 | OCH2CHCH2 | COOH | >300 | 144 | 111 | 30 | >300 | >10 | >10 | ||
| 70 | OCH2COOH | COOH | >300 | >300 | >300 | >300 | >300 | 1 | 1 | ||
| 71 | COOH | COOH | >300 | >300 | >300 | >300 | >300 | 1 | 1 | ||
IC50 values in μM were determined by fitting the Hill equation to average composite concentration-effect curves from 4-26 oocytes injected with NR1/NR2A, NR1/NR2B, NR1/NR2C, NR1/NR2D, GluR1 cRNA. Oocytes were obtained from 1-4 frogs. Compounds 7 and 46, previously shown in Table 3, are included to facilitate comparison. IC50 values greater than 300 μM were determined as described in the Experimental Methods.
(IC50 NR2A)/(IC50 NR2D)
(IC50 GluR1)/(IC50 NR2D)
1-10 mM 2-hydroxypropyl-β-cyclodextrin was included for 100 μM concentrations of test compound. See Figure S2 (Supporting Information) for full general structure.
Although compounds with R2 = NO2 (2) or R3 = NO2 (13) A ring substitution gave the best potency, improved selectivity for NMDA over AMPA receptors was noted for R1 = NO2 analogues (7 and 8 in Table 1), as well as modestly improved selectivity for NR2D over NR2A with R5 = COOH B ring substitution (11 in Table 1). This led to a systematic comparison of ring substituents among these two positions (A ring: R1, R2 = NO2; B ring: R4, R5 = COOH) for the most potent and selective ring C substitutions where R8 was iodo or methoxy. Table 5 summarizes data describing these compounds, fitted as described in the Methods. The best potency was obtained for compounds with para-carboxylic acid substitution on the B ring (R4 = COOH), in particular compounds 44, 46 (Fig. 2A). Among iodo-substituted compounds, 72, which contains a para-nitro A ring (R1 = NO2), inhibited NR2C/D containing receptors with the best apparent selectivity over NR2A, NR2B or AMPA receptors. We similarly saw somewhat improved selectivity with para-nitro in compound 41 (Table 3). Compound 72 had a maximum solubility of 10 μM, and thus required addition of 1 mM 2-hydroxypropyl-β-cyclodextrin at concentrations above 10 μM. We were unable to determine an IC50 value for NR2A, NR2B, and GluR1 AMPA receptors, but this value would theoretically be greater than 300 μM given the level of inhibition observed at 100 μM of compound 72. Inhibition by 72 at NR1/NR2C and NR1/NR2D was incomplete, leading us to fit the data with a variable minimum (see Methods), which was on average 31% and 26% of control, respectively (Fig. 2B).
Table 5.
Optimization of Ring A, Ring B, Ring substituents.
 |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number | R1 | R2 | R4 | R5 | R8 | NR2A IC50 |
NR2B IC50 |
NR2C IC50 |
NR2D IC50 |
GluR1 IC50 |
NR2Aa NR2D |
GluR1b NR2D |
| 72 * | NO2 | COOH | I | >300 | >300 | 2 c | 1 c | >300 | >150 | >220 | ||
| 44 | NO2 | COOH | I | 18 | 6 | 1 | 0.6 | 31 | 30 | 52 | ||
| 73 | NO2 | COOH | I | >300 | 279 | 9 | 8 | >300 | >37 | >37 | ||
| 74 | NO2 | COOH | I | 8 | 11 | 2 | 1 | 21 | 8 | 21 | ||
| 75 | NO2 | COOH | OCH3 | 197 | 206 | 13 | 7 | >300 | 28 | >42 | ||
| 46 * | NO2 | COOH | OCH3 | 229 | >300 | 6 | 3 | >300 | 76 | >100 | ||
| 76 | NO2 | COOH | OCH3 | 238 | 238 | 21 | 16 | >300 | 15 | >19 | ||
| 77 | NO2 | COOH | OCH3 | 208 | >300 | 24 | 13 | 289 | 16 | 22 | ||
IC50 values in μM were determined by fitting the Hill equation to average composite concentration-effect curves from 7-26 oocytes injected with NR1/NR2A, NR1/NR2B, NR1/NR2C, NR1/NR2D, GluR1 cRNA. Oocytes were obtained from 2-5 frogs. Compounds 44 and 46, previously shown in Table 3, are included to facilitate comparison. IC50 values greater than 300 μM were determined as described in the Experimental Methods.
(IC50 NR2A)/(IC50 NR2D)
(IC50 GluR1)/(IC50 NR2D)
indicates data fitted with variable minimum (see Fig 2B).
1 mM 2-hydroxypropyl-β-cyclodextrin was included for 30 and/or 100 μM concentrations of test compound. See Figure S2 (Supporting Information) for full general structure.
Figure 2.

Mean composite concentration-effect curves are shown for NR1/NR2A, NR1/NR2B, NR1/NR2C, NR1/NR2D, GluR1, GluR6 for compounds (A) 46 and (B) 72; error bars are SEM. 2-hydroxypropyl-β-cyclodextrin (1 mM) was included in solutions of 30 and 100 μM 72 and 100 μM for 46. For compound 46, recordings of NMDA and AMPA receptor responses were made from 16-28 oocytes per receptor; oocytes were isolated from 3-5 different frogs. For compound 72, recordings of NMDA and AMPA receptor responses were made from 11-15 oocytes from 3 batches of oocytes from different frogs. No significant effect was observed for 30 μM 46 or 72 on GluR6 responses (6 oocytes, 1-2 batches of oocytes from different frogs for each compound).
Effect of changes to the backbone on subunit selectivity and potency at NR2C/D receptors
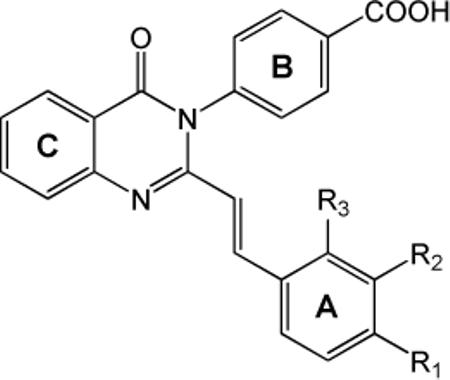
Modifications to the styryl A ring linker were also explored to determine the nature of the backbone structure in terms of both potency and subunit selectivity (Table 6). Complete removal of the ring, as in compound 78, which contains merely the quinazolin-4-one core structure, resulted in a total loss of inhibitory activity at concentrations up to 100 μM. We further investigated the saturation level of the styryl linker between the A ring and the quinazolin-4-one core structure. Reduction of the styryl linker of 80 (no A ring substitutions, B ring p-carboxylic acid, C ring R8 methyl; IC50 26 μM) to the fully saturated linker analogue 81 (fitted IC50 282 μM; Table 6) resulted in 10-fold potency reduction. These data suggest the geometry of the trans-alkene linker is preferred for activity. Incorporation of larger ring systems pendant to the C ring led to compounds with modest activity, as shown by the various napthyl derivatives (82, 83, and 84) and phenyl-1,4-dioxane (86; Table 6), suggesting the presence of an extended space in the binding pocket for larger hydrophobic groups. Replacement of the A ring with a 3-pyridyl system also produced active compounds. The 3-pyridyl A ring analogues with the most potent C ring substitution (R8 = iodine; Table 7) were subsequently evaluated. Compound 91 (R6 = OMe) exhibited reasonable potency (IC50 2.5 μM) with modest selectivity (10-fold) over NR2A and good selectivity over GluR1 (66-fold). Introduction of a carboxylic acid at position R4 (98) or R5 (99) gave reasonable potency; however, selectivity over GluR1 was more modest.
Table 6.
Optimization of the linker and Ring A.
 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Number | R | R8 | NR2A IC50 |
NR2B IC50 |
NR2C IC50 |
NR2D IC50 |
GluR1 IC50 |
NR2Aa NR2D |
GluR1b NR2D |
| 78 | CH3 | I | >300 | >300 | >300 | >300 | >300 | 1 | 1 |
| 79 |

|
I | 53 | 66 | 13 | 4 | 23 | 13 | 6 |
| 80 |

|
CH3 | 39 | 25 | 22 | 26 | 21 | 2 | 1 |
| 81 |

|
CH3 | >300 | 78 | 184 | 282 | >300 | 1 | 1 |
| 82 |
|
21 | 31 | 14 | 6 | >300 | 4 | >50 | |
| 83 |
|
63 | 36 | 20 | 11 | 103 | 6 | 9 | |
| 84 |

|
25 | 68 | 22 | 6 | 82 | 4 | 14 | |
| 85 |

|
>300 | 122 | 284 | 171 | >300 | 1 | 1 | |
| 86 * |
|
>300 | 262 | 26 | 16 | >300 | >18 | >18 | |
IC50 values in μM were determined by fitting the Hill equation to average composite concentration-effect curves from 3-12 oocytes injected with NR1/NR2A, NR1/NR2B, NR1/NR2C, NR1/NR2D, GluR1 cRNA. Oocytes were obtained from 1-2 frogs. IC50 values greater than 300 μM were determined as described in the Experimental Methods.
(IC50 NR2A)/(IC50 NR2D)
(IC50 GluR1)/(IC50 NR2D)
2-4 mM 2-hydroxypropyl-β-cyclodextrin was included for 1-100 μM concentrations of test compound. See Figure S2 (Supporting Information) for full general structure.
Table 7.
Optimization of Ring B substituents for pyridinyl analogues.
 |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Number | R4 | R5 | R6 | R8 | NR2A IC50 |
NR2B IC50 |
NR2C IC50 |
NR2D IC50 |
GluR1 IC50 |
IC50NR2A IC50NR2D |
IC50GluR1 IC50NR2D |
| 87 | 40 | 51 | 140 | 35 | 74 | 1 | 2 | ||||
| 88 | I | 132 | >300 | >300 | 108 | >300 | 1 | 3 | |||
| 89 * | OCH3 | I | >300 | >300 | >300 | >300 | >300 | 1 | 1 | ||
| 90 * | OCH3 | I | >300 | >300 | >300 | 200 | >300 | 1 | 1 | ||
| 91 * | OCH3 | I | 31 | 30 | 3 | 3 | 199 | 10 | 66 | ||
| 92 * | CH3 | I | >300 | >300 | >300 | >300 | >300 | 1 | 1 | ||
| 93 * | CH3 | I | >300 | >300 | 241 | >300 | >300 | 1 | 1 | ||
| 94 * | CH3 | I | >300 | 156 | 70 | 25 | 83 | 12 | 3 | ||
| 95 * | NO2 | I | >300 | >300 | 204 | 218 | >300 | 1 | 1 | ||
| 96 * | NO2 | I | >300 | >300 | >300 | >300 | >300 | 1 | 1 | ||
| 97 * | NO2 | I | 206 | 105 | 19 | 5 | >300 | 41 | 60 | ||
| 98 | COOH | I | 26 | 31 | 6 | 4 | 61 | 7 | 15 | ||
| 99 | COOH | I | 45 | 49 | 7 | 4 | 64 | 11 | 16 | ||
IC50 values in μM were determined by fitting the Hill equation to average composite concentration-effect curves from 3-20 oocytes injected with NR1/NR2A, NR1/NR2B, NR1/NR2C, NR1/NR2D, GluR1 cRNA. Oocytes were obtained from 1-3 frogs. IC50 values greater than 300 μM were determined as described in the Experimental Methods. 1-10 mM 2-hydroxypropyl-β-cyclodextrin was included for 10, 30, and/or 100 μM concentrations of test compounds. See Figure S2 (Supporting Information) for full general structure.
