

Abstract
A novel multi-domain biopolymer was designed and genetically engineered with the purpose to target and transfect cancer cells. The biopolymer contains at precise locations: 1) repeating units of arginine and histidine to condense pDNA and lyse endosome membranes, 2) a HER2 targeting affibody to target cancer cells, 3) a pH responsive fusogenic peptide to destabilize endosome membranes and enhance endosomolytic activity of histidine residues, and 4) a nuclear localization signal to enhance translocation of pDNA towards the cell nucleus. The results demonstrated that the biopolymer was able to condense pDNA into nanosize particles, protect pDNA from serum endonucleases, target HER2 positive cancer cells but not HER2 negative ones, efficiently disrupt endosomes, and effectively reach the cell nucleus of target cells to mediate gene expression. To reduce potential toxicity and enhance biodegradability, the biopolymer was designed to be susceptible to digestion by endogenous furin enzymes inside the cells. The results revealed no significant biopolymer related toxicity as determined by impact on cell viability.
Keywords: Biopolymer, Genetically engineered polymer, Biomimetic vector, Non-viral vector, Gene therapy
1. Introduction
Currently, an efficient non-viral gene delivery vehicle (vector), which mimics each step of the viral entry and infection, has yet to be developed. As a result, non-viral gene transfer technology remains stagnant due to the limitations of the existing vectors. Viral vectors are the current gene delivery vehicle of choice because of their high transfection efficiency and success in preclinical trials. However, immunogenicity and toxicity issues have arisen and clinical results have not translated into a successful commercial product. Lipoplexes are a viral alternative without the immunogenicity concerns; however, they have reproducibility and cytotoxicity issues [1]. Cationic polyplexes have less biocompatibility concerns, but their transfection efficiencies remain low [2]. For a non-viral vector to successfully overcome biological barriers, it must accurately mimic viral characteristics. Accurate mimicry of viral infection and ultimately full control over gene transfer processes requires a greater understanding of the natural mechanisms involved at the molecular scale and subsequent development of a class of biomaterials that would allow precise correlation of one dimensional design (amino acid sequence) with three-dimensional functionality.
Amino acid based polymers can be synthesized using genetic engineering techniques resulting in biopolymers with precise compositions, molecular weights, sterotacticity and specified functions [3,4). Compared to other conventional methods, the principal advantages are (i) monodisperse material, (ii) full control over polymer architecture at the molecular level, (iii) precise covalent attachment of functional moieties (e.g. targeting motifs), and from a manufacturing standpoint, (iv) elimination of the conjugation steps. Since most conventional polymers are synthesized using free radical addition or similar methods, the resulting polymer is heterodisperse. Heterogeneity in turn undermines conjugation or covalent coupling of other moieties at precise locations which complicates characterization and controlled drug delivery. In contrast, biopolymers can overcome these issues because of the fidelity associated with protein expression [5].
The overall objective of this research is to develop a gene delivery system that is customizable, easy to engineer, efficient and non-toxic. As a first step towards achieving the objective, a multifunctional biopolymer was designed and genetically engineered (Fig. 1). The architecture of this biopolymer is designed based on our current and past experience with genetically engineered biomimetic vectors [6–8]. It features a unique domain with repeating units of arginine-histidine (RH) with the general structure of (RRXRRXHHXHHX)n where X is any amino acid except D and E and n is the number of repeating units. This domain is named the DNA condensing and endosomolytic motif (DCE) where the ratio of R to H is kept constant at 50:50. Other domains include a fusogenic peptide (FP) for endosomal disruption [9], a C-terminal HER2 targeting motif (TM) [10] and a M9 nuclear localization signal (NLS) to enhance translocation of genetic material towards nucleus [11]. A cathepsin D enzyme substrate (CS) has also been engineered in between TM and NLS to facilitate dissociation of the targeting motif from the biopolymer inside late endosomes where cathepsin D is abundant [12]. To simplify, the biopolymer will be referred to as FP–(DCE)n–NLS–CS–TM. Biopolymers with the following structures were also genetically engineered (DCE)n–NLS–CS–TM [biopolymer without FP) and FP–(DCE)n–CS–TM (biopolymer without NLS) and used as controls.
Fig. 1.
