

Abstract
ArsD is a metallochaperone that delivers trivalent metalloids [As(III) or Sb(III)] to the ArsA ATPase, the catalytic subunit of the ArsAB pump encoded by the arsRDABC operon of Escherichia coli plasmid R773. Interaction with ArsD increases the affinity of ArsA for As(III), conferring resistance to environmental concentrations of arsenic. Previous genetic analysis suggested that ArsD residues Cys12, Cys13, and Cys18 are involved in the transfer of As(III) to ArsA. Here X-ray absorption spectroscopy was used to show that As(III) is coordinated with three sulfur atoms, consistent with the three cysteine residues forming the As(III) binding site. Two single-tryptophan derivatives of ArsD exhibited quenching of intrinsic protein fluorescence upon binding of As(III) or Sb(III), which allowed estimation of the rates of binding and affinities for metalloids. Substitution of Cys12, Cys13, or Cys18 decreased the affinity for As(III) more than 10-fold. Reduced glutathione greatly increased the rate of binding of As(III) to ArsD but did not affect binding of As(III) to ArsA. This suggests that in vivo cytosolic As(III) might be initially bound to GSH and transferred to ArsD and then to ArsAB, which pumps the metalloid out of the cell. The As(III) chelator dimercaptosuccinic acid did not block the transfer from ArsD to ArsA, consistent with channeling of the metalloid from one protein to the other, as opposed to release and rebinding of the metalloid. Finally, transfer of As(III) from ArsD to ArsA occurred in the presence of MgATP at 23 °C but not at 4 °C. Neither MgADP nor MgATP-γ-S could replace MgATP. These results suggest that transfer occurs with a conformation of ArsA that transiently forms during the catalytic cycle.
Arsenic is the most common toxic element in the environment, introduced from both geochemical and anthropogenic sources. The metalloid is a carcinogen, and is associated with cardio-vascular and peripheral vascular diseases, neurological disorders, and diabetes mellitus (1, 2). As a result of its ubiquity, health effects, and potential for human exposure, arsenic ranks first on the Centers for Disease Control’s Agency for Toxic Substances and Disease Registry Comprehensive Environmental Response, Compensation, and Liability Act Priority (Superfund) List of Hazardous Chemicals (http://www.atsdr.cdc.gov/cercla/07list.html). Consequently, arsenic detoxifying systems are found in nearly all organisms (3). In bacteria and archaea, various ars operons encode ArsAB ATPases that pump the trivalent metalloids As(III) and Sb(III) out of cells. In these operons, an arsD gene is almost always adjacent to the arsA gene, suggesting a related function. Most transition and heavy metal ions do not exist as free ions in the cytosol but are sequestered by a variety of proteins variously called metal ion chaperones, scaffolds, or intracellular carriers (4). ArsD was recently shown to be a chaperone for the transfer of cytosolic As(III) to the 583-residue ArsA ATPase, the catalytic subunit of the efflux pump (5, 6). ArsD is a 120-residue protein with three conserved cysteine residues, Cys12, Cys13, and Cys18, required for chaperone activity (6). ArsA exhibits a low, basal rate of ATPase activity in the absence of As(III) or Sb(III) and a higher, activated rate in their presence. ArsA has a high-affinity metalloid binding site composed of Cys113 and Cys422 (7), and a third residue, Cys172, that participates in high-affinity binding and activation of ATP hydrolysis (8). In analogy with the mechanism for the transfer of copper from chaperones to copper pumps or enzymes (9), we previously proposed a stepwise transfer from the cysteines of ArsD to the cysteines of ArsA (10). ArsD increases the affinity of ArsA for As(III), permitting detoxification of environmental concentrations of arsenic.
In this study, the properties of As(III) binding by ArsD and subsequent transfer to ArsA were examined. X-ray absorption spectroscopy was used to show that As(III) is coordinated with three sulfur atoms, consistent with Cys12, Cys13, and Cys18 forming the As(III) binding site. Assays with single-tryptophan derivatives of ArsA have been informative about As(III) or Sb(III) binding properties and catalysis (11–15), so an assay using intrinsic protein fluorescence was developed as a probe of metalloid binding to ArsD. Two single-tryptophan derivatives of ArsD were constructed by substitution of Thr15 or Val17 with tryptophan in a tryptophan-free background. Both exhibited quenching of fluorescence upon binding of As(III) or Sb(III), from which the apparent affinity for metalloid could be estimated. Since it is likely that cytosolic As(III) is bound to reduced glutathione (GSH)1 in the cytosol of cells, the effect of GSH on binding to ArsD was examined. GSH greatly increased the rate of binding of As(III) to ArsD, leading us to hypothesize that ArsD accepts a metalloid from the As(GS)3 complex in vivo. In contrast, GSH did not affect the As(III)-stimulated ArsA ATPase activity, suggesting that As(III) is directly transferred from ArsD to ArsA, as opposed to release from ArsD, binding to GSH, and then interaction of ArsA with the As(GS)3 complex. To differentiate between these two possibilities, the effect of the As(III) chelator dimercaptosuccinic acid(DMSA)was examined. The chelator did not affect transfer, indicating channeling of As(III) from ArsD to ArsA. Transfer occurs only under conditions where ArsA hydrolyzes ATP, suggesting that ArsD transfers As(III) to an ArsA conformation transiently formed during catalysis and not simply to the closed conformation that ArsA adopts when As(III) and MgATP are bound (13).
MATERIALS AND METHODS
Strains, Plasmids, and Media
Escherichia coli cells were grown in LB (16) at 37 °C Ampicillin (100 μg/mL) or kanamycin (40 μg/mL) was added as required. E. coli strain JM109 [recA1 supE44 endA1 hsdR17 gyrA96 relA1 thiΔ(lac-proAB) F′ (traD36 proAB+ lacIq lacZΔM15)] was used for molecular cloning. E. coli strain BL21(DE3) [F–ompT hsdSB (rB–mB–) gal dcm (DE3 [lacI lacUV5-T7 gene1 ind1 Sam7 nin5])] was used for protein expression and purification. ArsA with a C-terminal six-histidine tag was expressed from plasmid pAlter-dAhB (17). ArsD contains three vicinal cysteine pairs, of which the last two are not required for chaperone activity, so the last 11 residues containing those two cysteine pairs were removed, creating the ArsD109 truncation (6). The gene for the maltose binding protein (MBP) was fused to the 5′ end of the gene for ArsD109 in plasmid pMAL-ArsD109 (6). ArsD109 with a N-terminal six-histidine tag was expressed from plasmid pET28a-ArsD109. The codons for the two native ArsD tryptophan residues, Trp35 and Trp97, were mutated to tyrosine codons by site-directed mutagenesis in plasmid pET28a-ArsD109, generating pET28a-ArsD109W35/97Y. In this plasmid, Thr15 and Val17 codons were mutated to tryptophan codons, generating pET28a-ArsD109T15W and pET28a-ArsD109V17W, respectively. The codons for Cys12, Cys13, and Cys18 were individually mutated to glycine codons in pET28a-ArsD109, generating pET28a-ArsD109C12G, pET28a-ArsD109C13G, and pET28a-ArsD109C18G, respectively. The codons forCys12, Cys13, andCys18 were mutated to glycine codons in pET28a-ArsD109T15W, generating pET28a-ArsD109C12G,T15W, pET28a-ArsD109C13G,T15W, and pET28a-ArsD109C18G,T15W, respectively. Growth in liquid culture was estimated from the absorbance at 600 nm.