DISCUSSION AND CONCLUSIONS
These data show that a previously unknown and novel binding site exists on recombinant NMDA receptors expressed in heterologous systems at which modulators can act with clear preference for NR2C/D subunits. The two most potent and selective compounds in terms of inhibition of NR2C/D-containing receptors to emerge from these experiments are compounds 46 and 72 (Fig. 2). Both 46 and 72 are highly selective for NMDA receptors over GluR6 kainate receptors. Compound 46 is over 50-fold selective for NR2D over NR2A/B, and at least 100-fold selective over GluR1. Compound 72 also appears to be selective over NR2A/B-containing receptors. Figure 2 compares the concentration-effect curves for each of these compounds at five different glutamate receptors. Overall, a small subset of compounds within this class is characterized by poor aqueous solubility at or above 100 μM. Thus, the compounds discussed here may be more selective than we have conservatively reported. These compounds represent the first set of non-competitive antagonists with selectivity for recombinant NMDA receptors containing NR2C/D subunits over receptors containing NR2A/B subunits. These data suggest that native NMDA receptors may have binding sites for this class of modulators, and that the concentration-effect relationship associated with this class of compounds may show significant differences between the NR2A/B and NR2C/D receptors. Thus, this class of molecule may serve as a starting point for the development of potent and selective subunit selective NMDA receptor inhibitors.
BIOLOGY EXPERIMENTAL
Two-electrode voltage-clamp electrophysiology
Two-electrode voltage-clamp recordings were performed on Xenopus oocytes expressing recombinant rat NR1/NR2A, NR1/NR2B, NR1/NR2C, NR1/NR2D, GluR1, or GluR6 receptors. cDNAs for rat NR1-1a (GenBank accession numbers U11418 and U08261; hereafter NR1), NR2A (D13211), NR2B (U11419), NR2C (M91563), NR2D (L31611), GluR1 (X17184), GluR6 (Z11548) were provided by Drs. S. Heinemann (Salk Institute), S. Nakanishi (Kyoto University), and P. Seeburg (University of Heidelberg). Oocyte isolation and RNA injection were completed as described in detail elsewhere;59 all protocols involving Xenopus laevis were approved by the Emory University Institutional Animal Care and Use Committee. During two-electrode voltage-clamp recordings, oocytes were placed into a perfusion chamber and continually washed with recording solution containing (in mM) 90 NaCl, 1.0 KCl, 0.5 BaCl2, 0.005 EDTA, and 10 HEPES at pH 7.4 (23°C). Glass electrodes with a tip resistance of 0.5-2.5 MΩ were pulled from thin-walled glass capillary tubes and filled with 0.3-3.0 M KCl. An OC-725C amplifier (Warner Instrument Co) was used to hold the membrane potential of the oocytes at −40 mV during current recording. All compounds were made as 20 mM stock solutions in DMSO, and dissolved to reach the desired final concentration in recording solution containing 100 μM glutamate and 30 μM glycine for use on oocytes expressing NMDA receptors. Final DMSO content was 0.05-0.5% (vol/vol). Oocytes expressing GluR6 receptors were pre-treated with 10 μM concanavalin A for 10 minutes. Recombinant GluR1 and GluR6 receptors were activated by 100 μM glutamate. In order to prevent a gradual increase in current response over the course of the experiment, which appears to be a common feature of NR1/NR2A receptor responses in oocytes, some oocytes expressing NR1/NR2A were either pretreated with 50 μM BAPTA-AM (1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetraacetoxymethyl ester) for 10 minutes or injected with 50 nl of 2 mM K-BAPTA (potassium 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid).
For every test compound, we recorded 5-7 concentrations in at least 4 oocytes per concentration on each of five different receptors. We subsequently determined the IC50 (half-maximally effective concentration of inhibitor) by fitting the equation
| (1) |
to the mean composite concentration-response data normalized to the response in the absence of inhibitor (100%). N is the Hill slope, [ I ] is the inhibitor concentration, and minimum is the residual inhibition at saturating concentrations of ligand. Because inhibition was complete for most compounds tested, the minimum was fixed to 0 for all fitted curves, unless otherwise indicated. For a few compounds with bulky hydrophobic C ring substituents (49, 50, 72), minimum was allowed to vary. Compounds that showed a response greater than 75% of control in the presence of 100 μM test compound are reported as having an IC50 value >300 μM, which is theoretically predicted by the Hill equation for a slope of 1.
The maximum solubility was determined in a subset of 24 compounds representing various classes of structure using a BMG Labtech Nephelostar nephelometer (Offenburg, Germany), according to manufacturer's instructions. Maximum solubility of each test compound was determined in oocyte recording solution (components given below) and 1% DMSO. Only responses for concentrations below the experimentally determined limit of solubility were measured; whenever necessary we repeated experiments with 1-10 mM 2-hydroxypropyl-β-cyclodextrin added to the recording solution to ensure that the compounds remained in solution up to 100 μM. 2-hydroxypropyl-β-cyclodextrin had no detectable effect on NMDA response amplitude (data not shown).
CHEMISTRY EXPERIMENTAL
Compounds not described below were purchased from commercial vendors. Provided samples were of greater than 90% purity, as determined by the suppliers, via HPLC or NMR.
All reagents were obtained from commercial suppliers and used without further purification. Reaction progress was monitored by thin layer chromatography (TLC) on pre-coated glass plates (silica gel 60 F254, 0.25 mm). Proton and carbon NMR spectra were recorded on an INOVA-400 (400 MHz), VNMRS 400 (400 MHz), INOVA-600 (600 MHz), or Mercury 300 Vx (300 MHz). The spectra obtained were referenced to the residual solvent peak. Mass spectra were performed by the Emory University Mass Spectroscopy Center on either a VG 70-S Nier Johnson or JEOL instrument. Elemental analyses were performed by Atlantic Microlab Inc. C, H, N agreed with proposed structures within ±0.4% of theoretical values unless indicated. Flash chromatography was performed on a Teledyne ISCO Combiflash Companion with prepackaged Teledyne RediSep disposable normal phase silica columns. Melting temperatures were determined on a Mel-Temp apparatus and are uncorrected.
General procedure for synthesis of (E)-3-phenyl-2-styrylquinazolin-4(3H)-one products (Procedure C)
The quinazolinone (6, 1.0 equiv), benzaldehyde (1.33 equiv), and sodium acetate (1.63 equiv) were suspended in a mixture of glacial acetic acid (58 equiv) and acetic anhydride (7.0 equiv). The mixture was refluxed until TLC indicated the reaction was finished (generally 12-18 hours). After cooling to room temperature, the mixture was filtered and washed with methanol. Further purification was performed (chromatography or recrystallization) as necessary.
(E)-4-(2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (2)
Compound 2 was prepared via procedure C with compound 2b (0.100 g, 0.36 mmol) and meta-nitrobenzaldehyde (0.072 g, 0.48 mmol, 1.3 equiv). The crude material was purified via silica gel chromatography (ISCO, RediSep 12 g column, silica cake, 5-10% MeOH/DCM gradient) to give an off-white solid (0.030 g, 20%). 1H NMR (600 MHz, DMSO-d6) δ 8.26 (s, 1H), 8.17 (d, J=7.6 Hz, 4H), 8.01 (d, J=15.2 Hz, 1H), 7.92 (t, J=7.1 Hz, 1H), 7.86 (d, J=7.6 Hz, 1H), 7.81 (d, J=8.1 Hz, 1H), 7.65-7.62 (mult, 3H), 7.58 (t, J=7.6 Hz, 1H), 6.51 (d, J=15.7 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 166.7, 161.1, 150.6, 148.3, 147.2, 140.7, 136.7, 136.6, 135.0, 133.3, 131.6, 130.6, 130.5, 129.5, 127.4, 127.1, 126.5, 124.0, 122.8, 122.4, 120.7, 103.3. HRMS calcd for C23H15N3O5, 414.10911 [M+H]+; found, 414.10791 [M+H]+. Anal. (C23H15N3O5·0.25AcOH) C, H, N. MP > 260 °C.
(E)-2-(2-(4-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (9)
Compound 9 was prepared via procedure C with compound 9b (1.00 g, 3.57 mmol) and para-nitrobenzaldehyde (0.717 g, 4.75 mmol, 1.33 equiv) to give the title compound as a yellow solid (1.18 g, 80%). 1H NMR (400 MHz, DMSO-d6) δ 8.20-8.14 (mult, 4H), 7.96 (d, J= 15.9 Hz, 1H), 7.92-7.81 (mult, 3H), 7.74 (t, J= 7.6 Hz, 1H), 7.68 (d, J= 7.9 Hz, 2H), 7.61-7.56 (mult, 2H), 6.50 (d, J=15.6 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 165.7, 161.3, 151.0, 147.5, 147.3, 141.2, 136.6, 136.5, 134.9, 133.8, 131.8, 130.7, 130.0, 128.6, 127.4, 127.0, 126.5, 124.1, 124.0, 120.7. HRMS calcd for C23H15N3O5, 414.10911 [M+H]+; found, 414.10903 [M+H]+. Anal.(C23H15N3O5) C, H, N. MP > 260° C.
tert-Butyldimethylsilyl 4-(6-iodo-2-methyl-4-oxoquinazolin-3(4H)-yl)benzoate (10)
Carboxylic acid 78 (0.300 g, 0.74 mmol) was dissolved in THF (10.0 mL). To this solution was added N-methylmorpholine (0.081 mL, 0.74 mmol, 1.0 equiv) and TBDMSCl (0.111 g, 0.74 mmol, 1.0 equiv). The mixture was stirred at room temperature for 1 hour. The yellow suspension was concentrated in vacuo, and the resulting residue was partitioned between ethyl acetate and water. The mixture was extracted 3x with ethyl acetate. The combined organics were washed with brine, dried over MgSO4, and concentrated in vacuo to give a white solid. The crude material was purified using silica gel chromatography (ISCO, RediSep 12 g column, 100% EtOAc) to give a white foam (0.280 g, 73%). 1H NMR (400 MHz, CDCl3) δ 8.57 (d, J = 2.1 Hz, 1H), 8.23 (d, J = 8.8 Hz, 2H), 8.02 (dd, J1 = 8.7 Hz, J2 = 2.1 Hz, 1H), 7.41 (d, J = 8.6 Hz, 1H), 7.34 (d, J = 8.8 Hz, 2H), 2.22 (s, 3H), 1.05 (s, 9H), 0.41 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 165.5, 160.8, 154.3, 146.9, 143.7, 141.5, 135.9, 132.9, 132.1, 129.0, 128.4, 122.5, 91.3, 25.8, 24.6, 18.0, −4.6. HRMS calcd for C22H25IN2O3Si, 521.07585 [M+H]+; found, 521.07527 [M+H]+.