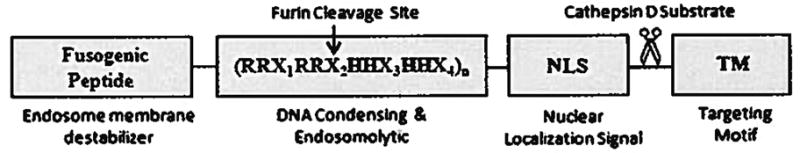
Schematic of the designed multidomain biopolymeric gene carrier composed of fusogenic peptide, DNA condensing and endosomolytic motif, nuclear localization signal and targeting motif. The corresponding amino acid sequence for each domain is also shown.
It is our central hypothesis that a biopolymer with multiple functional domains can be engineered to condense plasmid DNA (pDNA) into stable nanosize carriers, target model HER2 positive cancer cells, disrupt endosome membranes efficiently facilitating escape into the cytosol, and ultimately reach the nucleus to mediate gene expression.
2. Materials and methods
2.1. Cloning and expression of the biopolymer
The gene encoding FP–(DCE)3–NLS–CS–TM was designed, expression optimized, and synthesized by Integrated DNA Technologies (San Diego, CA) with N-terminal NdeI and C-terminal XhoI restriction sites. The synthesized gene was double digested with NdeI and XhoI (New England Biolabs, Ipswich, MA) restriction enzymes and cloned into a pET21b expression vector (EMD Biosciences, Gibbstown, NJ) to make pET21b:FP–(DCE)3–NLS–CS–TM. After cloning, the fidelity of the gene to its original design was verified by DNA sequencing. The expression vector was transformed into E coli BL21(DE3) pLysS (Novagen, San Diego, CA) and grown in a Barnstead-Labline MAX Q 4000 shaking incubator at 30 °C. Circlegrow media (MB Biomedicals, Solon, OH) starter cultures were gown overnight and used to inoculate 500 mL media containing 50 μg/mL carbenicillin. At OD600 of 0.6, gene expression was induced by the addition of IPTG to a final concentration of 0.4 mM at 30 °C for 4 h. Cells were collected at 5000 rpm for 10 min and frozen until further use. The pellet was suspended in lysis buffer (50 mM NaH2P04, 500 mM NaCl, 8 M urea, 12 mM 2-mercaptoethanol, 0.5% Triton X-100, 10 mM immidazole, pH 8.0) and centrifuged for 60 min at 30,000 ×g at 4 °C to pellet the insoluble fraction. The soluble fraction was incubated with 0.5 mL of Ni–NTA resin (Qiagen) equilibrated with lysis buffer. Incubation at room temperature with gentle mixing allowed for complete binding. The resin was centriguged at 1000 × g for 5 min. Supernatant was discarded and resin was loaded onto a 0.8 × 4 mL BioRad PolyPrep chromatography column. The column was washed with 40 volumes of wash buffer (50 mM NaH2P04, 1000 mM NaCl, 7 M urea, 12 mM 2-mercaptoethanol, 0.5% Triton X-100, 20 mM immidazole pH 8.0) and then eluted with 5 mL of elution buffer (50 mM NaH2P04, 250 mM NaCl, 5 M urea, 12 mM 2-mercaptoethanol, 250 mM immidazole pH 8.0). The purity and expression of the vector were confirmed by SDS-PAGE and western blot analysis using monoclonal mouse anti-6XHis antibody. The purified biopolymer was stored at −20 °C after adding glycerol to 40% final concentration. Mass spectrometry was used to determine the exact molecular weight of the purified biopolymer. Prior to use in further assays salts and buffer exchange was performed using a G-25 sepharose size exclusion resin. FP–(DCE)3–CS–TM (without NLS) and (DCE)3–NLS–CS–TM (without FP) were also purified as above.
22. Recognition of cathepsin D substrate by the cathepsin D enzyme
Cathepsin D enzyme (human liver) was purchased form Calbio-chem (Gibbstown, NJ) and dissolved as per manufacturer’s protocol in 0.1 M Glycine–HCl, 0.5% Triton X-100 and 150 mM NaCl pH 3.5 to a final concentration of 1 unit enzyme per 50 μL buffer. Purified biopolymer (10 μg) was incubated with 1 unit of enzyme for 3 h at 37 °C. Enzymatic reaction was stopped by addition of 2X Laemelli buffer (BioRad, Hercules, CA). Samples were boiled at 95 °C for 5 min and loaded onto 15% SDS-PAGE gel. Undigested biopolymer was used as control.