DNA Manipulations
Plasmid extraction, DNA restriction endonuclease analysis, ligation, and other general molecular biological procedures were performed as described previously (16). Transformation of E. coli cells was conducted using a Bio-Rad (Hercules, CA) MicroPluser. Site-directed mutagenesis was performed using a QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA).
Protein Expression and Purification
Cells bearing the indicated plasmids were grown in LB overnight at 37 °C and then diluted 50-fold into 1 L of the same medium. Proteins were expressed by induction with 0.3 mM isopropyl β-D-thiogalactopyranoside at an A600 of 0.6–0.8 for 3 h. MBP-ArsD109 was purified from E. coli strain BL21(DE3) bearing plasmid pMAL-ArsD109 as described previously (5). ArsA with a six-histidine tag at the C-terminus was purified from cells of strain BL21(DE3) expressing pAlter-1-dAhB plasmid, as described (14). ArsD109 and its derivatives with a six-histidine tag at the N-terminus were purified similarly. Purified proteins were stored at −80 °C until they were used, and their concentrations were determined according to the method of Bradford (18) or from the absorption at 280 nm (19).
Circular Dichroism Measurements
Circular dichroism (CD) spectra from 190 to 260 nm were recorded with a spectrometer from Olis Inc. (Bogart, GA) at 20 °C using a 0.2 cm path length cell at 1.75 nm intervals. Three scans were averaged for each spectrum. ArsDs were assayed at 10 μM in 10 mM potassium phosphate buffer (pH 7.5).
X-ray Absorption Spectroscopy (XAS)
XAS samples were prepared in 50 mM MOPS, 0.5 M NaCl, and 30% glycerol (pH 7.5). ArsD109 at 4 mM was incubated with 3.2 mM sodium arsenite for 1 h on ice, and unbound As(III) was removed with a Bio-Gel P6¯ column. Solution samples containing 30% glycerol were loaded in Lucite cells, wrapped in Kapton tape, and flash-frozen in liquid nitrogen.
XAS data were collected at the Stanford Synchrotron Radiation Laboratory (SSRL) on beamline 9-3, equipped with Si[220] double-crystal monochromator with a harmonic rejection mirror. Samples were maintained at 10 K using an Oxford Instruments continuous-flow liquid helium cryostat. Protein fluorescence excitation spectra were recorded using a 30-element Ge solid-state array detector. A germanium filter (0.6 mm in width) and solar slits were placed between the cryostat and detector to filter scattering fluorescence not associated with protein-bound arsenic signals. XAS spectra were recorded in 5 eV steps in the pre-edge region (11625–11825 eV), 0.25 eV steps in the edge region (11850–11900 eV), and 0.05Å−1 increments in the extended X-ray absorption fine structure (EXAFS) region out to a k range of 13 Å−1. The data were integrated from 2 to 20 s in a k-weighted manner in the EXAFS region for a total scan length of 45 min. X-ray energies were calibrated using an arsenic foil absorption spectrum collected simultaneously with the protein data. The first inflection point for the arsenic foil edge was assigned to 11867 eV. Each fluorescence channel of each scan was examined for spectral anomalies prior to averaging, and spectra were closely monitored for photodegradation. The data represent an average of six to seven scans.
XAS data were processed using the Macintosh OS X version of the EXAFSPAK software suite (available on the World Wide Web) integrated with Feff version 7 (20) for theoretical model generation. Data reduction followed a previously published protocol for a spectral resolution in bond lengths of 0.13 Å (21). EXAFS fitting analysis was performed on raw unfiltered data. Protein EXAFS data were fit using single-scattering Feff version 7 theoretical models, calculated for coordination of carbon, oxygen, sulfur, and copper to simulate arsenic–ligand environments, with values for the scale factors (Sc) and E0 calibrated by fitting crystallographically characterized arsenic model compounds. All spectra were fit using identical protocols, first by distinguishing the best single-shell fit to the data and then by progressively adding extra scattering environments to the fit. Criteria for judging the best-fit EXAFS simulations utilized both the lowest mean-square deviation between data and fit, corrected for the number of degrees of freedom (F′), and reasonable Debye–Waller factors (σ2<0.006Å2). The best fits are given in the Supporting Information (Supplemental Table 1).
Fluorescence Measurements
Fluorescence measurements were conducted with a QuantaMaster UV VIS QM-4 steady-state spectrofluorometer (Photon Technology International, Birmingham, NJ) at room temperature. For time-based measurements, the excitation wavelength was 295 nm and the emission wavelength was 345 nm. Emission scans were made between 310 and 390 nm. As(III) or Sb(III) was added as sodium arsenite or potassium antimonyl tartrate, respectively, at the indicated concentrations to a cuvette containing 2 mL of 1 μM ArsD109 in a buffer consisting of 50mMMOPS-KOH(pH7.5) and 0.25mMEDTA. GSH was added to the indicated concentrations. Additions were made from concentrated solutions, and fluorescence was corrected for dilution. To examine the effect of pH, buffers containing 50 mM sodium acetate (pH 4.0), 50 mM sodium acetate (pH 5.0), 50mMMES (pH 6.0), 50mMMOPS (pH 7.0), 50mM Tris (pH 8.0), or 50 mM CHES (pH 9.0) were used.