(E)-2-(2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (12)
Compound 12 was prepared via procedure C with compound 9b (0.250 g, 0.89 mmol) and meta-nitrobenzaldehyde (0.179 g, 1.19 mmol, 1.33 equiv) to give the title compound as a yellow solid (0.202 g, 55%). 1H NMR (400 MHz, DMSO-d6) δ 8.22-8.13 (mult, 4H), 7.99 (d, J=15.2 Hz, 1H), 7.93-7.80 (mult, 4H), 7.75 (t, J=7.3 Hz, 1H), 7.65-7.60 (mult, 2H), 7.57 (t, J=7.0 Hz, 1H), 6.47 (d, J=15.6 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 165.7, 161.3, 151.1, 148.3, 147.4, 136.6, 136.6, 136.6, 134.9, 133.8, 133.3, 131.8, 130.7, 130.5, 129.9, 129.4, 127.3, 126.8, 126.5, 124.0, 122.6, 122.1, 120.7. HRMS calcd for C23H15N3O5, 414.10911 [M+H]+; found, 414.10822 [M+H]+. Anal. (C23H15N3O5) C, H, N. MP > 260° C.
(E)-4-(5-chloro-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (36)
Compound 36 was prepared via procedure C using compound 36b (0.300 g, 0.95 mmol) and meta-nitrobenzaldehyde (0.192 g, 1.27 mmol, 1.33 equiv) to give the title compound as a bright yellow solid (0.251 g, 59%). 1H NMR (400 MHz, DMSO-d6) δ 13.31 (bs, 1H), 8.22 (s, 2H), 8.19-8.15 (mult, 2H), 7.99 (d, J=15.7 Hz, 1H), 7.83-7.78 (mult, 2H), 7.65 (d, J=8.6 Hz, 2H), 7.61 (t, J=7.8 Hz, 2H), 7.55 (d, J=7.4 Hz, 1H), 6.42 (d, J=15.7 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.7, 159.1, 151.2, 149.7, 148.2, 140.6, 137.3, 136.3, 134.7, 133.3, 132.9, 131.6, 130.7, 130.5, 129.5, 129.3, 126.9, 124.1, 122.4, 122.3, 117.6. HRMS calcd for C23H14ClN3O5, 448.07014 [M+H]+; found, 448.06970 [M+H]+. Anal.(C23H14ClN3O5) C, H, N. MP > 260 °C.
(E)-4-(6-chloro-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (37)
Compound 37 was prepared via procedure C using 37b (0.300 g, 0.950 mmol) and meta-nitrobenzaldehyde (0.192 g, 1.30 mmol, 1.33 equiv). The crude material was purified using recrystallization with methanol and dioxane. Further trituration with water yielded the title compound as a yellow solid (0.060 g, 14%). 1H NMR (400 MHz, DMSO-d6) δ 8.24 (s, 1H), 8.19-1.66 (mult, 3H), 8.05 (d, J=2.2 Hz, 1H), 8.01 (d, J=15.6 Hz, 1H), 7.94 (dd, J1=8.7 Hz, J2=2.2 Hz, 1H), 7.85-7.81 (mult, 2H), 7.66-7.61 (mult, 3H), 6.48 (d, J=15.6 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 167.4, 160.9, 151.8, 148.9, 146.6, 141.0, 137.9, 137.1, 135.7, 134.0, 133.0, 132.4, 131.9, 131.4, 131.8, 130.2, 130.0, 126.1, 124.8, 123.1, 122.6. HRMS calcd for C23H14ClN3O5, 446.05426 [M-H]+; found, 446.05511 [M-H]+. Anal.(C23H14ClN3O5·0.75 H2O) C, H, N. MP > 260 °C.
(E)-4-(7-chloro-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (38)
Compound 38 was prepared via procedure C using compound 38b (0.200 g, 0.640 mmol) and meta-nitrobenzaldehyde (0.128 g, 0.850 mmol, 1.33 equiv) to give the title compound as a yellow solid (0.135 g, 47%). 1H NMR (400 MHz, DMSO-d6) δ 8.24 (s, 1H), 8.18-8.13 (mult, 4H), 8.00 (d, J=15.6 Hz, 1H), 7.85 (d, J=8.3 Hz, 1H), 7.83 (d, J=1.9 Hz, 1H), 7.65 (d, J=8.6 Hz, 3H), 7.60 (dd, J1=8.3 Hz, J2=1.9 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.7, 160.6, 152.0, 148.3, 148.2, 140.4, 139.5, 137.6, 136.3, 133.4, 131.7, 130.7, 130.6, 130.5, 129.4, 128.6, 127.2, 126.3, 124.2, 122.4, 119.5 HRMS calcd for C23H14ClN3O5, 448.07014 [M+H]+; found, 448.06890 [M+H]+. Anal.( C23H14ClN3O5) C, H, N. MP > 260 °C.
(E)-4-(8-chloro-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (39)
Compound 39 was prepared via procedure C using compound 39b (0.400 g, 1.27 mmol) and meta-nitrobenzaldehyde (0.255 g, 1.69 mmol, 1.33 equiv) to yield the title compound as a yellow solid (0.071 g, 12%). 1H NMR (400 MHz, DMSO-d6) δ 13.24 (bs, 1H), 8.26 (s, 1H), 8.17 (d, J=8.2 Hz, 3H), 8.10-8.02 (mult, 3H), 7.86 (d, J=7.8 Hz, 1H), 7.67-7.62 (mult, 3H), 7.53 (t, J=7.8 Hz, 1H), 6.51 (d, J=15.6 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ1 67.3, 161.4, 151.9, 148.9, 144.3, 141.0, 138.4, 137.0, 135.5, 134.0, 132.3, 131.5, 131.4, 131.1, 130.0, 127.9, 126.2, 124.8, 123.2, 123.0. HRMS calcd for C23H14ClN3O5, 448.07014 [M+H]+; found 448.07015 [M+H]+. Anal.( C23H14ClN3O5) C, H, N. MP > 260 °C.
(E)-4-(6,8-dichloro-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (40)
Compound 40 was prepared via procedure C using 41b (0.250 g, 0.716 mmol) and meta-nitrobenzaldehyde (0.144 g, 0.952 mmol, 1.3 equiv) to give the title compound as a yellow solid, which was obtained by filtration (0.139 g, 40%). 1H NMR (400 MHz, DMSO-d6) δ 13.34 (bs, 1H), 8.29 (s, 1H), 8.24 (d, J=2.4 Hz, 1H), 8.16-8.20 (m, 3H), 8.08 (s, 1H), 8.04-8.05 (m, 1H) 7.88 (d, J=7.8 Hz, 1H), 7.62-7.78 (m, 3H), 6.51 (d, J=15.6 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.7, 159.9, 151.8, 148.3, 142.8, 140.2, 138.2, 136.3, 134.5, 133.4, 131.8, 130.8, 130.7, 130.6, 129.3, 124.7, 124.3, 123.2, 122.8, 122.4. HRMS calcd for C23H13Cl2N3O5, 482.03114 [M+H]+; found 482.03068 [M+H]+. Anal. (C23H13Cl2N3O5) C, H, N. MP > 260°C.
(E)-4-(6,8-dichloro-2-(4-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (41)
Compound 41 was prepared via procedure C using compound 41b (0.250 g, 0.716 mmol) and para-nitrobenzaldehyde (0.144 g, 0.952 mmol, 1.3 equiv) to yield the title compound as a yellow solid (0.050 g, 14%). 1H NMR (400 MHz, DMSO-d6) δ 13.34 (bs, 1H), 8.24 (d, J=2.7, 1H) 8.16-8.20 (mult, 4H), 8.05 (d, J=2.4, 1H), 8.02 (d, J=15.7, 1H), 7.71 (d, J=9.0, 2H), 7.65 (d, J=8.6, 2H), 6.53 (d, J=15.3, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.7, 159.9, 151.6, 147.7, 142.7, 140.8, 140.1, 138.0, 134.5, 132.4, 131.8, 130.9, 130.8, 129.3, 129.0, 124.7, 124.2, 123.7, 123.2. HRMS calcd for C23H13Cl2N3O5, 482.03114 [M+H]+; found, 482.0974 [M+H]+. Anal. (C23H13Cl2N3O5) C, H, N. MP > 260 °C.
(E)-4-(6-fluoro-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (42)
Compound 42 was prepared via procedure C using 42b (0.400 g, 1.34 mmol) and meta-nitrobenzaldehyde (0.270 g, 1.78 mmol, 1.33 equiv). The crude material was purified by recrystallization with methanol and dioxane and further purification by trituration with methanol and ethyl acetate to remove residual dioxane to yield the title compound as a pale yellow solid (0.268 h, 43%). 1H NMR (600 MHz, DMSO-d6) δ 8.24 (s, 1H), 8.18-8.16 (mult, 3H), 7.99 (d, J=15.5 Hz, 1H), 7.89-7.80 (mult, 4H), 7.66-7.62 (mult, 3H), 6.49 (d, J=15.7, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.7, 160.5, 159.8 (d, J=245 Hz), 150.2, 148.3, 144.1, 140.5,136.8, 136.5, 133.3, 131.6, 130.7, 130.5, 130.3, 130.0, 129.4, 124.0, 123.3 (d, J=24 Hz), 122.4 (d, J=15 Hz) 121.9 (d, J=8.4 Hz), 111.2 (d, J=24 Hz). HRMS calcd for C23H14FN3O5, 432.09969 [M+H]+; found, 432.09850 [M+H]+. Anal.(C23H14FN3O5) C, H, N. MP > 260 °C.
(E)-4-(6-bromo-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (43)
Compound 43 was prepared via procedure C using compound 43b (0.300 g, 0.840 mmol) and meta-nitrobenzaldehyde (0.168 g, 1.10 mmol, 1.33 equiv). The crude material was recrystallized using methanol and dioxane and further purified by hot trituration with methanol and ethyl acetate to remove residual dioxane to yield the title compound as a yellow solid (0.120 g, 30%). 1H NMR (300 MHz, DMSO-d6) δ 8.22 (s, 1H), 8.19-8.14 (mult, 4H), 8.04 (dd, J1= 8.5 Hz, J2=2.3 Hz, 1H), 8.00 (d, J=2.3 Hz, 1H), 7.82 (d, J=7.6 Hz, 1H), 7.73 (d, J=8.5 Hz, 1H), 7.64-7.59 (mult, 3H), 6.46 (d, J=15.5 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.8, 160.0, 151.1, 148.2, 146.1, 140.0, 137.7, 137.2, 136.4, 133.2, 132.5, 130.7, 130.5, 129.6, 129.2, 128.5, 124.1, 122.4, 122.2, 119.3. HRMS calcd for C23H14BrN3O5, 492.01962 [M+H]+; found, 492.00228 [M+H]+. Anal. (C23H14BrN3O5) C, H, N. MP > 260 °C.
(E)-4-(6-methyl-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (45)
Compound 45 was prepared via procedure C using compound 45b (0.507 g, 1.72 mmol) and meta-nitrobenzaldehyde (0.346 g, 2.29 mmol, 1.33 equiv) to yield the title compound as a yellow solid (0.498 g, 68%). 1H NMR (400 MHz, DMSO-d6) δ 13.34 (bs, 1H), 8.22 (s, 1H), 8.18-8.14 (mult, 3H), 7.96 (d, J=15.6 Hz, 1H), 7.93 (s, 1H), 7.83 (d, J=7.8 Hz, 1H), 7.73-7.77 (mult, 2H), 7.64-7.60 (mult, 3H), 6.47 (d, J=15.6 Hz, 1H), 2.48 (s, 3H). 13C (100 MHz, DMSO-d6) δ 166.7, 161.0, 149.8, 148.2, 145.2, 140.8, 136.9, 136.6, 136.3, 133.3, 131.6, 130.7, 130.5, 129.5, 127.2, 125.8, 123.9, 122.8, 122.3, 120.4, 20.9. HRMS calcd for C24H17N3O5, 428.12476 [M+H]+; found 428.12422 [M+H]+. Anal.(C24H17N3O5) C, H, N. MP > 260 °C.