2.3. Susceptibility of biopolymer to proteolytic activity of furin
Furin protease was purchased from New England Biolabs (Ipswich, MA). Biopolymer (10 μg) was prepared in 50 μL of buffer (100 mM sodium phosphate, 0.5% Triton X-100, 1 mM CaCl2, 1 mM 2-mercaptoethanol) and pH was adjusted to 7.2, 6.0 and 5.5. One unit of furin enzyme was added and incubated at room temperature for 3 h. Reaction was stopped by addition of 2X Laemelli buffer (Sigma). Samples were boiled at 95 °C for 5 min and loaded onto SDS-PAGE gel.
2.4. Hemolysis assay
Two milliliters of sheep red blood cells (Innovative Research, Novi, MI) were washed 2 times with Dulbeccos Phosphate Buffered Saline (Invitrogen, Carlsbad, CA). Cell numbers were adjusted to 10 × 108 cells/mL in DPBS at pH 7.4 or 6.0. Various amounts (0.5, 1.0 or 20 μg) of the biopolymer or biopolymer without fusogenic peptide were added to the cell suspension and incubated at 37 °C for 1 h in a shaking incubator set at 60 rpm. Cells were pelleted by centrifugation and the absorbance of the supernatant was measured at 541 nm. Triton X-100 (1%) was used as a positive control while buffers only (pH 7.4 and 6.0) were the negative controls. The percentage of hemolysis was reported relative to the positive Triton X-100 control which was defined as 100%. Data is reported as mean ± s.d. n = 3. Statistical significance was evaluated using t-tests (p<0.05).
2.5. DNA neutralization at different pH
Gel mobility assays were performed to examine the neutralization of the pDNA negative charges by the biopolymer. pEGFP (1 μg) was complexed with the biopolymer at various N:P ratios (Nitrogen to Phosphate) at pH 7.4 and pH 5.5. N:P ratios were calculated based on the number of arginine, histidine and lysine residues in the biopolymer sequence. After incubation at room temperature for 15 min, the mobility of the pDNA was visualized on an agarose gel by ethidium bromide.
2.6. Particle size and charge analysis
Various amounts of biopolymer in 20 mM Tris–HCl buffer at pH 5.5 were added to 1 μg pEGFP to form complexes at a range of N:P ratios in a total volume of 100 μL. For example, at N:P ratio of 1, 1.5 μg of biopolymer was used to condense 1 μg of pEGFP. After incubation at room temperature for 15 min, the mean hydronamic particle size and charge measurements were performed using Dynamic Light Scattering (DLS) and Laser Doppler Velociometry (LDV) on a Malvern Nano ZS90 instrument running DTS software (Malvern Instruments, UK). Size and zeta potential were measured and reported as mean ± SEM (n = 3). Each mean is the average of 15 measurements and n represents the number of separate batches prepared for the measurements.
2.7. Particle stability in the presence of serum
One μg plasmid DNA (pEGFP) was complexed with the biopolymer at N:P ratio of 14 and incubated for 15 min. After stable particles had formed, fetal bovine serum (Invitrogen, CA, USA) was added to a final concentration of 10% (v/v). Complexes were incubated for 30 min at 37 °C. Subsequently, 1% heparin (Sigma) was added to decomplex pDNA from biopolymer. Samples were then electrophoresed on a 1% agarose gel and visualized with ethidium bromide.
