

In 2007, two simultaneous publications from Sibi and MacMillan described the progression of organocatalysis beyond closed shell bond-forming pathways, as each report detailed the α-functionalization of aldehydes via a radical cation intermediate.1 Our laboratory has further developed this catalysis manifold, termed SOMO-activation, to enable a wide range of transformations that include aldehyde α-alkylation,1a enolation,2a vinylation,2b nitroalkylation,2c arylation,2d carbooxidation,2e and polyene cyclization.2f During the course of these studies, however, we have continually met with failure when attempting to translate the Sibi oxidative conditions3 – FeCl3, NaNO2, and O2 – to catalytic bond constructions beyond the initial report. This is intriguing given that the key activated intermediate should be identical in each case (an imidazolidinone-derived radical cation); yet, the observed reactivity of these species appears related to the manner in which they were generated. In an effort to understand this mechanistic discrepancy, we initiated an investigation into the role of TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) as a purported SOMO-phile in the α-oxyamination catalytic cycle. Surprisingly, our findings revealed that the Sibi reaction does not operate by a SOMO-activation mechanism but instead via a more traditional enamine catalysis pathway.
The α-oxyamination mechanism proposed by Sibi (Scheme 1) begins with condensation of imidazolidinone catalyst 1 and an aldehyde to generate electron-rich enamine 2. Oxidation of this intermediate with FeCl3 was proposed to produce radical cation 3 which would then combine with TEMPO to yield iminium ion 4. Hydrolysis of such a species would liberate the observed α-oxygenated product 5, while an oxygen atmosphere, assisted by catalytic NaNO2, was believed to replenish the requisite Fe(III) species.
Scheme 1.


Proposed Mechanism for FeCl3-Catalyzed Oxidation
Given that the capture of α-carbonyl radicals by TEMPO has been known for more than a decade, this catalytic cycle appears more than reasonable.4 Moreover, the α-oxyamination of aldehydes with TEMPO has been performed recently using both photoredox catalysis5 and electrochemical techniques,6 and in both cases an identical mechanism was suggested. However, a wealth of mechanistic studies pertaining to CuCl2/TEMPO catalyzed alcohol oxidations7 has clearly demonstrated the capacity of TEMPO to undergo metal complexation8 to form the reactive oxidant. Specifically, the recent theoretical work of Baerends outlines that CuX2•TEMPO can behave as an ionic electrophile via single electron pairing of CuX2 and TEMPO,9 while Sheldon's kinetic isotope studies10 show that copper-TEMPO complexes do not generate free oxoammonium itself ([R2N=O]+). As such, an alternative pathway for the α-oxyamination reaction must be considered wherein enamine addition to an Fe(III)-bound TEMPO species is operative (see Alternative mechanism). With this in mind, we have undertaken several mechanistic studies to determine whether the Sibi reaction functions via a SOMO-activation pathway involving a radical cation (as originally proposed), or by direct enamine addition to an iron–TEMPO complex.
Design Plan
We recognized that implementation of a suitable radical clock-containing substrate should allow the detection of a SOMO-activated pathway (Scheme 2).11 The cis-cyclopropane substituted aldehyde 6 was chosen as a mechanistic probe given that the corresponding cyclopropylcarbinyl radical 7 should undergo rapid ring opening and ring closing12 prior to C–O bond formation (an equilibration step that would predominantly lead to trans-cyclopropyl oxyamination products). In contrast, such radical intermediates would be absent from an enamine addition mechanism, and therefore only cis-substituted cyclopropane products should be observed if an ionic pathway dominates.
Scheme 2.

Differentiating SOMO-Activation and Enamine Addition
Results
To test the validity of this radical clock study, we exposed the cis-cyclopropyl substrate 6 to established SOMO-activation conditions (specifically [FeCp2][PF6] in THF, a reliable outer-sphere oxidant) (Scheme 3). It should be noted that this oxidation system was initially employed in the original Sibi publication,1b however, it did not become the optimized or preferred protocol. As expected, initial experiments with high concentrations of TEMPO (4.0 M) delivered a mixture of non-isomerized cis-product 8a (71%) and trans-product 8b (29%). As the concentration of TEMPO is decreased to 0.75 M, direct radical trapping to yield cis-8a becomes a minor pathway (10%) while cyclopropyl rearrangement to the trans-adduct 8b dominates. Having validated the use of formyl cyclopropane 6 as a mechanistic probe, we next focused on the Sibi protocol. Indeed, when the optimal conditions from the initial study were employed (FeCl3/NaNO2, DMF, 2.0 M TEMPO) we observed that 95% of cis-8a is delivered. In contrast, the use of [FeCp2][PF6] in THF yields only a minor amount (35%) of the cis adduct.
Scheme 3.