(E)-4-(6-methoxy-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (46)
Compound 46 was prepared via procedure C using compound 46b (0.120 g, 0.390 mmol) and meta-nitrobenzaldehyde (0.078 g, 0.51 mmol, 1.3 equiv) to yield the title compound as a yellow solid (0.094 g, 55%). 1H NMR (400 MHz, DMSO-d6) δ 8.24 (s, 1H), 8.16 (d, J=8.3 Hz, 3H), 7.94 (d, J=15.6 Hz, 1H), 7.84 (d, J=7.9 Hz, 2H), 7.62 (d, J=9.8 Hz, 1H), 7.65-7.61 (mult, 3H), 7.54-7.53 (mult, 2H), 6.49 (d, J=15.6 Hz, 1H), 3.83 (s, 3H). 13C NMR (100 MHz, DMSO-d6).δ167.7, 161.5, 158.8, 149.2, 148.9, 142.5, 140.9, 137.4, 136.4, 133.8, 133.7, 131.2, 129.9, 129.8, 129.7, 125.2, 124.5, 123.4, 122.9, 122.2, 56.4. HRMS calcd for C24H17N3O6, 444.11968 [M+H]+; found, 444.11849 [M+H]+. Anal.(C24H17N3O6 ·0.50 H2O) C, H, N. MP > 260 °C.
(E)-4-(6-nitro-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (47)
Compound 47 was prepared via procedure C using compound 47b (0.250 g, 0.769 mmol) and 3-nitrobenzaldehyde (0.154 g, 1.33 equiv, 1.02 mmol). The crude material was purified by recrystallization with ethyl acetate and hexanes to give a yellow solid (0.321 g, 91%). 1H NMR (400 MHz, DMSO-d6) δ 13.35 (bs, 1H), 8.84 (d, J=3.0 Hz, 1H), 8.65 (dd, J1=9.0 Hz, J2=2.6 Hz, 1H), 8.32 (s, 1H), 8.15-8.21 (mult, 4H), 7.98 (d, J=9.0 Hz, 1H), 7.91 (d, J=8.1 Hz, 1H), 7.63-7.77 (mult, 3H), 6.54 (d, J=15.4 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.7, 160.6, 153.9, 151.5, 148.3, 144.8, 140.0, 139.1, 136.2, 133.6, 131.9, 130.8, 130.6, 129.3, 129.0, 128.9, 124.5, 122.9, 122.6, 122.2, 120.8. HRMS calcd for C23H14N4O7, 459.09414 [M+H]+; found 459.09363 [M+H]+. Anal.(C23H14N4O7) C, H, N. MP > 260 °C.
(E)-4-(6-hydroxy-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (48)
Compound 48 was prepared via procedure C using compound 48b (0.262 g, 0.770 mmol) and meta-nitrobenzaldehyde (0.156 g, 1.0 mmol, 1.33 equiv). The crude material was purified by recrystallization with methanol and dioxane, and further purified by hot gravity filtration after trituration with ethyl acetate and methanol (0.095 g, 29%). 1H NMR (400 MHz, DMSO-d6) δ 8.23 (s, 1H), 8.15 (d, J=8.6 Hz, 3H), 7.90 (d, J=15.6 Hz, 1H), 7.83 (d, J=7.8 Hz, 1H), 7.69 (d, J=9.0 Hz, 1H), 7.62-7.59 (mult, 3H), 7.45 (d, J=3.1 Hz, 1H), 7.36 (dd, J1=8.6 Hz, J2=2.7 Hz, 1H), 6.47 (d, H=15.7 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.7, 160.8, 156.7, 148.3, 147.6, 140.9, 140.4, 136.8, 135.3, 133.2, 131.4, 130.6, 130.5, 129.5, 129.2, 124.5, 123.7, 122.8, 122.2, 121.7, 109.4. HRMS calcd for C23H15N3O6, 430.10404 [M+H]+; found, 430.10455 [M+H]+ Anal. (C23H15N3O6·0.25 H2O) C, H, N. MP > 260 °C.
(E)-4-(2-(3-nitrostyryl)-4-oxo-6-phenylquinazolin-3(4H)-yl)benzoic acid (49)
Compound 49 was prepared via procedure C using 49b (0.200 g, 0.560 mmol) and meta-nitrobenzaldehyde (0.113 g, 0.750 g, 1.33 equiv) to yield the title compound as a yellow solid (0.210 g, 76%). 1H NMR (400 MHz, DMSO-d6) δ 8.35 (d, J=2.0 Hz, 1H), 8.26-8.25 (mult, 1H), 8.23 (d, J=2.3 Hz, 1H), 8.19-8.15 (mult, 3H), 8.03 (d, J=15.7 Hz, 1H), 7.89 (d, J=8.2 Hz, 2H), 7.86 (d, J=7.8 Hz, 2H), 7.67-7.65 (mult, 3H), 7.53 (t, J=7.4 Hz, 2H), 7.43 (t, J=7.0 Hz, 1H), 6.51 (d, J=15.7 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.8, 161.1, 150.6, 148.2, 146.5, 140.4, 138.7, 138.6, 136.8, 136.5, 133.4, 133.3, 132.2, 130.7, 130.5, 129.4, 129.2, 128.1, 126.8, 124.0, 123.7, 122.7, 122.4, 121.0. HRMS calcd for C29H19N3O5, 490.14044 [M+H]+; found, 490.13965 [M+H]+. Anal.(C29H19N3O5) C, H, N. MP > 260 °C.
(E)-4-(2-(3-nitrostyryl)-4-oxobenzo[g]quinazolin-3(4H)-yl)benzoic acid (50)
Compound 50 was prepared via procedure C using compound 50b (0.250 g, 0.760 mmol) and meta-nitrobenzaldehyde (0.152 g, 1.01 mmol, 1.33 equiv). The crude material was purified by hot gravity filtration with methanol (0.194 g, 55%). 1H NMR (400 MHz, DMSO-d6) δ 13.31 (bs, 1H), 8.87 (s, 1H), 8.28 (s, 1H), 8.25 (mult, 2H), 8.20-8.16 (mult, 4H), 8.02 (d, J=15.7 Hz, 1H), 7.86 (d, J=7.8 Hz, 1H), 7.33-7.59 (mult, 5H), 6.52 (d, J=15.7 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.8, 161.6, 149.6, 148.3, 142.6, 140.9, 136.7, 136.5, 133.3, 131.5, 131.2, 130.7, 130.5, 129.7, 129.4, 128.8, 128.0, 126.6, 124.9, 124.0, 123.0, 122.3, 119.8. HRMS calcd for C27H17N3O5, 464.12474 [M+H]+ ; found, 464.125661 [M+H]+. Anal. (C27H17N3O5) C, H, N. MP > 260 °C.
(E)-4-(2-(3-nitrostyryl)-4-oxo-7,8-dihydro-[1,4]dioxino[2,3-g]quinazolin-3(4H)-yl)benzoic acid (51)
Compound 51 was prepared via procedure C using compound 51b (0.150 g, 0.44 mmol) and meta-nitrobenzaldehyde (0.089 g, 0.59 mmol, 1.33 equiv). The crude material was purified by hot gravity filtration with methanol to give a yellow solid (0.056 g, 27%). 1H NMR (400 MHz, DMSO-d6) δ 8.19 (s, 1H), 8.14 (d, J = 8.2 Hz, 1H), 8.03 (d, J = 8.2 Hz, 2H), 7.89 (d, J = 15.7 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.64 (t, J = 7.8 Hz, 1H), 7.51 (s, 1H), 7.26 (d, J = 8.2 Hz, 2H), 7.21 (s, 1H), 6.49 (d, J = 15.7 Hz, 1H), 4.44-4.38 (mult, 4H). 13C NMR (100 MHz, DMSO-d6) δ 168.1, 160.3, 149.8, 149.7, 148.3, 143.7, 142.7, 142.0, 136.8, 136.7, 135.5, 132.9, 130.6, 130.1, 127.6, 123.8, 123.0, 122.1, 115.0, 114.2, 113.4, 112.6, 64.4, 64.3. HRMS calcd for C25H17N3O7, 472.11459 [M+H]+; found, 472.11429 [M+H]+. Anal.(C25H17N3O7) C, H, N. MP > 260 C.
(E)-4-(6-(3-nitrostyryl)-8-oxo-[1,3]dioxolo[4,5-g]quinazolin-7(8H)-yl)benzoic acid (52)
Compound 52 was prepared via procedure C using compound 52b (0.100 g, 0.31 mmol) and meta-nitrobenzaldehyde (0.062 g, 0.41 mmol, 1.33 equiv). The crude material was purified by hot gravity filtration with methanol to give a yellow solid (0.048 g, 34%). 1H NMR (400 MHz, d6-DMSO) δ 8.21 (s, 1H), 8.15 (d, J = 8.6 Hz, 1H), 8.00 (d, J = 8.3 Hz, 2H), 7.91 (d, J = 15.6 Hz, 1H), 7.76 (d, J = 7.0 Hz, 1H), 7.65 (t, J = 7.9 Hz, 1H), 7.46 (s, 1H), 7.28 (d, J = 8.3 Hz, 2H), 7.24 (s, 1H), 6.49 (d, J = 15.6 Hz, 1H), 6.27 (s, 2H). HRMS calcd for C24H15N3O7, 458.09894 [M+H]+; found, 458.09772 [M+H]+. Anal.(C24H15N3O7) C, H, N. MP > 260°C.
(E)-4-(6-isopropyl-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (53)
Compound 53 was prepared via procedure C using 53b (0.150 g, 0.47 mmol) and meta-nitrobenzaldehyde (0.094 g, 0.62 mmol, 1.3 equiv) to yield the title compound as a yellow solid (0.137 g, 65%). 1H NMR (400 MHz, DMSOd6) δ 13.34 (bs, 1H), 8.26 (d, 1H), 8.16 (d, J=8.6 Hz, 2H), 8.00 (d, J=15.7 Hz, 1H), 7.99 (d, J=2.0 Hz, 1H), 7.87-7.83 (mult, 3H), 7.75 (d, J=8.2 Hz, 1H), 7.66-7.61 (mult, 3H), 6.49 (d, J=15.7 Hz, 1H), 3.11 (sept, J=7.0 Hz, 1H), 1.29 (d, J=7.0 Hz, 6H). 13C NMR (100 MHz, DMSO-d6) δ 166.8, 161.1, 149.9, 148.3, 147.6, 145.6, 140.7, 136.6, 136.3, 133.8, 133.3, 131.8, 130.7, 130.5, 129.4, 127.5, 123.9, 123.1, 122.7, 122.3, 120.4, 33.3, 23.7. HRMS calcd for C26H21N3O5, 456.15604 [M+H]+; found, 456.15557 [M+H]+. Anal. (C26H21N3O5) C, H, N. MP > 260 °C.