2.8. Cell culture and transgene expression
SK-OV-3 cells (human ovarian cancer) with high levels of HER2 expression and MDA-MB-231 (human breast cancer) and PC-3 (human prostate cancer) cells with low levels of HER2 expression were seeded in a 96-well plate at 2.0 × 104 cells per well. Cells were incubated overnight at 37 °C until 80–90% confluent. Cells were transfected with biopolymer/pEGFP complexes at various N:P ratios in the presence of McCoy media supplemented with antibiotic, transferrin, selenium, ovalbumin, dexamethasone, and fibronectin. Three hours after transfection the media was removed and replaced with fresh McCoy media supplemented with 10% serum. If used, 100 μM chloroquine or 100 nM bafilomycin (Sigma, Milwaukee, WI) were added 5 min prior to transfection. 10 μM nocodazole (Sigma, Milwaukee, WI) was added 20 min prior to transfection when used. An epifluorescent microscope (Carl Zeiss) was used to qualitatively visualize expression of the green fluorescent protein (GFP). Total green fluorescence intensity and percent transfected cells were measured using a FACS Calibur flowcytometer (BD Biosciences). The total fluorescence intensity of positive cells was normalized against the total fluorescence intensity of untransfected cells (control) to account for cellular auto-fluorescence. The total green fluorescence intensity is an indicator of overall GFP expression levels. The data are presented as mean ± s.d, n = 3. Statistical significance was evaluated using t-test (p<0.05).
2.9. Inhibition assay
The details of this method has been reported previously [6,7]. In brief, SKOV-3 cells were pre-treated with the competitive inhibitor (HER2 targeting motif) at various concentrations (2.5, 50, and 100 μg/ml) followed by transfection with biopolymer/pEGFP complexes at N:P ratio of 14 in serum free media. After 3 h of incubation at 37 °C, media was removed and replaced with media supplemented with 10% serum. Cells were collected 48 h post transfection and total fluorescence intensity was measured using flow cytometry. Untreated SKOV-3 cells (0 μg) were transfected with biopolymer/pEGFP complexes at N:P ratio of 14 and used as control. The data are presented as mean ± s.d, n = 3.
2.10. Toxicity assay
SK-OV-3 cells were seeded in 96-well plates at 2 × 104 cells per well in McCoy media plus 10% serum. Cells were treated with various amounts of biopolymer/pEGFP complexes or phosphate buffer saline (PBS) for 3 h. The media was removed and replaced with fresh media supplemented with 10% serum followed by overnight incubation at 37 °C humidified C02 atmosphere. Twenty four hours after incubation with biopolymer/pEGFP complexes, WST-1 reagent (Roche Applied Science, Indianapolis, IN) was added, incubated for 4 h followed by measuring absorbance at 440 nm. The measured absorbance for test groups is expressed as percent of the control where the control is defined as %100 viable. The data are reported as mean ± s.d., n = 3. The statistical significance was evaluated using a t-test (p<0.05).
3. Results
3.1. Cloning, expression, and characterization of biopolymer
Using the cloning strategy shown in Fig. 2a, the biopolymer DNA sequence was cloned into pET21b expression vector. The fidelity of the DNA sequence to its original design was confirmed by DNA sequencing. The biopolymer was expressed in E. coli and purified with a 10 mg/L yield. SDS-PAGE and western blot analysis confirmed the expression and high purity of the biopolymer (Fig. 2b). The exact molecular weight of the biopolymer was determined to be 22,682 Da which is in close agreement with the theoretical value of 22,613 Da (Fig. 2c).
Fig. 2.

Cloning, expression, and characterization of the purified biopolymer. a) An overview of the cloning strategy used to clone FP–(DCE)3–NLS–CS–TM gene into pET21b expression vector, b) SDS-PAGE (left panel) and western blot analysis (right panel) of purified biopolymer. c) The MALDI-TOF spectra of the purified biopolymer. The observed molecular weight was 22,682 Da.
Furin and cathepsin D substrates were incorporated into the biopolymer architecture to enable biopolymer digestion upon entry into the cell. The accessibility of the furin substrate in the biopolymer structure to the enzyme was evaluated at different pH conditions (Fig. 3a, lanes 2,3 and 4). The results demonstrate that biopolymer can be digested by furin at various pH values with optimum digestion at pH 5.5.
Fig. 3.