ATPase Assays
ATPase activity was estimated using a coupled assay (22), as described previously (23). ArsA was added to a final concentration of 0.3 μM into an assay mixture containing 5 mM ATP, 1.25 mM phosphoenolpyruvate, 0.25 mM NADH, 10 units of pyruvate kinase, and lactate dehydrogenase with or without the indicated concentrations of potassium antimonyl tartrate or sodium arsenite. ArsD109 was added at the indicated concentrations. The mixture was prewarmed to 37 °C, and the reaction was initiated by addition of 2.5 mM MgCl2 and assessed at 340 nm. The linear steady-state rate of ATP hydrolysis was used to calculate specific activity. The reaction volume was 0.2 mL each in 96-well microplates, and the reactions were monitored by microplate reader SPECTRA max 340PC (Molecular Devices). GSH or DMSA was added as indicated.
Metalloid Transfer Assays
Transfer assays were performed as described previously (10) except that purifiedMBP-ArsD109 was used in place of cytosol fractions containing MBP-ArsD109. A 0.5 mL amylose column pre-equilibrated with 4 mL of buffer consisting of 50 mM MOPS-KOH and 0.5 mM NaCl (pH 7.5) (column buffer) was used. Purified MBP-ArsD109 was dialyzed against column buffer supplemented with 5 mM β-mercaptoethanol and 0.5 mM dithiolthreitol and loaded into the column, which was then washed with 1 mL of column buffer. Next 0.5mL of 1mMsodium arsenite was added to the column, which was then washed with 4 mL of column buffer. BSA or purified ArsA (0.5 mL of a 20 μM solution of either protein) was applied to the columns with 5 mM MgATP, MgADP, or MgATPγS, as indicated. The column was eluted with 1 mL of column buffer supplemented with the appropriate nucleotide and then 2 mL of column buffer. Finally, the column was eluted with 1.5 mL of 10mMmaltose. Ten fractions of 0.5 mL were collected and analyzed by sodium dodecyl sulfate—polyacrylamide gel electrophoresis (SDS–PAGE) (24). For assays conducted at 4 °C, everything was precooled to 4 °C for 30 min. The protein concentration of each fraction was determined according to the method of Bradford (18). The arsenic concentration was quantified by inductively coupled plasma mass spectroscopy (ICP-MS).
RESULTS
Cys12, Cys13, and Cys18 Form a Three-Coordinate As(III) Binding Site in ArsD
R773 ArsD has three cysteine pairs, Cys12-Cys13, Cys112-Cys113, and Cys119-Cys120, plus an additional cysteine at residue 18. However, only cysteine residues 12, 13, and 18 are conserved in known homologues, and truncation of the last two vicinal pairs has no effect on ArsD chaperone activity. In this study, an ArsD truncation of the last 11 residues, including the two C-terminal vicinal pairs, was used to avoid the interference of metal binding at these two sites, and this 109-residue variant with an N-terminal six-histidine tag was used as the parental protein for further analysis (6). It should be pointed out that six-histidine tags do not bind As(III) (25). The nature of the binding site was investigated in structural detail using XAS. XANES analysis indicated arsenic is As(III) (Figure 1A). The observed value for the first inflection point energy is 11868 eV, consistent with observed values for As(III) in protein and model systems (26).
Figure 1.

(A) Normalized XANES spectra of ArsD109. (B) ArsD EXAFS data and simulations. Raw EXAFS data (left) and phase-shifted Fourier transforms (right) of As(III) bound to ArsD109. Raw unfiltered data are shown as a solid line, while the best-fit simulated data are shown as a dotted line.
The EXAFS fitting results are most consistent with trivalent arsenic being maintained in a three-coordinate all-sulfur structural environment inArsD109 (Figure 1B). The structural environment is dominated by a symmetric three-sulfur coordination environment to the As(III) with an average bond length of 2.24 Å. Addition of a small oxygen/nitrogen scattering contribution to the As–S3 fits for the parental protein suggests a possible low-percentage partial dissociation of the bound metalloid (Supplemental Table 1 of the Supporting Information). No sulfur ligation was observed with the C12G, C13G, or C18G derivatives (data not shown).
Tryptophan Fluorescence Reports Trivalent Metalloid Binding by ArsD
Previous assays for quantifying arsenic binding by parental and mutant ArsDs used ICP-MS (5, 6). To develop a real-time assay for arsenic binding, single-tryptophan derivatives of ArsD109 were constructed. ArsD has two native tryptophan residues, Trp35 and Trp97. Trp35 does not respond to As(III), but the fluorescence of Trp97 is quenched when As(III) binds to the Cys112-Cys113 vicinal pair but not to the Cys12-Cys13-Cys18 As(III) binding site (27). A tryptophan-free derivative of ArsD109, which lacks the last two vicinal cysteine pairs, was constructed (W35/97Y). Two derivatives with single tryptophan residues near the As(III) binding site were then constructed via substitution of Thr15 or Val17 with tryptophan. Introduction of either tryptophan residue had no apparent effect on ArsD structure or function. The overall secondary structures of these two mutants, T15W and V17W, were similar to that of parental ArsD109, as determined by circular dichroism (Supplemental Figure 1 of the Supporting Information), and both stimulated ArsA ATPase activity nearly as well as the parental ArsD109 (Supplemental Figure 2 of the Supporting Information; data not shown for V17W). Tryptophan-freeW35/97Y exhibited very low background fluorescence (Figure 2A). Both single-tryptophan derivatives exhibited considerable intrinsic protein fluorescence compared with W35/97Y, with emission maxima of approximately 340 nm compared with an emission maximum of approximately 355 nm for free tryptophan in solution. Denaturation of the proteins with guanidine red-shifted the emission maxima to approximately 352 nm. While metalloids do not collisionally quench the fluorescence of tryptophan in solution (data not shown), the fluorescence of either T15Wor V17Wwas quenched approximately 50%by addition of high concentrations of As(III) or Sb(III) (Figure 2B).
Figure 2.

Protein fluorescence of single-tryptophan derivatives T15W and V17W reports metalloid binding. Emission scans were performed with excitation at 295 nm, as described in Materials and Methods: (A) (▼) 1 μM tryptophan, (●) tryptophan-free W35/97Y, (■) T15W, (▲) V17W, (○) W35/97Y with 6 M guanidine-HCl, (□) T15W with 6Mguanidine-HCl, and (△) V17Wwith 6Mguanidine-HCl and (B) (○)T15W, (▽)T15Wwith 5mMAs(III), (□) T15Wwith 0.1 mM Sb(III), (●) V17W, (▼) V17Wwith 5mMAs(III), (■) V17W with 0.1 mM Sb(III), and (◇) tryptophan-free W35/97Y.