(E)-4-(2-(3-nitrostyryl)-4-oxo-6-propylquinazolin-3(4H)-yl)benzoic acid (54)
Compound 54 was prepared via procedure C using compound 54b (0.578 g, 1.79 mmol) and meta-nitrobenzaldehyde (0.360 g, 2.38 mmol). The crude material was purified by silica gel chromatography (ISCO, RediSep 24 g column, silica cake, 0-10% MeOH/DCM gradient) to give a yellow solid (0.086 g, 11%). 1H NMR (400 MHz, DMSO-d6) δ 8.26 (s, 1H), 8.16 (d, J=8.6 Hz, 3H), 7.96 (d, J=15.6, 2H), 7.85 (d, J=7.6 Hz, 1H), 7.79-7.73 (mult, 3H), 7.64 (t, J=7.9 Hz, 3H), 6.50 (d, J=15.6 Hz, 1H), 2.75 (t, J=7.3 Hz, 2H), 1.67 (sext, J=7.3 Hz, 2H), 0.93 (t, J=7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.8, 161.1, 149.9, 148.3, 145.5, 141.5, 140.8, 136.6, 136.3, 135.7, 133.3, 131.6, 130.7, 130.5, 129.5, 127.3, 125.4, 123.9, 122.8, 122.3, 120.4, 36.8, 24.1, 13.5. HRMS calcd for C26H21N3o5, 456.15604 [M+H]+; found, 456.15568 [M+H]+. Anal.(C26H21N3O5) C, H, N. MP > 260 °C.
(E)-4-(2-(3-nitrostyryl)-4-oxo-6-(thiophen-2-yl)quinazolin-3(4H)-yl)benzoic acid (55)
Compound 55a (0.077 g, 0.212 mmol) was dissolved in a mixture of acetic acid (0.71 mL, 12.3 mmol, 58 equiv) and acetic anhydride (0.14 mL, 1.4 mmol, 7.0 equiv). To this solution was added meta-nitrobenzaldehyde (0.043 g, 0.283 mmol, 1.33 equiv) and sodium acetate (0.028 g, 0.35 mmol, 1.63 equiv). The mixture was refluxed for 6 hours. After cooling to room temperature, the resultant yellow solid was collected by filtration and washed with methanol. The crude material was purified using silica gel chromatography (silica cake, ISCO, RediSep 24 g column, 0-10% MeOH/DCM gradient) to give a yellow solid (0.049 g, 48%). 1H NMR (400 MHz, d6-DMSO) δ 13.35 (bs, 1H), 8.31-8.25 (mult, 3H), 8.17 (d, J = 8.2 Hz, 3H), 8.03 (d, J = 15.7 Hz, 1H), 7.87 (t, J = 7.4 Hz, 2H), 7.74 (dd, J1 = 3.5 Hz, J2=1.2 Hz, 1H), 7.68-7.63 (mult, 4H), 7.21 (dd, J1 = 5.1 Hz, J2 = 3.51 Hz, 1H), 6.51 (d, J = 15.7 Hz, 1H). 13C NMR (150 MHz, d6-DMSO) δ 166.7, 160.9, 150.5, 148.3, 146.4, 141.9, 140.6, 136.8, 136.5, 131.3, 132.3, 131.9, 131.7, 130.7, 130.5, 129.4, 129.0, 128.3,126.8, 124.9, 124.0, 122.6, 122.4, 121.9, 121.2. HRMS calcd for C27H17N3O5S, 496.09683 [M+H]+; found, 496.10655 [M+H]+. Anal. (C27H17N3O5S) C, H, N. MP > 260°C.
4-(2-(3-Nitrostyryl)-4-oxo-6-styrylquinazolin-3(4H)-yl)benzoic acid (56)
Compound 56a (0.219 g, 0.57 mmol) was dissolved in a mixture of acetic acid (1.9 mL, 33 mmol, 58 equiv) and acetic anhydride (0.38 mL, 4.0 mmol, 7.0 equiv). To this solution was added meta-nitrobenzaldehyde (0.115 g, 0.76 mmol, 1.33 equiv) and sodium acetate (0.077 g, 0.93 mmol, 1.63 equiv). The mixture was refluxed for 6 hours. The mixture was cooled to room temperature and the resulting yellow solid was collected by filtration and washed with methanol. The crude material was purified using silica gel chromatography (silica cake, ISCO, RediSep 24 g column, 0-10% MeOH/DCM gradient) to give a yellow solid (0.146 g, 50%). 1H NMR (400 MHz, CDCl3) δ 8.27 (s, 1H), 8.17 (d, J=7.8 Hz, 1H), 8.11 (d, J=8.2 Hz, 2H), 8.03 (d, J=15.3 Hz, 1H), 7.97 (s, 1H), 7.85-7.82 (mult, 2H), 7.65 (t, J=8.2 Hz, 2H), 7.52 (d, J=8.6 Hz, 2H), 7.44-7.24 (mult, 3H), 7.37-7.35 (mult, 2H), 6.52 (d, J=15.6 Hz, 1H). HRMS calcd for C31H21N3O5, 516.15606 [M+H]+; found, 516.14182 [M+H]+. Anal. (C31H21N3O5) C, H, N. MP > 260 °C
(E)-4-(6-methoxy-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzonitrile (58)
Compound 58 was prepared via procedure C using compound 58a (0.150 g, 0.520 mmol) and meta-nitrobenzaldehyde (0.103 g, 0.690 mmol, 1.33 equiv). The crude material was purified by recrystallization with hexanes and chloroform to yield the title compound as a bright yellow solid (0.112 g, 51%). 1H NMR (400 MHz, CDCl3) δ 8.19-8.17 (mult, 2H), 8.00 (d, J=15.7 Hz, 1H), 7.93 (d, J=8.6 Hz, 2H), 7.76 (d, J=9.0 Hz, 1H), 7.66 (d, J=2.7 Hz, 1H), 7.63-7.61 (mult, 1H), 7.54 (d, J=8.6 Hz, 1H), 7.51 (d, J=8.6 Hz, 2H), 7.45 (dd, J1=9.0 Hz, 1H), 3.95 (s, 3H). 13C NMR (400 MHz, CDCl3) δ 161.8, 159.3, 148.9, 147.2, 142.1, 141.2, 137.3, 137.0, 134.1, 132.9, 130.3, 130.1, 129.6, 125.7, 124.3, 122.8, 122.1, 121.8, 118.0, 114.0, 106.9, 56.1. HRMS calcd for C24H16N4O4, 425.12514 [M+H]+; found, 425.12473 [M+H]+. Anal.(C24H16N4O4) C, H, N. MP > 260 °C
(E)-4-(6-methoxy-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzamide (59)
Compound 59 was prepared via procedure C using compound 59a (0.200 g, 0.650 mmol) and meta-nitrobenzaldehyde (0.130 g, 0.860 mmol, 1.33 equiv). The crude material was purified by hot gravity filtration after boiling with methanol and collection of the title compound as the resulting yellow solid (0.162 g, 57%). 1H NMR (400 MHz, DMSO-d6) δ 8.24 (s, 1H), 8.16 (d, J=8.6 Hz, 2H), 8.09 (d, J=8.6 Hz, 2H), 7.94 (d, J=15.7 Hz, 1H), 7.83 (d, J=7.8 Hz, 1H), 7.77 (d, J=9.8 Hz, 1H), 7.64 (t, J=7.8 Hz, 1H), 7.58 (d, J=8.6 Hz, 2H), 7.54-7.51 (mult, 3H), 6.49 (d, J=15.7 Hz, 1H), 3.91 (s, 3H). 13C NMR (100 MHz, , DMSO-d6) δ 167.1, 160.9, 158.1, 148.6, 148.3, 141.7, 139.4, 136.7, 135.7, 134.9, 133.1, 130.5, 129.2, 129.0, 128.9, 124.5, 123.8, 122.8, 122.2, 121.6, 55.8. HRMS calcd for C24H18N4O5, 443.13564; found, 443.13529 [M+H]+. Anal.( C24H18N4O5) C, H, N. MP > 260 °C.
(E)-methyl 4-(6-methoxy-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoate (60)
Compound 60 was prepared via procedure C using compound 60a (0.200 g, 0.650 mmol) and meta-nitrobenzaldehyde (0.124 g, 0.860 mmol, 1.33 equiv) to yield the title compound as a yellow solid (0.143 g, 51%). 1H NMR (400 MHz, DMSO-d6) δ 8.29 (d, J=8.2 Hz, 2H), 8.15 (d, J=6.7 Hz, 2H), 7.96 (d, J=15.7 Hz, 1H), 7.76 (d, J=9.0 Hz, 1H), 7.67 (d, J=2.7 Hz, 1H), 7.59 (d, J=7.8 Hz, 1H), 7.50 (t, J=7.8 Hz, 1H), 7.45-7.43 (mult, 3H), 6.42 (d, J=15.7 Hz, 1H), 4.01 (s, 3H), 3.95 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.3, 161.9, 159.1, 148.8, 148.3, 142.2, 141.1, 137.2, 136.8, 132.9, 131.6, 131.5, 130.0, 129.5, 129.2, 125.5, 124.1, 122.7, 122.6, 121.9, 106.8, 56.4, 53.1. HRMS calcd for C25H19N3O6, 458.13534 [M+H]+; found, 458.13473 [M+H]+. Anal. (C25H19N3O6) C, H, N. MP > 260 °C.
(E)-4-(6-methoxy-2-(3-methoxystyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (61)
Compound 61 was prepared via procedure C using compound 46b (0.200 g, 0.650 mmol) and meta-anisaldehyde (0.104 mL, 0.860 mmol, 1.33 equiv) to yield the title compound as a yellow solid (0.150 g, 54%). 1H NMR (300 MHz, DMSO-d6) δ 13.32 (bs, 1H), 8.16 (d, J=8.5 Hz, 2H), 7.78 (d, J=15.2 Hz, 1H), 7.75 (d, J=9.4 Hz, 1H), 7.52-7.49 (mult, 2H), 7.26 (t, J=7.9 Hz, 1H), 6.92 (d, J=8.5 Hz, 3H), 6.28 (d, J=15.5 Hz, 1H), 3.90 (s, 3H), 3.72 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.7, 160.9, 159.5 157.7, 148.8, 141.9, 141.0, 138.0, 136.3, 131.6, 130.6, 130.1, 129.5, 129.0, 124.5, 121.3, 120.2, 119.1, 115.1, 113.5, 106.5, 55.7, 55.1. HRMS calcd for C25H20N2O5, 429.14514 [M+H]+; found, 429.14404 [M+H]+. Anal. (C25H20N2O5) C, H, N. MP > 260 °C.
(E)-3-(2-(3-(4-carboxyphenyl)-6-methoxy-4-oxo-3,4-dihydroquinazolin-2-yl)vinyl)benzoic acid (62)
Compound 62 was prepared via procedure C using compound 46b (0.200 g, 0.650 mmol) and meta-formylbenzoic acid (0.129 g, 0.860 mmol, 1.33 equiv) to yield the title compound as a yellow solid (0.259 g, 45%). 1H NMR (400 MHz, DMSO-d6) δ 8.16 (d, J=8.6 Hz, 2H), 7.89 (mult, 3H), 7.76 (d, J=9.5 Hz, 1H), 7.63 (d, J=8.6 Hz, 2H), 7.53-7.50 (mult, 2H), 7.58 (t, J=7.9 Hz, 1H), 6.37 (d, J=15.6 Hz, 1H), 3.90 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.8, 166.7, 160.9, 158.1, 148.7, 141.8, 141.0, 137.2, 135.3, 131.52, 131.49, 130.7, 130.2, 129.5, 129.1, 128.1, 124.5, 121.4, 120.9, 106.5, 55.8. HRMS calcd for C25H18N2O6, 443.12444 [M+H]+; found, 443.12393 [M+H]+. Anal.(C25H18N2O6) C, H, N. MP > 260 °C.