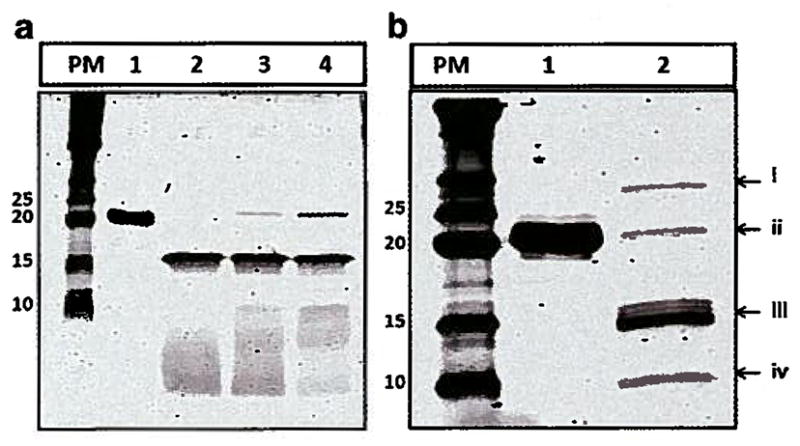
Digestion of biopolymer by furin and cathepsin D. left) Furin cleavage; PM: Protein Marker, lane 1: undigested biopolymer, lane 2: furin digestion at pH 5.5, lane 3: furin digestion at pH 6, lane 4: furin Digestion at pH 7. b) Cathespin D cleavage; PM: Protein Marker, lane 1: undigested biopolymer, lane 2: cathepsin D digestion, i – cathepsin D fragment (~30 kDa), ii – uncut biopolymer (~22.6 kDa), iii – biopolymer fragment (~ 14.4 kDa) and cathepsin D fragment (~15 kDa), iv – biopolymer fragment ~8.4 kDa.
The results of biopolymer digestion by cathepsin D illustrates that the substrate is readily available to the protease (Fig. 3b, lane 2). Digestion by cathepsin D results in bands at 14.2 kDa (iii) and 8.4 (iv) kDa. The bands at 30 kDa (i) and 15 kDa (iii) are a result of autolysis of the cathepsin D enzyme at the conditions used [13].
3.2. Effect of pH on DNA neutralization
The gel mobility assays were performed at pH 7.4 and 5.5 to evaluate the effect of partially charged histidine residues on DNA neutralization. At pH 5.5, 15 μg of biopolymer (N:P~ 10) was sufficient to fully neutralize 1 μg of pEGFP. However, at pH 7.4, 37 μg of biopolymer (N:P~ 25) was needed to fully neutralize the same amount of pEGFP (Fig. 4a and b).
Fig. 4.
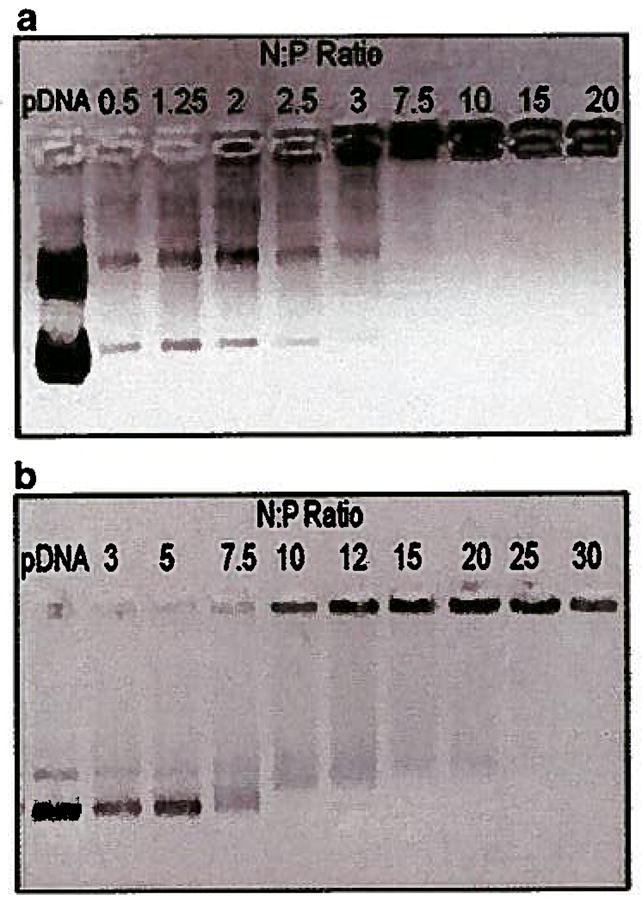
DNA neutralization by biopolymer at two different pH values. a) Gel mobility assay at pH 5.5 at various N:P ratios. b) Gel mobility assay at pH 7.4 at various N:P ratios.