Radical Clock Investigation of Oxygenation Mechanism
These results can be explained by one of three scenarios:
(i) The identity of the solvent (DMF vs. THF) has a significant effect on the relative rates of TEMPO-radical trapping versus cyclopropane isomerization
The direct comparison of [FeCp2][PF6] and FeCl3 in the same solvent is complicated by two factors: FeCl3 delivers only traces of product in THF,1b while ferrocenium species are known to steadily decompose in DMF,13 liberating Fe(III) which in turn could activate TEMPO towards enamine addition (resulting in a net increase in cis-8a formation). We have now been able to qualitatively account for the liberation of free iron in the following comparison experiment (Scheme 4). [FeCp2][PF6] and catalyst 1•HBF4 were aged in DMF for various periods of time prior to the addition of TEMPO and aldehyde 6. Given that the amount of free iron should increase with the duration of premixing, we would assume that the amount of direct enamine trapping to generate cis-8a should increase accordingly. Indeed, this is exactly observed. However, when no aging step is employed, there is almost no difference in product distribution between the use of ferrocenium in DMF or THF (15% 8a in THF, 22% 8a in DMF).
Scheme 4.

Product Distribution Using [FeCp2][PF6] in DMF
(ii) The 2.0 M ferrocenium case involves a diminished concentration of free TEMPO in comparison to the 2.0 M Sibi protocol
ReactIR experiments have clearly demonstrated that the in situ concentration of free TEMPO is effectively 2.0 M for both the FeCl3/NaNO2/DMF and [FeCp2][PF6]/THF cases (see Supporting Information).
(iii) The FeCl3 system acts by enamine addition, rather than a SOMO-type pathway
These combined results clearly indicate that the ferrocenium case predominantly leads to radical clock opening in DMF whereas the preferred Sibi protocol does not, and by inference, does not involve a radical cation as the major reaction pathway. Importantly, the observation of small amounts of rearranged product when using FeCl3 at low TEMPO concentration (15% trans at 0.75 M TEMPO) has been shown to arise from increased content of the unbound iron oxidant (see Supporting Information).
Evidence for a TEMPO complexation/enamine addition pathway was further substantiated by consideration of the oxidation potentials of the reagents and intermediates involved (Figure 1). It is well established that DMF solvation of FeCl3 induces disproportionation to [Fe(dmf)3Cl2][FeCl4],14a a salt which is less oxidizing than FeCl3 by ~0.5 V.14b Having measured the irreversible oxidation potential of enamine 2 (derived from catalyst 1), we have found that it is well-matched for oxidation with [FeCp2][PF6], while oxidation by [Fe(dmf)3Cl2][FeCl4] is endergonic by ~0.5 V.
Figure 1.

Cyclic Voltammetry Measurements in DMF
Given that our mechanistic picture for enamine addition was founded upon previous studies of TEMPO-metal coordination,7-10 a number of further predictions can be made. First, metal complexes known to bind TEMPO in a manner similar to [Fe(dmf)3Cl2]+ (including CuCl2) should effect the desired transformation. Second, Lewis acids that do not have an empty d-orbital, and thereby cannot participate in TEMPO binding, should not be effective. As shown in Table 1, these predictions were comprehensively confirmed. Most notably, the use of TEMPO or oxoammonium without metal additives15 gave similar results to the Sc(OTf)3 and Zn(NTf2)2 cases.
Table 1.
Other Systems for Catalytic Aldehyde α-Oxyamination
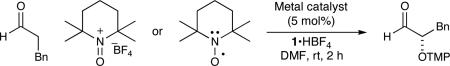 | |||
|---|---|---|---|
| entry | oxygen source | metal catalyst | % yielda |
| 1 | TEMPO | none | 11 |
| 2 | Oxoammoniumb | none | 19 |
| 3 | TEMPO | Sc(OTf)3 | 17 |
| 4 | TEMPO | Zn(NTf2)2 | 12 |
| 5 | TEMPO | [Fe(dmf)3Cl2][FeCl4] | 72 |
| 6 | TEMPO | Co(salen) | 75 |
| 7 | TEMPO | CuCl2 | 82 |
Yield determined by 1H NMR analysis relative to internal standard.
Added as a solution in DMF by syringe pump over 1h.
Finally, kinetic measurements have revealed that the initial rate of reaction depends exclusively on amine catalyst and aldehyde concentration, indicating that enamine formation is rate-determining (Scheme 5). Moreover, while FeCl3 is essential, identical levels of conversion were obtained without NaNO2 and O2 (the oxidant for the Sibi protocol is likely TEMPO,16 which is known to reoxidize Cu(I) to Cu(II) in related alcohol oxidations17). The sum of our results indicate that a significant revision to the proposed mechanism for the FeCl3-catalyzed α-oxyamination of aldehydes is required. The mechanistic picture that best fits literature precedent and the results contained herein is shown in Scheme 5.18 While the results of our investigations suggest that FeCl3/NaNO2/O2 will not find application in SOMO-catalysis, we expect that activation of TEMPO by metal complexation will lead to several new and efficient oxygenation protocols.
Scheme 5.