(E)-4-(6,7-dimethoxy-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (63)
Compound 63 was prepared via procedure C using compound 63b (0.200 g, 0.59 mmol) and meta-nitrobenzaldehyde (0.118 g, 0.780 mmol, 1.33 equiv). The crude material was purified by hot gravity filtration after boiling with methanol to yield the title compound as a yellow solid (0.165 g, 59%). 1H NMR (400 MHz, DMSO-d6) δ 13.31 (bs, 1H), 8.18-8.14 (mult, 4H), 7.94 (d, J=15.2 Hz, 1H), 7.81 (d, J=7.8 Hz, 1H), 7.64-7.60 (mult, 3H), 7.42 (s, 1H), 7.26 (s, 1H), 6.45 (d, J=15.2 Hz, 1H), 3.97 (s, 3H), 3.88 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 172.1, 167.3, 160.3, 155.0, 149.4, 149.0, 148.3, 143.3, 136.7, 135.6, 133.0, 130.5, 130.3, 123.8, 122.9, 122.1, 114.2, 107.8, 105.8, 56.1, 55.8. HRMS calcd for C25H19N3O7, 474.13024 [M+H]+; found, 474.13077 [M+H]+. Anal.(C25H19N3O7·0.25 H2O) C, H, N. MP > 260 °C.
(E)-4-(2-(3,4-diacetoxystyryl)-6-methoxy-4-oxoquinazolin-3(4H)-yl)benzoic acid (64)
Compound 64 was prepared via procedure C using 46b (0.400 g, 1.29 mmol) and 3,4-dihydroxybenzaldehyde (0.237 g, 1.71 g, 1.33 equiv). The crude material was purified by silica gel chromatography (ISCO, RediSep 40 g column, silica cake, 0-10% MeOH/DCM gradient) to yield the title compound as a yellow solid (0.221 g, 33%). 1H NMR (400 MHz, DMSO-d6) δ 8.15 (d, J=8.3 Hz, 2H), 7.91 (d, J=15.6 Hz, 1H), 7.76 (d, J=7.6 Hz, 1H), 7.60 (d, J=8.3 Hz, 2H), 7.52 (d, J=7.3 Hz, 2H), 7.25 (d, J=8.3 Hz, 1H), 6.32 (d, J=15.6 Hz, 1H), 3.90 (s, 3H), 2.27 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 168.81, 168.76, 161.5, 158.6, 149.4, 143.3, 143.0, 142.5, 141.5, 137.2, 134.4, 132.3, 131.3, 130.0, 129.8, 126.3, 125.1, 124.9, 123.4, 123.3, 122.0, 121.6, 107.2, 56.4, 21.02, 20.98. HRMS calcd for C28H22N2O8, 515.1510 [M]+; found, 515.14526 [M]+. Anal.(C28H22N2O8) C, H, N. MP 247-252°C.
(E)-4-(6-iodo-2-(4-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (72)
Compound 72 was prepared via procedure C using compound 72b (0.200 g, 0.490 mmol) and para-nitrobenzaldehyde (0.099 g, 0.65 mmol, 1.3 equiv) to yield the title compound as a yellow solid (0.174 g, 66%). 1H NMR (400 MHz, DMSOd6) δ 8.32 (s, 1H), 8.16-8.12 (mult, 4H), 8.05 (s, 1H), 7.98 (d, J=15.6 Hz, 1H), 7.77-7.68 (mult, 3H), 7.61-7.54 (mult, 2H), 6.41 (d, J=15.6 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 166.6, 159.9, 151.4, 148.2, 146.4, 143.1, 137.0, 136.5, 136.4, 134.6, 133.3, 130.5, 130.1, 129.8, 129.3, 124.0, 122.5, 122.4, 122.1, 91.9.HRMS calcd for C23H14IN3O5, 540.00574 [M+H]+; found, 540.00559 [M+H]+. Anal. (C23H14IN3O5) C, H, N. MP > 260 °C.
(E)-3-(6-iodo-2-(4-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (73)
Compound 73 was prepared via procedure C using compound 73b (0.800 g, 1.97 mmol) and para-nitrobenzaldehyde (0.396 g, 2.62 mmol, 1.33 equiv) to yield the title compound as a yellow solid (0.572 g, 54%). 1H NMR (400 MHz, DMSO-d6) δ 13.29 (bs, 1H), 8.32 (mult, 1H), 8.16-8.09 (mult, 5H), 7.94 (d, J=15.3 Hz, 1H), 7.77 (s, 2H), 7.65-7.54 (mult, 3H), 6.47 (d, J=15.3 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 166.8, 158.7, 151.6, 148.7, 147.2, 142.8, 136.7, 136.6, 135.9, 134.3, 133.2, 130.8, 130.6, 129.4, 128.9, 128.8, 123.9, 123.5, 123.4, 122.1, 92.2 HRMS calcd for C23H14IN3O5, 540.00574 [M+H]+.; found, 540.00548 [M+H]+. Anal. (C23H14IN3O5) C, H, N. MP > 260 °C.
(E)-3-(6-iodo-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (74)
Compound 74 was prepared via procedure C using compound 73b (0.800 g, 1.97 mmol) and meta-nitrobenzaldehyde (0.396 g, 2.62 mmol, 1.33 equiv) to yield the title compound as a yellow solid (0.626 g, 59%). 1H NMR (400 MHz, DMSO-d6) δ 8.32 (s, 1H), 8.16-8.12 (mult, 4H), 8.05 (s, 1H), 7.98 (d, J=15.6 Hz, 1H), 7.77-7.68 (mult, 3H), 7.61-7.54 (mult, 2H), 6.41 (d, J=15.6 Hz, 1H).13C NMR (150 MHz, DMSO-d6) δ 166.6, 159.9, 151.4, 148.2, 146.4, 143.1, 137.0, 136.5, 136.4, 134.6, 133.3, 130.5, 130.1, 129.8, 129.3, 124.0, 122.5, 122.4, 122.1, 91.9.HRMS calcd for C23H14IN3O5, 540.00574 [M+H]+; found, 540.00546 [M+H]+. Anal. (C23H14IN3O5), C, H, N. MP > 260 °C.
(E)-4-(6-methoxy-2-(4-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (75)
Compound 75 was prepared via procedure C using compound 46b (0.730 g, 2.35 mmol) and para-nitrobenzaldehyde (0.473 g, 3.13 mmol, 1.3 equiv) to yield the title compound as a yellow solid (0.559 g, 54%). 1H NMR (400 MHz, DMSO-d6) δ 13.32 (bs, 1H), 8.17 (t, J=8.8 Hz, 4H), 7.92 (d, J=15.3, 1H), 7.78 (dd, J1=10 Hz, J2=4 Hz, 1H), 7.68 (d, J=8.9 Hz, 1H), 7.63 (d, J=8.2 Hz, 1H) 7.58-7.52 (mult, 2H), 6.51 (d, J=15.7 Hz, 1H), 3.90 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.7, 158.4, 148.3, 147.4, 141.7, 141.3, 140.7, 135.7, 130.7, 129.4, 128.6, 124.5, 124.2, 106.6, 55.8. HRMS calc for C24H17N2O6, 444.11964 [M+H]+; found, 444.11978 [M+H]+. Anal.(C24H17N2O6) C, H, N. MP > 260 °C.
(E)-3-(6-methoxy-2-(4-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (76)
Compound 76 was prepared via procedure C using compound 76a (0.730 g, 2.35 mmol) and para-nitrobenzaldehyde (0.473 g, 3.13 mmol, 1.3 equiv) to yield the title compound as a yellow solid (0.728g, 70%). 1H NMR (400 MHz, DMSO-d6) δ 13.34 (bs, 1H), 8.13-8.18 (mult, 3H), 8.04 (s, 1H), 7.91 (d, J=15.7 Hz, 1H), 7.74-7.79 (mult, 3H), 7.65 (d, J=9.0 Hz, 2H), 7.51-7.54 (mult, 2H), 6.50 (d, J=15.7 Hz, 1H), 3.90 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.5, 161, 158.2, 148.6, 147.4, 141.7, 141.4, 137.1, 135.5, 133.4, 132.5, 130.1, 130, 129.2, 128.5, 124.4, 124.2, 124.2, 121.7, 106.6, 55.8. HRMS calcd for C24H17N2O6, 444.11964 [M+H]+; found, 444.11978 [M+H]+. Anal.(C24H17N2O6) C, H, N. MP > 260 °C.
(E)-3-(6-methoxy-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid (77)
Compound 77 was prepared via procedure C using compound 76a (0.194 g, 0.626 mmol) and meta-nitrobenzaldehyde (0.126 g, 0.833 mmol, 1.3 equiv.) to yield the title compound as a yellow solid (0.087g, 33%). 1H NMR (400 MHz, DMSO-d6) δ 13.32 (bs, 1H), 8.21 (s, 1H), 8.13-8.15 (mult, 2H), 8.03 (s, 1H), 7.92 (d, J=15.7 Hz, 1H), 7.82 (d, J=7.8 Hz, 1H), 7.73-7.77 (mult, 3H), 7.61 (t, J=8.0 Hz, 1H), 7.51-7.53 (mult, 2H), 6.47 (d, J=15.7 Hz, 1H), 3.89 (s, 3H). (100 MHz, DMSO-d6) δ 166.6, 161.0, 158.1, 148.7, 148.3, 141.7, 137.2, 136.8, 135.7, 133.5, 133.2, 132.4, 130.5, 130.1, 130, 129.2, 124.4, 123.8, 122.9, 122.1, 121.6, 106.6, 55.8. HRMS calcd for C24H17N2O6, 444.11964 [M+H]+; found, 444.11957 [M+H]+. Anal.(C24H17N2O6) C, H, N. MP > 260 °C.
4-(6-Iodo-2-methyl-4-oxoquinazolin-3(4H)-yl)benzoic acid (78)
Compound 78 was prepared via procedure B using compound 78a (2.00 g, 7.00 mmol) and para-aminobenzoic acid (1.15 g, 8.40 mmol, 1.20 equiv). The crude material was recrystallized with ethyl acetate and methanol to give an off-white solid (1.27 g, 45%). 1H NMR (400 MHz, DMSO-d6) δ 13.27 (bs, 1H), 8.36 (d, J=2.0 Hz, 1H), 8.15-8.11 (mult, 3H), 7.61 (d, J=8.5 Hz, 2H), 7.47 (d, J=8.5 Hz, 1H), 2.11 (s, 3H).13C NMR (100 MHz, DMSO-d6) δ 166.7, 160.0, 1547, 146.6, 143.0, 141.5, 134.5, 131.5, 130.7, 128.9, 122.3, 91.3, 24.2. HRMS calcd for C16H11IN2O3, 406.98934 [M+H]+; found, 406.98893 [M+H]+.
(E)-4-(6-Iodo-4-oxo-2-styrylquinazolin-3(4H)-yl)benzoic acid (79)
Compound 79 was prepared via procedure C using compound 45b (0.300 g, 0.740 mol) and benzaldehyde (0.100 mL, 0.980 mmol, 1.33 equiv). The crude material was purified by hot gravity filtration of the title compound as a yellow solid after boiling in methanol (0.070 g, 19%). 1H NMR (400 MHz, DMSO-d6) δ 8.40 (d, J=1.9 Hz, 1H), 8.19-8.15 (mult, 3H), 7.92 (d, J=15.5 Hz, 1H), 7.62-7.57 (mult, 3H), 7.39-7.35 (mult, 5H), 6.30 (d, J=15.5 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 166.8, 160.0, 151.6, 146.7, 143.2, 140.4, 139.7, 134.6, 132.3, 130.7, 130.0,129.3, 129.2, 129.0, 127.7, 122.4, 119.5, 114.0, 91.5. HRMS calcd for C23H15IN2O3, 495.02064; found, 495.01987 [M+H]+. Anal.(C23H15IN2O3) C, H, N. MP > 260 °C.