3.3. Evaluation of particle size, charge, and stability
Biopolymer/pEGFP complexes were formed at various N:P ratios ranging from 1 to 20 at pH 5.0 followed by the size and zeta potential measurements. At N:P ratios greater than 5.5, nanoparticles with sizes below 100 nm were obtained (Fig. 5a). For example, at N:P ratio of 15, the size of the nanoparticles were 78 ± 4 nm with a zeta potential of + 10 ± 1 mV.
Fig. 5.

Size, charge, and serum stability of biopolymer/pEGFP complexes: a) Sizes and zeta potentials of complexes at various N:P ratios b) Lane 1: pEGFP only. Lane 2: pEGFP incubated with serum, Lane 3: Bipolymer/pEGFP in the absence of serum, Lane 4: Biopolymer/pEGFP incubated with Serum, Lane 5: released pEGFP from the vector/pEGFP complexes after incubation with serum.
The ability of biopolymer to shield the genetic material from serum endonucleases was examined by incubating the biopolymer/pEGFP with serum. The results demonstrated that the nanoparticles were stable in the presence of serum and the pEGFP was protected (Fig. 5b, lanes 4 and 5). Plain pEGFP was fully degraded by the serum nucleases (Fig. 5b, lane 2).
3.4. Determination of optimum N:P ratio for cell transfection
SKOV-3 cells were transfected with biopolymer/pEGFP at various N:P ratios ranging from 5 through 15. The results revealed that at N:P ratio of 14 the highest level of transfection efficiency was achieved (Fig. 6a). At N:P ratio of 14, 21 ± 1.0% of cells were transfected with total green fluorescence intensity of 724,000 ± 65,000. At N:P 15, the total green fluorescence intensity and the percentage of transfected cells significantly decreased.
Fig. 6.

Determination of optimum N:P ratio and biopolymer related toxicity. a) Transfection efficiency of biopolymer/pEGFP at various N:P ratios in SKOV-3 cells. b) WST-1 cell toxicity assay for SK-OV-3 cells treated with various concentrations of biopolymer/pEGFP complexes equivalent to 0 to 110 μg/ml of biopolymer.
A toxicity assay was also performed to examine the potential toxicity of biopolymer and its effect on cell transfection. The results showed no significant toxicity in the SKOV-3 cells at up to 110 μg/mL concentrations (Fig. 6b).
3.5. Evaluation of the functionality of targeting motif
To show targetability for HER2, SKOV-3 (high HER2 expression), MDA-MB231 and PC-3 (low HER2 expression) cells were transfected with biopolymer/pEGFP complexes at N:P ratio of 14. The results of flowcytometry showed 21 ± 2 percent cell transfection in SKOV-3 versus 2 ± 0.5 and 0.1 ± 0.02 percent for MDA-MB-231 and PC-3, respectively (Fig. 7a). Lipofectamine in complex with pEGFP was able to transfect all three cell lines non-selectively with relatively high efficiency.
Fig. 7.

Evaluation of the functionality of targeting motif: a) Epifluorescent images and corresponding percent transfected cells by biopolymer/pEGFP and lipofectamine. Percent transfection was determined by flow cytometry. b) Inhibition assay: SKOV-3 cells were pre-treated with various amounts of targeting peptide (competitive inhibitor) followed by transfection with biopolymer/pDNA complexes.
Receptor mediated endocytosis of nanoparticles via HER2 was examined by using an inhibition assay. The results of this assay showed that as the concentration of the targeting motif in the solution (competitive inhibitor) increased the levels of gene expression shown by total fluorescence intensity significantly decreased (Fig. 7b).
3.6. Functionality of the fusogenic peptide
pH dependent membrane disrupting activity of the fusogenic peptide was examined by hemolytic assay. The results of this assay revealed that the biopolymer was significantly lytic at pH 6.0 and only at high concentration (20 μg) (Fig. 8a). At pH 6.0 and low concentrations the biopolyemer did not show significant hemolytic activity. At pH 7.4 and 20 μg concentration, slight hemolytic activity was observed. Biopolymer without fusogenic peptide [biopolymer (–) FP] did not show any significant hemolytic activity at any concentration at pH 6.
Fig. 8.