In the absence of added thiols, the rate of fluorescence quenching, presumably proportional to the rate of As(III) binding, was very slow, requiring more than 2 h to attain a steady-state level of fluorescence when 1 μM protein was mixed with an equimolar amount of As(III). From the As(III) concentration dependence of fluorescence quenching, apparent binding constants for the two single-tryptophan proteins could be calculated from the relationship kobs=koff + kon[As(III)] (28), where kobs is the rate of quenching fitted with SigmaPlot 9.0 (Figure 3). For V17W, koff was calculated to be (4±2)×10−4 s−1 and kon was calculated to be (2±0.06)×10−4 s−1 μM−1. From the relationship koff/kon=Kd, a binding affinity of 2 ± 1 μM was calculated. For T15W, koff was estimated to be (3 ± 1) × 10−4 s−1, kon to be (2 ± 0.05) × 10−4 s−1 μM−1, and Kd to be 1.5 ± 0.5 μM. Thus, the values for the two single-tryptophan derivatives are in agreement.
Figure 3.

Binding of As(III) by single-tryptophan derivatives T15W andV17Wand cysteine mutants. The rate of fluorescence quenching, Kobs, was determined as a function of arsenite concentration with excitation and emission wavelengths of 295 and 345 nm, respectively: (●) T15W, (○) V17W, (■) T15W/C12G, (△) T15W/C13G, and (▼) T15W/C18G. The data were fitted using SigmaPlot 9.0, with error bars representing the standard deviation from three assays.
Effect of Cysteine Substitutions on As(III) Binding
Substitutions of the three cysteines in ArsD109 previously resulted in the loss of Sb(III) binding ability in gel filtration assays (6). However, gel filtration assays for binding are as sensitive as fluorescence assays and cannot measure low-affinity binding. As(III) binding parameters were estimated from quenching of T15W fluorescence of each cysteine mutant (Figure 3). C18G/T15W, which retains the Cys12 and Cys13 vicinal pair, has a kon of (5 ± 0.1) × 10−5 s−1 μM−1, a koff of (1.4 ± 0.1) × 10−3 s−1, and a Kd of 28 ± 3 μM. C12G/T15W, which retains Cys13 and Cys18, has a kon of (5 ± 0.1) × 10−5 s−1 μM−1, a koff of (2.4 ± 0.1) × 10−3 s−1, and a Kd of 48 ± 3 μM. C13G/T15W, which retains Cys12 and Cys18, has a kon of (4 ±0.2) × 10−5 s−1 μM−1, a koff of (2.7 ±0.1) × 10−3 s−1, and a Kd of 68 ± 6 μM. Thus, the affinity of each of the three cysteine mutants decreased by 20-fold or more compared with the parental protein containing Cys12, Cys13, and Cys18.
Reduced Glutathione Increases the Rate of Binding of As(III) to ArsD but Not ArsA
In vivo, As(III) is nearly completely complexed with GSH (29). As GSH is the major intracellular thiol in E. coli, cytosolic As(III) might be expected to be primarily in the form of As(GS)3. For that reason, the effect of 5 mM GSH on the rate of binding of 50 μM sodium arsenite to 1 μM V17Wwas examined over a pH range of 4.0–9.0 (Figure 4). The rate of fluorescence quenching increased with an increase in pH with or without GSH. In the absence of GSH, the rate of fluorescence quenching was ~10-fold faster at pH 9 than at pH7. Addition of GSH accelerated the rate by 10-fold at pH 7.5, and the rate was too fast to measure accurately at pH ≥8. The pH dependence in the absence of GSH indicates binding of As(III) by thiolates of the three ArsD cysteines but could also reflect dissociation of As(OH)3 to As(OH)2−1 (pKa = 9.23). Acceleration by GSH is consistent with binding of As(III) by thiolates of GS−1 (pKa=8.7), which is highly pH-dependent (30). Although the possibility that another thiol donor exists cannot be ruled out, we hypothesize that in vivo intermolecular transfer from As(GS)3 to ArsD is a major pathway of intracellular metalloid movement.
Figure 4.
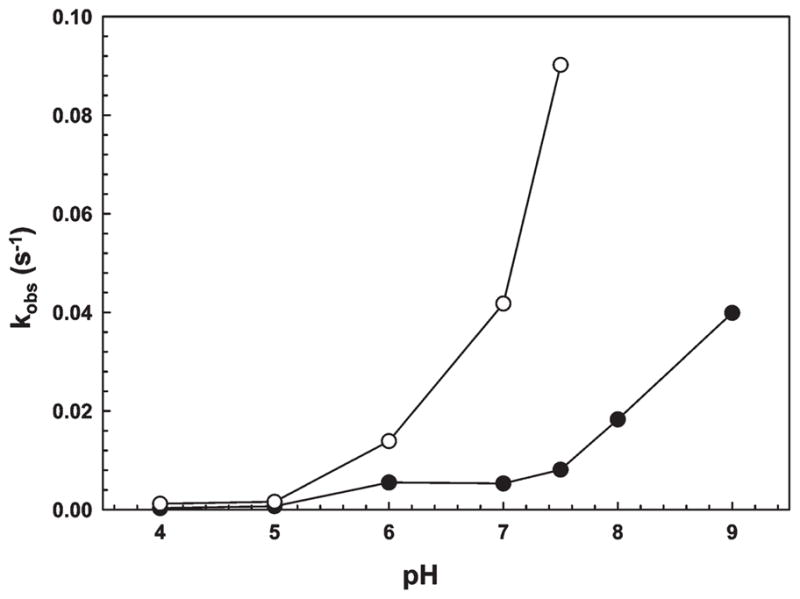
GSH accelerates binding of As(III) by V17W. The rate of fluorescence quenching, kobs, was determined as a function of pH in the presence (○) or absence (●) of 5 mM GSH.