4-(6-Methyl-4-oxo-2-phenethylquinazolin-3(4H)-yl)benzoic acid (81)
Compound 80 (0.100 g, 0.26 mmol) was dissolved in DMF (5.0 mL). Palladium on carbon (10%, 0.10 g, 10 wt %) was added. A balloon filled with hydrogen was added and the mixture was stirred at room temperature for 1 hour. The mixture was filtered over a pad of celite, and the resulting solution was concentrated in vacuo to give a white solid. The solid was triturated with DCM and obtained by filtration (0.016 g, 16%). 1H NMR (400 MHz, DMSO-d6) δ 13.27 (bs, 1H), 8.09 (d, J=8.2 Hz, 2H), 7.93 (s, 1H), 7.71 (d, J=8.6 Hz, 1H), 7.65 (d, J=8.2 Hz, 1H), 7.54 (d, J=8.1 Hz, 2H), 7.22 (t, J=7.4 Hz, 2H), 7.14 (t, J=7.0 Hz, 1H), 7.06 (d, J=7.0 Hz, 2H), 2.98 (t, J=7.4 Hz, 2H), 2.58 (t, J=7.4 Hz, 2H), 2.47 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.7, 161.2, 154.6, 145.1, 141.3, 140.8, 136.4, 136.1, 131.5, 130.5, 129.2, 128.4, 128.2, 126.9, 126.1, 125.7, 120.2, 37.1, 32.0, 20.8. HRMS calcd for C24H20N2O3, 385.15534 [M+H]+; found, 385.15459 [M+H]+. Anal.(C24H20N2O3) C: 72.02, H: 5.03, N: 7.19. MP > 260 °C
(E)-4-(6-iodo-4-oxo-2-(2-(pyridin-3-yl)vinyl)quinazolin-3(4H)-yl)benzoic acid (98)
Compound 98 was prepared via procedure C using compound 72b (0.240 g, 0.591 mmol) and nicotinaldehyde (0.084 g, 0.074 mL, 0.786 mmol, 1.33 equiv). The reaction mixture was filtered and washed with MeOH to give the title compound as a yellow solid (0.170 g, 58%). %). 1H NMR (400 MHz, DMSO-d6) δ 13.31 (bs, 1H), 8.67 (d, J=2.4 Hz, 1H), 8.52 (dd, J1=4.7 Hz, J2=1.6 Hz, 1H), 8.41 (d, J=2.0 Hz, 1H), 8.19 (dd, J1=8.4 Hz, J2=8.4 Hz, 1H), 8.15 (d, J=8.6 Hz, 2H), 7.93 (d, J=15.7 Hz, 1H), 7.79 (d, J=8.2 Hz, 1H), 7.58-7.63 (mult, 3H), 7.37 (dd, J1=8.0 Hz, J2=4.9 Hz, 1H), 6.43 (d, J=15.7 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.7, 159.9, 151.3, 149.5, 146.6, 143.3, 140.5, 136.3, 133.9, 131.7, 130.7, 130.5, 129.3, 124.1, 122.5, 121.6, 91.9. HRMS calcd for C22H14IN3O3,494.00006 [M-H]+; found, 494.00150 [M-H]+. Anal. (C22H14IN3O3) C, H, N. MP > 260 °C.
(E)-3-(6-iodo-4-oxo-2-(2-(pyridin-3-yl)vinyl)quinazolin-3(4H)-yl)benzoic acid (99)
Compound 99 was prepared via procedure C using compound 73b (0.300 g, 0.739 mmol) and nicotinaldehyde (0.105 g, 0.092 mL, 0.982 mmol, 1.3 equiv.) The reaction mixture was recrystallized from ethyl acetate and hexanes to give the title compound as a yellow solid (0.111 g, 30%). 1H NMR (400 MHz, DMSO-d6) δ 13.31 (bs, 1H), 8.65 (s, 1H), 8.51 (d, J=3.9 Hz, 1H), 8.39 (s, 1H), 8.16 (t, J=9.4 Hz, 2H), 8.05 (s, 1H), 7.92 (d, J=15.7 Hz, 1H), 7.74-7.78 (mult, 3H), 7.58 (d, J=8.6 Hz, 1H), 7.34-7.37 (mult, 1H), 6.42 (d, J=15.7 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.5, 160.1, 151.6, 150.5, 149.5, 146.6, 143.2, 136.9, 136.1, 134.7, 133.9, 133.4, 132.4, 130.5, 130.2, 130.1, 129.9, 129.4, 124.1, 122.6, 121.7, 91.8. HRMS calcd for C22H14IN3O3 494.00006 [M-H]+; found, 494.00165 [M-H]+. Anal. (C22H14IN3O3) C: 45.18, H: 2.23, N: 7.16. MP > 260 °C.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Serdar Kurtkaya for helpful discussions. This work was supported by the Michael J. Fox Foundation (SFT), the Alfred Benzon Foundation (KBH), the Villum Kann Rasmussen Foundation (KBH), the Lundbeck Foundation (KBH), the NIH (NS065371, NS036654 ST), Pharmacological Sciences Training grant (T32 GM008602, TA), and a research grant from Pfizer (ST).
ABBREVIATIONS
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- BAPTA-AM
1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetraacetoxymethyl ester
- cDNA
complementary DNA
- DMSO
dimethyl sulfoxide
- HPLC
high performance liquid chromatography
- IC50
concentration of a test compound that produces half maximal inhibition
- K-BAPTA
potassium 1,2-bis(oaminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- NMDA
N-methyl-D-aspartate
- NMR
nuclear magnetic resonance
Footnotes
The authors declare no competing financial interests.
SUPPORTING INFORMATON
Supporting information is available online
REFERENCES
- 1.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. doi: 10.1124/pr.109.002451. Manuscript in press. DOI 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rudhard Y, Kneussel M, Nassar MA, Rast GF, Annala AJ, Chen PE, Tigaret CM, Dean I, Roes J, Gibb AJ, Hunt SP, Schoepfer R. Absence of Whisker-related pattern formation in mice with NMDA receptors lacking coincidence detection properties and calcium signaling. J. Neurosci. 2003;23:2323–2332. doi: 10.1523/JNEUROSCI.23-06-02323.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colonnese MT, Shi J. Constantine-Paton, M., Chronic NMDA receptor blockade from birth delays the maturation of NMDA currents, but does not affect AMPA/kainate currents. J. Neurophysiol. 2003;89:57–68. doi: 10.1152/jn.00049.2002. [DOI] [PubMed] [Google Scholar]
- 4.Waters KA, Machaalani R. Role of NMDA receptors in development of respiratory control. Respir. Physiol. Neurobiol. 2005;149:123–130. doi: 10.1016/j.resp.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 5.Nacher J, McEwen BS. The role of N-methyl-D-asparate receptors in neurogenesis. Hippocampus. 2006;16:267–270. doi: 10.1002/hipo.20160. [DOI] [PubMed] [Google Scholar]
- 6.Lisman J. Long-term potentiation: outstanding questions and attempted synthesis. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2003;358:829–842. doi: 10.1098/rstb.2002.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyamoto E. Molecular mechanism of neuronal plasticity: induction and maintenance of long-term potentiation in the hippocampus. J. Pharmacol. Sci. 2006;100:433–442. doi: 10.1254/jphs.cpj06007x. [DOI] [PubMed] [Google Scholar]
- 8.Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, Stray SM, Rippey CF, Roccanova P, Makarov V, Lakshmi B, Findling RL, Sikich L, Stromberg T, Merriman B, Gogtay N, Butler P, Eckstrand K, Noory L, Gochman P, Long R, Chen Z, Davis S, Baker C, Eichler EE, Meltzer PS, Nelson SF, Singleton AB, Lee MK, Rapoport JL, King MC, Sebat J. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- 9.Lindahl JS, Keifer J. Glutamate receptor subunits are altered in forebrain and cerebellum in rats chronically exposed to the NMDA receptor antagonist phencyclidine. Neuropsychopharmacology. 2004;29:2065–2073. doi: 10.1038/sj.npp.1300485. [DOI] [PubMed] [Google Scholar]
- 10.Whetsell WO., Jr. Current concepts of excitotoxicity. J. Neuropathol. Exp. Neurol. 1996;55:1–13. doi: 10.1097/00005072-199601000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 12.Brauner-Osborne H, Egebjerg J, Nielsen EO, Madsen U, Krogsgaard-Larsen P. Ligands for glutamate receptors: design and therapeutic prospects. J. Med. Chem. 2000;43:2609–2645. doi: 10.1021/jm000007r. [DOI] [PubMed] [Google Scholar]
- 13.Wang CX, Shuaib A. NMDA/NR2B selective antagonists in the treatment of ischemic brain injury. Curr. Drug Targets CNS Neurol. Disord. 2005;4:143–151. doi: 10.2174/1568007053544183. [DOI] [PubMed] [Google Scholar]
- 14.Miyabe M, Kirsch JR, Nishikawa T, Koehler RC, Traystman RJ. Comparative analysis of brain protection by N-methyl-D-aspartate receptor antagonists after transient focal ischemia in cats. Crit. Care Med. 1997;25:1037–1043. doi: 10.1097/00003246-199706000-00022. [DOI] [PubMed] [Google Scholar]
- 15.Preskorn SH. How multiple medication use evolves and the importance of therapeutic trials: the slippery slide. J. Psychiatr. Pract. 2008;14:170–175. doi: 10.1097/01.pra.0000320116.15564.65. [DOI] [PubMed] [Google Scholar]
- 16.Stern P, Behe P, Schoepfer R, Colquhoun D. Single-channel conductances of NMDA receptors expressed from cloned cDNAs: comparison with native receptors. Proc. Biol. Sci. 1992;250:271–277. doi: 10.1098/rspb.1992.0159. [DOI] [PubMed] [Google Scholar]
- 17.Erreger K, Chen PE, Wyllie DJ, Traynelis SF. Glutamate receptor gating. Crit. Rev. Neurobiol. 2004;16:187–224. doi: 10.1615/critrevneurobiol.v16.i3.10. [DOI] [PubMed] [Google Scholar]
- 18.Erreger K, Dravid SM, Banke TG, Wyllie DJ, Traynelis SF. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J. Physiol. 2005;563:345–358. doi: 10.1113/jphysiol.2004.080028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erreger K, Geballe MT, Dravid SM, Snyder JP, Wyllie DJ, Traynelis SF. Mechanism of partial agonism at NMDA receptors for a conformationally restricted glutamate analog. J. Neurosci. 2005;25:7858–7866. doi: 10.1523/JNEUROSCI.1613-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- 21.Wyllie DJ, Behe P, Colquhoun D. Single-channel activations and concentration jumps: comparison of recombinant NR1a/NR2A and NR1a/NR2D NMDA receptors. J. Physiol. 1998;510:1–18. doi: 10.1111/j.1469-7793.1998.001bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR. Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. J. Neurophysiol. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- 23.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 24.Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, Mizuno N. Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J. Comp. Neurol. 1994;347:150–160. doi: 10.1002/cne.903470112. [DOI] [PubMed] [Google Scholar]
- 25.Buller AL, Larson HC, Schneider BE, Beaton JA, Morrisett RA, Monaghan DT. The molecular basis of NMDA receptor subtypes: native receptor diversity is predicted by subunit composition. J. Neurosci. 1994;14:5471–5484. doi: 10.1523/JNEUROSCI.14-09-05471.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunah AW, Luo J, Wang YH, Yasuda RP, Wolfe BB. Subunit composition of N-methyl-D-aspartate receptors in the central nervous system that contain the NR2D subunit. Mol. Pharmacol. 1998;53:429–437. doi: 10.1124/mol.53.3.429. [DOI] [PubMed] [Google Scholar]