Revised Mechanism for α-Oxygenation of Aldehydes
Supplementary Material
Acknowledgement
Financial support was provided by NIHGMS (R01 01 GM093213-01) and gifts from Merck and Amgen. J. F. VH. thanks the Natural Sciences and Engineering Research Council (NSERC) for a predoctoral fellowship (PGS D).
Footnotes
Supporting Information Available: Experimental procedures, kinetic investigations, and spectral data for all new compounds are provided (38 pages) (PDF).
References
- 1.Beeson TD, Mastracchio A, Hong J, Ashton K, MacMillan DWC. Science. 2007;316:582.Sibi MP, Hasegawa M. J. Am. Chem. Soc. 2007;129:4124. doi: 10.1021/ja069245n.Narasaka K, Okauchi T, Tanaka K, Murakami M. Chem. Lett. 1992:2099. For a stoichiometric, non-catalytic example, see:
- 2.a Jang H–Y, Hong J–B, MacMillan DWC. J. Am. Chem. Soc. 2007;129:7004. doi: 10.1021/ja0719428. [DOI] [PubMed] [Google Scholar]; b Kim H, MacMillan DWC. J. Am. Chem. Soc. 2008;130:398. doi: 10.1021/ja077212h. [DOI] [PubMed] [Google Scholar]; c Wilson JE, Casarez AD, MacMillan DWC. J. Am. Chem. Soc. 2009;131:11332. doi: 10.1021/ja904504j. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Conrad JC, Kong J, Laforteza BN, MacMillan DWC. J. Am. Chem. Soc. 2009;131:11640. doi: 10.1021/ja9026902. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Graham TH, Jones CM, Jui NT, MacMillan DWC. J. Am. Chem. Soc. 2008;130:16494. doi: 10.1021/ja8075633. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Rendler S, MacMillan DWC. J. Am. Chem. Soc. 2010;132:5027. doi: 10.1021/ja100185p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang N, Liu R, Chen J, Liang X. Chem. Commun. 2005:5322. doi: 10.1039/b509167e. [DOI] [PubMed] [Google Scholar]
- 4.a Braslau R, Burrill LC, II, Siano M, Naik N, Howden RK, Mahal LK. Macromolecules. 1997;30:6445. [Google Scholar]; b Ullrich J. J. Org. Chem. 1998;63:7130. doi: 10.1021/jo981180m. [DOI] [PubMed] [Google Scholar]
- 5.Koike T, Akita M. Chem. Lett. 2009;38:166. [Google Scholar]
- 6.a Bui N–N, Ho X–H, Mho S–I, Jang H–Y. Eur. J. Org. Chem. 2009:5309. [Google Scholar]; b Schämann M, Schäfer HJ. Electrochimica Acta. 2005;50:4956. [Google Scholar]
- 7.a Semmelhack MF, Schmid CR, Cortes DA. Tetrahedron Lett. 1986;27:1119. [Google Scholar]; b Vogler T, Studer A. Synthesis. 2008:1979. [Google Scholar]
- 8.a Laugier J, Latour J, Caneschi A, Rey P. Inorg. Chem. 1991;30:4474. [Google Scholar]; b Pervukhina NV, Romanenko GV, Podberezskaya NV. J. Struct. Chem. 1994;35:367. [Google Scholar]
- 9.Michel C, Belanzoni P, Gamez P, Reedjik J, Baerends EJ. Inorg. Chem. 2009;48:11909. doi: 10.1021/ic902155m. [DOI] [PubMed] [Google Scholar]
- 10.Dijksman A, Arends IWCE, Sheldon RA. Org. Biomol. Chem. 2003;1:3232. doi: 10.1039/b305941c. [DOI] [PubMed] [Google Scholar]
- 11.Newcomb M. Tetrahedron. 1993;49:1151. [Google Scholar]
- 12.Cyclopropyl radical ring opening/re-closure prior to productive bond formation has been previously observed: Benkovics T, Du J, Guzie IA, Yoon TP. J. Org. Chem. 2009;74:5545. doi: 10.1021/jo900902k.Spence EL, Langley J, Bugg TDH. J. Am. Chem. Soc. 1996;118:8343.
- 13.Prins R, Korswagen AR, Kortbeek AGTG. J. Organomet. Chem. 1972;39:335. [Google Scholar]
- 14.a Tobinaga S, Kotani E. J. Am. Chem. Soc. 1972;94:309. [Google Scholar]; b Safavi A, Fotouhi L. Microchemical Journal. 1998;60:224. [Google Scholar]
- 15.A background reaction with aldehyde and oxoammonium (which can also be formed in situ from TEMPO) was also observed.
- 16.Two equivalents of TEMPO allows for its action as both the oxygen atom source and stoichiometric reoxidant for iron. See reference 1b.
- 17.Sheldon RA, Arends IWCE. Adv. Synth. Catal. 2004;346:1051. [Google Scholar]
- 18.Enamine/metal-TEMPO electron transfer, followed by solvent cage radical combination, cannot be excluded based on the studies herein.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.