The effect of GSH was investigated in more detail. In the absence of GSH, the fluorescence of V17Wdecreased very slowly when 50 μM sodium arsenite was added at pH 7.5 (Figure 5). Addition of 0.2 mM GSH had little effect, but 5 mM GSH increased the rate substantially. Addition of 5 mM β-mercaptoethanol or L-cysteine also increased the rate of quenching with As(III) (Supplemental Figure 3 of the Supporting Information). While ArsD might be able to accept As(III) from complexes with other monothiols, only GSH is present intracellularly at concentrations sufficiently high to be involved physiologically. In contrast, 2.5 μM potassium antimonyl tartrate rapidly quenched fluorescence in the absence of added monothiols, consistent with the fact that ArsD has a higher affinity for Sb(III) than for As(III) (27). Addition of 5 mM tris(2-carboxyethyl)phosphine (TCEP), which is a stronger reductant than GSH but is not a monothiol, did not increase the rate of quenching, indicating that the effect of GSH is not simply to protect either ArsD or As(III) from oxidation. Since formation of As(GS)3 is slow at pH 7.5 (30), 4 mM sodium arsenite was preincubated with 16 mM GSH for 10 min to allow for formation of the complex. When preformed As(GS)3 was added at a concentration of 50 μM, the rate of quenching was too fast to quantify, supporting our hypothesis that As(GS)3 is a direct donor of metalloid to ArsD.
Figure 5.
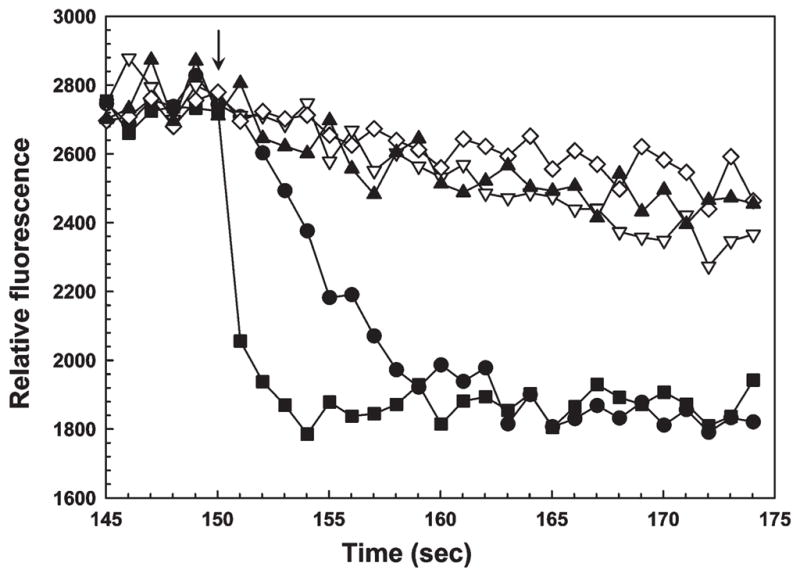
As(GS)3 can donate a metalloid to ArsD. The fluorescence of V17W was measured with excitation and emission wave-lengths of 295 and 345 nm, respectively. At the arrow the following additions were made: (▲) 50 μM As(III), (▽) 0.2 mM GSH followed by 50 μM As(III), (◇) 5 mM TCEP followed by 50 μM As(III), (●) 5 mM GSH followed by 50 μM As(III), and (■) 50 μM As(GS)3.
To examine whether GSH also facilitates the binding of As(III) to ArsA, the stimulation of ArsA activity by As(III) was determined in the presence or absence of GSH (Supplemental Figure 4 of the Supporting Information). ArsA exhibited an apparent affinity of 0.63 mM for As(III) with or without GSH. These results indicate that ArsA accepts either free As(III) or As(GS)3 with low affinity, but As(GS)3 does not replace ArsD as an intracellular metallochaperone for high-affinity As(III) binding.
ArsD Transfers As(III) Directly to ArsA
From the results of yeast two-hybrid analysis, it is clear that ArsD and ArsA physically interact (5). How is As(III) transferred during this interaction? One possibility is that the metalloid dissociates from ArsD and reassociates with ArsA. A second possibility is that there is direct transfer from the thiols of ArsD to the thiols of ArsA in a stepwise fashion (6), similar to the mechanism of transfer of copper from the yeast Atx1p chaperone to the Ccc2p copper efflux pump (31).
Metabolic trapping is often used to examine channeling (32, 33). To differentiate between channeling and a dissociation—reassociation mechanism, the As(III) chelator DMSA was added to the reaction mixture. DMSA has an apparent affinity of approximately 20 μM for As(III) (34), which is between the ArsD and ArsA affinities. Direct transfer of As(III) from ArsD to ArsA would be expected to be insensitive to DMSA. In contrast, if As(III) dissociates from ArsD before binding to ArsA, an excess of DMSA should prevent transfer. Using the stimulation of ArsA ATPase by the ArsD109–As(III) complex, the effect of DMSA was examined (Figure 6). DMSA did not affect the basal activity of ArsA, but the As(III)-stimulated rate of ArsA activity was inhibited by DMSA. In that experiment, ArsA with As(III) was titrated with DMSA, and the stimulated ATPase activity returned to the basal level at higher concentrations of DMSA. When ArsD109 was preincubated with As(III) and then added to ArsA, the rate of ATP hydrolysis was stimulated by low concentrations of As(III), reflecting the chaperone activity of ArsD. Titration of that mixture with DMSA resulted in an initial decrease in activity as free As(III) was chelated, but the activity plateaued at a level corresponding to the activated rate. Since ArsD has a higher affinity for As(III) than DMSA, it is able to retain and transfer the metalloid even at higher concentrations of DMSA. These results are consistent with direct transfer of As(III) from ArsD to ArsA.
Figure 6.
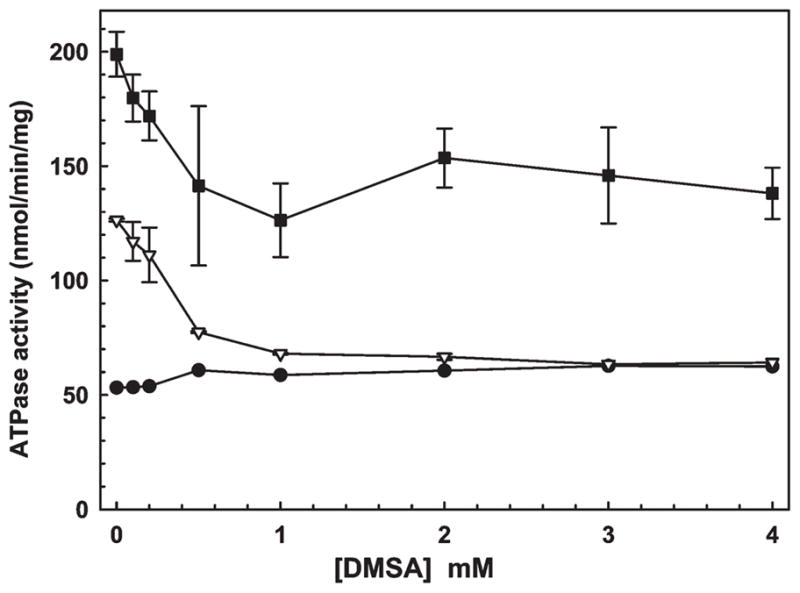
ArsD channels As(III) to ArsA. The effect of the As(III) chelator DMSA on the transfer of a metalloid from ArsD to ArsA was estimated from the activation of ATPase activity of 0.3 μM ArsA, as described in Materials and Methods. ATP hydrolysis was assessed in the presence of the indicated concentrations of DMSA with (●) ArsA alone, (▽) ArsA and 0.5 mM sodium arsenite, or (■) ArsA and 0.5 mM sodium arsenite with 6 μM ArsD109. The error bars represent the standard deviation from three assays.