- 27.Thompson CL, Drewery DL, Atkins HD, Stephenson FA, Chazot PL. Immunohistochemical localization of N-methyl-D-aspartate receptor subunits in the adult murine hippocampal formation: evidence for a unique role of the NR2D subunit. Mol. Brain. Res. 2002;102:55–61. doi: 10.1016/s0169-328x(02)00183-3. [DOI] [PubMed] [Google Scholar]
- 28.Lau WK, Lui PW, Wong CK, Chan YS, Yung KK. Differential expression of N-methyl-D-aspartate receptor subunit messenger ribonucleic acids and immunoreactivity in the rat neostriatum during postnatal development. Neurochem. Int. 2003;43:47–65. doi: 10.1016/s0197-0186(02)00191-2. [DOI] [PubMed] [Google Scholar]
- 29.Lopez de Armentia M, Sah P. Development and subunit composition of synaptic NMDA receptors in the amygdala: NR2B synapses in the adult central amygdala. J. Neurosci. 2003;23:6876–6883. doi: 10.1523/JNEUROSCI.23-17-06876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dunah AW, Standaert DG. Subcellular segregation of distinct heteromeric NMDA glutamate receptors in the striatum. J. Neurochem. 2003;85:935–943. doi: 10.1046/j.1471-4159.2003.01744.x. [DOI] [PubMed] [Google Scholar]
- 31.Dunah AW, Yasuda RP, Wang YH, Luo J, Davila-Garcia M, Gbadegesin M, Vicini S, Wolfe BB. Regional and ontogenic expression of the NMDA receptor subunit NR2D protein in rat brain using a subunit-specific antibody. J. Neurochem. 1996;67:2335–2345. doi: 10.1046/j.1471-4159.1996.67062335.x. [DOI] [PubMed] [Google Scholar]
- 32.Hallett PJ, Standaert DG. Rationale for and use of NMDA receptor antagonists in Parkinson's disease. Pharmacol. Ther. 2004;102:155–174. doi: 10.1016/j.pharmthera.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 33.Salter MG, Fern R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature. 2005;438:1167–1171. doi: 10.1038/nature04301. [DOI] [PubMed] [Google Scholar]
- 34.Karadottir R, Cavelier P, Bergersen LH, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature. 2005;438:1162–1166. doi: 10.1038/nature04302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karavanova I, Vasudevan K, Cheng J, Buonanno A. Novel regional and developmental NMDA receptor expression patterns uncovered in NR2C subunit-beta-galactosidase knock-in mice. Mol. Cell. Neurosci. 2007;34:468–480. doi: 10.1016/j.mcn.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paquet M, Tremblay M, Soghomonian JJ, Smith Y. AMPA and NMDA glutamate receptor subunits in midbrain dopaminergic neurons in the squirrel monkey: an immunohistochemical and in situ hybridization study. J. Neurosci. 1997;17:1377–1396. doi: 10.1523/JNEUROSCI.17-04-01377.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carter C, Benavides J, Legendre P, Vincent JD, Noel F, Thuret F, Lloyd KG, Arbilla S, Zivkovic B, Mackenzie ET, Scatton B, Langer SZ. Ifenprodil and Sl-82.0715 as Cerebral Anti-Ischemic Agents .2. Evidence for N-Methyl-D-Aspartate Receptor Antagonist Properties. J. Pharmacol. Exp. Ther. 1988;247:1222–1232. [PubMed] [Google Scholar]
- 38.Gotti B, Duverger D, Bertin J, Carter C, Dupont R, Frost J, Gaudilliere B, Mackenzie ET, Rousseau J, Scatton B, Wick A. Ifenprodil and Sl-82.0715 as Cerebral Anti-Ischemic Agents .1. Evidence for Efficacy in Models of Focal Cerebral-Ischemia. J. Pharmacol. Exp. Ther. 1988;247:1211–1221. [PubMed] [Google Scholar]
- 39.Williams K. Ifenprodil discriminates subtypes of the N-methyl-D-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol. Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- 40.Hess SD, Daggett LP, Deal C, Lu CC, Johnson EC, Velicelebi G. Functional characterization of human N-methyl-D-aspartate subtype 1A/2D receptors. J. Neurochem. 1998;70:1269–1279. doi: 10.1046/j.1471-4159.1998.70031269.x. [DOI] [PubMed] [Google Scholar]
- 41.Feng B, Tse HW, Skifter DA, Morley R, Jane DE, Monaghan DT. Structure-activity analysis of a novel NR2C/NR2D-preferring NMDA receptor antagonist: 1-(phenanthrene-2-carbonyl) piperazine-2,3-dicarboxylic acid. Br. J. Pharmacol. 2004;141:508–516. doi: 10.1038/sj.bjp.0705644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morley RM, Tse HW, Feng B, Miller JC, Monaghan DT, Jane DE. Synthesis and pharmacology of N1-substituted piperazine-2,3-dicarboxylic acid derivatives acting as NMDA receptor antagonists. J. Med. Chem. 2005;48:2627–2637. doi: 10.1021/jm0492498. [DOI] [PubMed] [Google Scholar]
- 43.Frizelle PA, Chen PE, Wyllie DJ. Equilibrium constants for (R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydroquino xalin-5-yl)-methyl]-phosphonic acid (NVP-AAM077) acting at recombinant NR1/NR2A and NR1/NR2B N-methyl-D-aspartate receptors: Implications for studies of synaptic transmission. Mol. Pharmacol. 2006;70:1022–1032. doi: 10.1124/mol.106.024042. [DOI] [PubMed] [Google Scholar]
- 44.Neyton J, Paoletti P. Relating NMDA receptor function to receptor subunit composition: limitations of the pharmacological approach. J. Neurosci. 2006;26:1331–1333. doi: 10.1523/JNEUROSCI.5242-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brothwell SL, Barber JL, Monaghan DT, Jane DE, Gibb AJ, Jones S. NR2B- and NR2D-containing synaptic NMDA receptors in developing rat substantia nigra pars compacta dopaminergic neurones. J. Physiol. 2008;586:739–750. doi: 10.1113/jphysiol.2007.144618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mayer ML, Armstrong N. Structure and function of glutamate receptor ion channels. Annu. Rev. Physiol. 2004;66:161–181. doi: 10.1146/annurev.physiol.66.050802.084104. [DOI] [PubMed] [Google Scholar]
- 47.Chen PE, Geballe MT, Stansfeld PJ, Johnston AR, Yuan H, Jacob AL, Snyder JP, Traynelis SF, Wyllie DJ. Structural features of the glutamate binding site in recombinant NR1/NR2A N-methyl-D-aspartate receptors determined by site-directed mutagenesis and molecular modeling. Mol. Pharmacol. 2005;67:1470–1484. doi: 10.1124/mol.104.008185. [DOI] [PubMed] [Google Scholar]
- 48.Dravid SM, Erreger K, Yuan H, Nicholson K, Le P, Lyuboslavsky P, Almonte A, Murray E, Mosely C, Barber J, French A, Balster R, Murray TF, Traynelis SF. Subunit-specific mechanisms and proton sensitivity of NMDA receptor channel block. J. Physiol. 2007;581:107–128. doi: 10.1113/jphysiol.2006.124958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuan H, Hansen KB, Vance KM, Ogden KK, Traynelis SF. Control of N-methyl-D-aspartate Receptor Function by the NR2 Subunit Amino-Terminal Domain. J. Neurosci. 2009;29:12045–12058. doi: 10.1523/JNEUROSCI.1365-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sobolevsky AI, Koshelev SG, Khodorov BI. Probing of NMDA channels with fast blockers. J. Neurosci. 1999;19:10611–10626. doi: 10.1523/JNEUROSCI.19-24-10611.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Furukawa H, Singh SK, Mancusso R, Gouaux E. Subunit arrangement and function in NMDA receptors. Nature. 2005;438:185–192. doi: 10.1038/nature04089. [DOI] [PubMed] [Google Scholar]
- 52.Karakas E, Simorowski N, Furukawa H. Structure of the zinc-bound amino-terminal domain of the NMDA rececptor NR2B subunit. EMBO J. doi: 10.1038/emboj.2009.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hansen KB, Brauner-Osborne H, Egebjeg J. Pharmacological Characterization of Ligands at Recombinant NMDA Receptor Subtypes by Electrophysiological Recordings and Intracellular Calcium Measurements. Comb. Chem. High. Throughput Screen. 2009;11:304–315. doi: 10.2174/138620708784246040. [DOI] [PubMed] [Google Scholar]
- 54.Menniti FS, Chenard BL, Collins MB, Ducat MF, Elliott ML, Ewing FE, Huang JI, Kelly KA, Lazzaro JT, Pagnozzi MJ, Weeks JL, Welch WM, White WF. Characterization of the binding site for a novel class of noncompetitive alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor antagonists. Mol. Pharmacol. 2000;58:1310–1317. doi: 10.1124/mol.58.6.1310. [DOI] [PubMed] [Google Scholar]
- 55.Welch WM, Ewing FE, Huang J, Menniti FS, Pagnozzi MJ, Kelly K, Seymour PA, Guanowsky V, Guhan S, Guinn MR, Critchett D, Lazzaro J, Ganong AH, DeVries KM, Staigers TL, Chenard BL. Atropisomeric quinazolin-4-one derivatives are potent noncompetitive alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonists. Bioorg. Med. Chem. Lett. 2001;11:177–181. doi: 10.1016/s0960-894x(00)00622-3. [DOI] [PubMed] [Google Scholar]
- 56.Lazzaro JT, Paternain AV, Lerma J, Chenard BL, Ewing FE, Huang J, Welch WM, Ganong AH, Menniti FS. Functional characterization of CP-465,022, a selective, noncompetitive AMPA receptor antagonist. Neuropharmacology. 2002;42:143–153. doi: 10.1016/s0028-3908(01)00170-8. [DOI] [PubMed] [Google Scholar]
- 57.Perich JW, Johns RB. An Improved One-Pot Synthesis of N-Alpha-(Tert-Butoxycarbonyl)-O-(O′,O″-Dialkylphosphoro)-L-Tyrosines Using Dialkyl N,N-Diethylphosphoramidites. Synthesis. 1989:701–703. [Google Scholar]
- 58.Hansen KB, Mullasseril P, Dawit S, Kurtkaya NL, Yuan H, Vance KM, Orr AG, Kvist T, Ogden KK, Le P, Vellano KM, Lewis I, Kurtkaya S, Du Y, Qui M, Murphy TJ, Snyder JP, Brauner-Osborne H, Traynelis SF. J. Pharmacol. Exp. Ther. 2010;333:650–662. doi: 10.1124/jpet.110.166256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Traynelis SF, Burgess MF, Zheng F, Lyuboslavsky P, Powers JL. Control of voltage-independent zinc inhibition of NMDA receptors by the NR1 subunit. J. Neurosci. 1998;18:6163–6175. doi: 10.1523/JNEUROSCI.18-16-06163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.