Transfer of As(III) from ArsD to ArsA Requires Catalysis
We have previously shown that ArsD transfers Sb(III) to ArsA in the presence of MgATP (6). Here transfer of As(III) from ArsD109 to ArsA was compared under catalytic and noncatalytic conditions. Purified MBP-ArsD109 was bound to an amylose column. Sufficient As(III) was added to the column to saturate ArsD, after which free As(III) was washed off. Next either ArsA or BSA preincubated with nucleotides was passed through the column. When ArsA preincubated with MgATP was passed through the column at room temperature, conditions under which ArsA hydrolyzes ATP, nearly all of the As(III) eluted in the ArsA-containing fractions, with almost none in the MBP-ArsD109-containing fractions (Figure 7A,B). In contrast, when BSA preincubated with MgATP was passed through the column, almost none of the As(III) eluted in the BSA-containing fractions, but it did elute in theMBP-ArsD109-containing fractions (Figure 7C).
Figure 7.

Transfer of the metalloid from ArsD occurs during ArsA catalysis. As(III) transfer assays were performed as described in Materials and Methods. MBP-ArsD109 was bound to an amylose resin, and then the column was charged with sodium arsenite to saturate ArsD109. Next purified ArsA or BSA was applied to the columns with the indicated nucleotides (5 mM) at room temperature unless otherwise noted. (A) Ten fractions were collected and analyzed by SDS–PAGE. Nearly all of the BSA or ArsA eluted in fraction 2, and fraction 9 contained nearly all of theMBP-ArsD109. Nucleotides: (B) ArsA with MgATP,(C) BSA with MgATP,(D) ArsA with MgADP, (E)ArsA with MgATP-γ-S, and(F)ArsA with MgATP at 4 °C. The concentration of protein (open bars) and As(III) (●) was quantified in each fraction. The error bars represent the standard deviation from three assays.
When MgADP was used instead of MgATP, As(III) eluted with MBP-ArsD109 and not with ArsA (Figure 7D). Similarly, the MgATP-γ-S, a nonhydrolyzable nucleotide, did not promote transfer (Figure 7E). When the reaction with MgATP and column elution were conducted at 4 °C, where ArsA hydrolyzes ATP very slowly, almost no As(III) eluted with ArsA, but it did remain bound to MBP-ArsD109 (Figure 7F). These data demonstrate that ATP hydrolysis is required for the transfer of As(III) from ArsD to ArsA.
DISCUSSION
In vivo results quite clearly demonstrate that ArsD is an As(III)/Sb(III) chaperone that transfers a metalloid to the ArsA ATPase (5, 6), but the molecular details of the process are lacking. In this study, the questions of the nature of the metalloid binding site in ArsD and requirements for the transfer reaction were addressed.
From genetic analysis and conservation of residues in homologues, it seemed likely that the metalloid binding site in ArsD involved the three conserved cysteines, residues 12, 13, and 18. Here the EXAFS data with wild-type ArsD unambiguously demonstrate that the binding site is a three-coordinate sulfur-containing site, similar in nature but not sequence to the ArsA As(III) binding site (35) and the three convergently evolved sites of three different ArsR repressors (25, 36, 37). This suggests that a common type of As(III) binding site in proteins is composed of three cysteine residues, with similar As–S bond distances of 2.24 Å and probably similar geometries.
While EXAFS is useful for determining the environment surrounding the bound metalloid, a more complete analysis requires quantitative measurements of As(III) binding. Previous assays using gel filtration yielded stoichiometries, but it is difficult to extract more detailed information from analysis conducted after the reaction is complete. Changes in intrinsic protein fluorescence can provide real-time information about ligand binding or catalysis. Full-length ArsD has three vicinal cysteine pairs, Cys12-Cys13, Cys112-Cys113, and Cys119-Cys120, and two tryptophan residues, Trp35 and Trp97. Trp97 fluorescence reports binding of Sb(III) or As(III) to the vicinal Cys112-Cys113 pair (27). However, only the N-terminal Cys12-Cys13 pair is involved in chaperone activity; the C-terminal pairs are dispensable and not present in the constructs used for this study. For that reason, the two endogenous tryptophan residues were altered by site-directed mutagenesis, and new tryptophan residues introduced singly at positions 15 and 17, near the Cys12-Cys13-Cys18 As(III) binding site. The fluorescence of either single-tryptophan derivative was blue-shifted approximately 14 nm compared with that of free tryptophan. The fluorescence was red-shifted and decreased by denaturation with guanidine. These results indicate that the two tryptophan residues are in a less aqueous environment compared with free tryptophan. These two tryptophans are located among the three cysteines that form the As(III) binding site. Both single-tryptophan derivatives of ArsD109 exhibited substantial quenching of protein fluorescence upon addition of either As(III) or Sb(III), consistent with the region of ArsD containing the metalloid binding site undergoing a conformational change when the site is filled.
However, the rate of quenching with As(III) was slower than might be expected for a simple binding reaction. If in vivo binding to ArsD were slower than transfer to ArsA, then free arsenite might accumulate in the cytosol, negating the value of a chaperone. As(III) likely forms a As(GS)3 complex with cytosolic GSH, the major reduced thiol in E. coli, where it can accumulate to concentrations as high as 10 mM (38, 39). However, ArsA accepts a metalloid from As(GS)3 no faster than it binds free As(III), so even though GSH might serve as an As(III) buffer, it does not aid in detoxification via the ArsAB pump. In contrast, ArsD109 accepts a metalloid from As(GS)3 in vitro at a rate so fast that it cannot be determined with our instrumentation. These considerations lead us to propose that the physiological donor to ArsD might be As(GS)3 (Figure 8). As arsenite enters E. coli cells via the aquaglyceroporin GlpF as As(OH)3, the high concentration of GSH would by mass action rapidly drive exchange of hydroxyls for thiols.
Figure 8.

Model of transfer from As(GS)3 to ArsD to ArsA. In the first step, As(OH)3 forms As(GS)3 with intracellular GSH. In the next step, ArsD extracts As(III) from As(GS)3 by thiol exchange with residues Cys12, Cys13, and Cys18. In a stepwise series of thiol exchanges (with an unknown order), the bound As(III) is transferred to thiolates Cys113, Cys172, and Cys422 of ArsA when the two nucleotide binding domains, NBD1 and NBD2, hydrolyze ATP. Binding of As(III) and ATP induces a series of conformational changes in ArsA, resulting in activation of catalysis, with final extrusion of the metalloid from the cells.
The next series of steps is proposed to involve the stepwise transfer of As(III) from ArsD thiolates to ArsA thiolates. The lack of an effect of the As(III) chelator DMSA on the transfer reaction indicates that transfer is direct channeling from one protein to the other, rather than dissociation from ArsD and reassociation with ArsA. The transfer reaction itself apparently requires ArsA to be undergoing catalysis. As(III) is not transferred from ArsD109 to ArsA in the presence of MgADP or MgATP-γ-S, but only in the presence of MgATP, and then only at a temperature at which hydrolysis occurs. During the catalytic cycle, ArsA undergoes a series of conformational changes in which the A1 and A2 halves are open relative to each other to a tightly closed conformation in which the product MgADP is trapped (13, 40). In our model, ArsD would interact with one of the transitional conformations, with transfer of As(III) integral to formation of the closed conformation. Further details of the transfer reaction will require structural information about ArsD and the ArsD–ArsA complex.
Supplementary Material
Acknowledgments
A portion of the research was conducted at the Stanford Synchrotron Radiation Laboratory (SSRL). SSRL is a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program.
Footnotes
Abbreviations: LB, Luria-Bertani broth; MBP, maltose binding protein; GSH, reduced glutathione; DMSA, dimercaptosuccinic acid; SDS, sodium dodecyl sulfate; PAGE, polyacrylamide gel electrophoresis; NBD, nucleotide binding site.
This study was supported by National Institutes of Health Grants AI043428 to B.P.R. and DK068139 to T.L.S.
SUPPORTING INFORMATION AVAILABLE
Additional figures and table. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Abernathy CO, Thomas DJ, Calderon RL. Health effects and risk assessment of arsenic. J Nutr. 2003;133:1536S–1538S. doi: 10.1093/jn/133.5.1536S. [DOI] [PubMed] [Google Scholar]
- 2.Beane Freeman LE, Dennis LK, Lynch CF, Thorne PS, Just CL. Toenail arsenic content and cutaneous melanoma in Iowa. Am J Epidemiol. 2004;160:679–687. doi: 10.1093/aje/kwh267. [DOI] [PubMed] [Google Scholar]
- 3.Bhattacharjee H, Rosen BP. Arsenic metabolism in prokaryotic and eukaryotic microbes. In: Nies DH, Silver S, editors. Molecular microbiology of heavy metals. Springer-Verlag; Heidelberg, Germany: 2007. pp. 371–406. [Google Scholar]
- 4.Field LS, Luk E, Culotta VC. Copper chaperones: Personal escorts for metal ions. J Bioenerg Biomembr. 2002;34:373–379. doi: 10.1023/a:1021202119942. [DOI] [PubMed] [Google Scholar]
- 5.Lin YF, Walmsley AR, Rosen BP. An arsenic metallochaperone for an arsenic detoxification pump. Proc Natl Acad Sci USA. 2006;103:15617–15622. doi: 10.1073/pnas.0603974103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin YF, Yang J, Rosen BP. ArsD residues Cys12, Cys13, and Cys18 form an As(III)-binding site required for arsenic metallochaperone activity. J Biol Chem. 2007;282:16783–16791. doi: 10.1074/jbc.M700886200. [DOI] [PubMed] [Google Scholar]
- 7.Ruan X, Bhattacharjee H, Rosen BP. Cys-113 and Cys-422 form a high affinity metalloid binding site in the ArsA ATPase. J Biol Chem. 2006;281:9925–9934. doi: 10.1074/jbc.M600125200. [DOI] [PubMed] [Google Scholar]
- 8.Ruan X, Bhattacharjee H, Rosen BP. Characterization of the metalloactivation domain of an arsenite/antimonite resistance pump. Mol Microbiol. 2008;67:392–402. doi: 10.1111/j.1365-2958.2007.06049.x. [DOI] [PubMed] [Google Scholar]
- 9.Boal AK, Rosenzweig AC. Structural biology of copper trafficking. Chem Rev. 2009;109:4760–4779. doi: 10.1021/cr900104z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin YF, Yang J, Rosen BP. ArsD: An As(III) metallochaperone for the ArsAB As(III)-translocating ATPase. J Bioenerg Biomembr. 2007;39:453–458. doi: 10.1007/s10863-007-9113-y. [DOI] [PubMed] [Google Scholar]
- 11.Walmsley AR, Zhou T, Borges-Walmsley MI, Rosen BP. The ATPase mechanism of ArsA, the catalytic subunit of the arsenite pump. J Biol Chem. 1999;274:16153–16161. doi: 10.1074/jbc.274.23.16153. [DOI] [PubMed] [Google Scholar]
- 12.Walmsley AR, Zhou T, Borges-Walmsley MI, Rosen BP. A kinetic model for the action of a resistance efflux pump. J Biol Chem. 2001;276:6378–6391. doi: 10.1074/jbc.M008105200. [DOI] [PubMed] [Google Scholar]
- 13.Zhou T, Radaev S, Rosen BP, Gatti DL. Conformational changes in four regions of the Escherichia coli ArsA ATPase link ATP hydrolysis to ion translocation. J Biol Chem. 2001;276:30414–30422. doi: 10.1074/jbc.M103671200. [DOI] [PubMed] [Google Scholar]
- 14.Zhou T, Rosen BP. Tryptophan fluorescence reports nucleotide-induced conformational changes in a domain of the ArsA ATPase. J Biol Chem. 1997;272:19731–19737. doi: 10.1074/jbc.272.32.19731. [DOI] [PubMed] [Google Scholar]
- 15.Zhou T, Shen J, Liu Y, Rosen BP. Unisite and multisite catalysis in the ArsA ATPase. J Biol Chem. 2002;8:23815–23820. doi: 10.1074/jbc.M203432200. [DOI] [PubMed] [Google Scholar]
- 16.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: Alaboratory manual. Cold Spring Harbor Laboratory Press; Plainview, NY: 1989. [Google Scholar]
- 17.Li J, Rosen BP. The linker peptide of the ArsA ATPase. Mol Microbiol. 2000;35:361–367. doi: 10.1046/j.1365-2958.2000.01696.x. [DOI] [PubMed] [Google Scholar]
- 18.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 19.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 20.Ankudinov AL, Rehr JJ. Phys Rev B: Condens Matter Mater Phys. 1997;56:R1712–R1715. [Google Scholar]
- 21.Lieberman RL, Kondapalli KC, Shrestha DB, Hakemian AS, Smith SM, Telser J, Kuzelka J, Gupta R, Borovik AS, Lippard SJ, Hoffman BM, Rosenzweig AC, Stemmler TL. Characterization of the particulate methane monooxygenase metal centers in multiple redox states by X-ray absorption spectroscopy. Inorg Chem. 2006;45:8372–8381. doi: 10.1021/ic060739v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogel G, Steinhart R. ATPase of Escherichia coli: Purification, dissociation, and reconstitution of the active complex from the isolated subunits. Biochemistry. 1976;15:208–216. doi: 10.1021/bi00646a032. [DOI] [PubMed] [Google Scholar]
- 23.Hsu CM, Rosen BP. Characterization of the catalytic subunit of an anion pump. J Biol Chem. 1989;264:17349–17354. [PubMed] [Google Scholar]
- 24.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 25.Qin J, Fu HL, Ye J, Bencze KZ, Stemmler TL, Rawlings DE, Rosen BP. Convergent evolution of a new arsenic binding site in the ArsR/SmtB family of metalloregulators. J Biol Chem. 2007;282:34346–34355. doi: 10.1074/jbc.M706565200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramírez-Solis A, Mukopadhyay R, Rosen BP, Stemmler TL. Experimental and theoretical characterization of arsenite in water: Insights into the coordination environment of As-O. Inorg Chem. 2004;43:2954–2959. doi: 10.1021/ic0351592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li S, Rosen BP, Borges-Walmsley MI, Walmsley AR. Evidence for cooperativity between the four binding sites of dimeric ArsD, an As(III)-responsive transcriptional regulator. J Biol Chem. 2002;277:25992–26002. doi: 10.1074/jbc.M201619200. [DOI] [PubMed] [Google Scholar]
- 28.Tamerler C, Oren EE, Duman M, Venkatasubramanian E, Sarikaya M. Adsorption kinetics of an engineered gold binding peptide by surface plasmon resonance spectroscopy and a quartz crystal microbalance. Langmuir. 2006;22:7712–7718. doi: 10.1021/la0606897. [DOI] [PubMed] [Google Scholar]
- 29.Delnomdedieu M, Basti MM, Otvos JD, Thomas DJ. Reduction and binding of arsenate and dimethylarsinate by glutathione: A magnetic resonance study. Chem-Biol Interact. 1994;90:139–155. doi: 10.1016/0009-2797(94)90099-x. [DOI] [PubMed] [Google Scholar]
- 30.Yehiayan L, Pattabiraman M, Konstantinos K, Wang X, Boise LH, Cai Y. Speciation, formation, stability and analytical challenges of human arsenic metabolites. J Anal At Spectrom. 2009;24:1397–1405. doi: 10.1039/B910943A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pufahl RA, Singer CP, Peariso KL, Lin SJ, Schmidt PJ, Fahrni CJ, Culotta VC, Penner-Hahn JE, O’Halloran TV. Metal ion chaperone function of the soluble Cu(I) receptor Atx1. Science. 1997;278:853–856. doi: 10.1126/science.278.5339.853. [DOI] [PubMed] [Google Scholar]
- 32.Anderson KS. Fundamental mechanisms of substrate channeling. Methods Enzymol. 1999;308:111–145. doi: 10.1016/s0076-6879(99)08008-8. [DOI] [PubMed] [Google Scholar]
- 33.Kwok EY, Severance S, Kosman DJ. Evidence for iron channeling in the Fet3p-Ftr1p high-affinity iron uptake complex in the yeast plasma membrane. Biochemistry. 2006;45:6317–6327. doi: 10.1021/bi052173c. [DOI] [PubMed] [Google Scholar]
- 34.Spuches AM, Kruszyna HG, Rich AM, Wilcox DE. Thermodynamics of the As(III)-thiol interaction: Arsenite and monomethylarsenite complexes with glutathione, dihydrolipoic acid, and other thiol ligands. Inorg Chem. 2005;44:2964–2972. doi: 10.1021/ic048694q. [DOI] [PubMed] [Google Scholar]
- 35.Bhattacharjee H, Li J, Ksenzenko MY, Rosen BP. Role of cysteinyl residues in metalloactivation of the oxyanion-translocating ArsA ATPase. J Biol Chem. 1995;270:11245–11250. doi: 10.1074/jbc.270.19.11245. [DOI] [PubMed] [Google Scholar]
- 36.Shi W, Dong J, Scott RA, Ksenzenko MY, Rosen BP. The role of arsenic-thiol interactions in metalloregulation of the ars operon. J Biol Chem. 1996;271:9291–9297. doi: 10.1074/jbc.271.16.9291. [DOI] [PubMed] [Google Scholar]
- 37.Ordóñez E, Thiyagarajan S, Cook JD, Stemmler TL, Gil JA, Mateos LM, Rosen BP. Evolution of metal(loid) binding sites in transcriptional regulators. J Biol Chem. 2008;283:25706–25714. doi: 10.1074/jbc.M803209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fahey RC, Sundquist AR. Evolution of glutathione metabolism. In: Meister A, editor. Advances in Enzymology and Related Areas of Molecular Biology. John Wiley and Sons, Inc; New York: 1991. pp. 1–53. [DOI] [PubMed] [Google Scholar]
- 39.Apontoweil P, Berends W. Glutathione biosynthesis in Escherichia coli K 12. Properties of the enzymes and regulation. Biochim Biophys Acta. 1975;399:1–9. doi: 10.1016/0304-4165(75)90205-6. [DOI] [PubMed] [Google Scholar]
- 40.Zhou T, Radaev S, Rosen BP, Gatti DL. Structure of the ArsA ATPase: The catalytic subunit of a heavy metal resistance pump. EMBO J. 2000;19:1–8. doi: 10.1093/emboj/19.17.4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.