Evaluation of the functionality of fusogenic peptide: a) The hemolytic activity of biopolymer and biopolymer (–) FP were evaluated at different pH (i.e., 7.4 and 6.0) and concentrations. Triton X-100 was used as positive control and DPBS buffer as the negative control. b) SKOV-3 cell transfection with biopolymer, biopolymer plus chloroquine, biopolymer plus bafilomycin, and biopolymer without fusogenic peptide [biopolymer (–) FP].
The ability of the biopolymer to efficiently disrupt endosome membranes was evaluated by transfecting SKOV-3 cells with the biopolymer/pEGFP complexes in the presence and absence of chloroquine and bafilomycin at N:P 14. Significant reduction in total green fluorescent protein expression was observed in cells treated with bafilomycin (Fig. 8b). However, no significant difference in total green fluorescent protein expression was observed in cells transfected in the presence or absence of chloroquine. In comparison to biopolymer, a significant reduction in green fluorescence intensity was observed when cells were transfected with biopolymer without fusogenic peptide.
3.7. Effect of nuclear localization signal on gene transfer
The effect of the NLS on gene transfer efficiency was examined by transfecting SKOV-3 cells with biopolymer and biopolymer without NLS. Comparison of the transfection efficiencies in these two constructs showed a marked decrease in total fluorescence intensity and percent cell transfection in the NLS deficient biopolymer (Fig. 9).
Fig. 9.
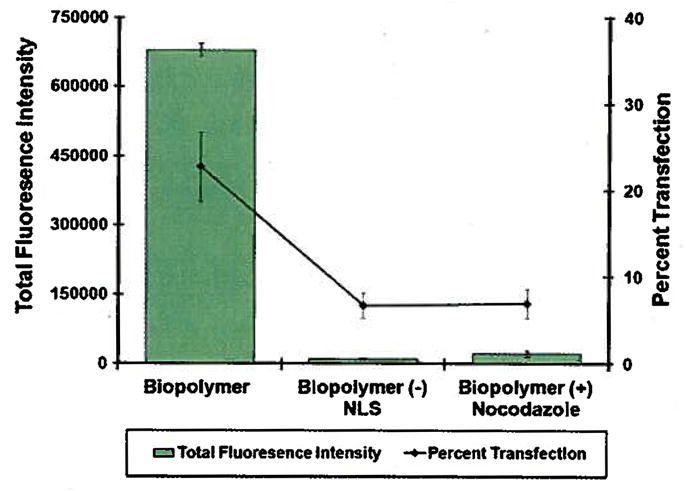
Evaluation of the functionality of nuclear localization signal. Biopolymer and biopolymer without NLS [(FP–(DCE)3–CS–TM) were complexed with pEGFP at N:P 14 and used to transfect SKOV-3 cells. Cells pretreated with nocodazole were also transfected with biopolymer/pEGFP complexes at N:P 14.
The effect of the microtubules network on gene transfer efficiency was evaluated by transfecting SKOV-3 cells in the presence of nocodazole (microtubule depolymerizer). Treatment with nocodazole showed a significant decrease in transfection efficiency (Fig. 9). At the concentration used (10 μM), nocodazole did not show any significant cell toxicity which could impact transfection efficiency. This concentration is in agreement with the literature [8,14].
4. Discussion
In order to facilitate correlation of vector structure with function and help identify the rate limiting steps to gene transfer by the targeted vector in this study, a chimeric biopolymer was designed. It is composed of multiple functional domains with potential to mimic some of the major viral characteristics including pDNA condensation, cell targeting, endosome disruption, and gene expression mediation.
The biopolymer was genetically engineered to contain at precise locations four major domains (Fig. 1). The first is a DNA condensing and endosomolytic (DCE) motif comprised of three repeating units of RRX1RRX2HHX3HHX4, where X can be any amino acid except aspartic acid (D) or glutatimic acid (E) as these negatively charged residues could interfere with DNA condensation. The role of the DCE is to primarily condense pDNA, and secondarily to disrupt endosomes via the proton sponge effect [15]. Histidine residues will become protonated in the low pH environment of the endosome resulting in swelling and eventual bursting of endosomes releasing the contents into the cytosol. In the DCE motif (RRX1RRX2HHX3HHX4) the arginine and histidine residues were arranged in clusters based on previous work by our group which found that a clustered arrangement in lysine-histidine biopolymers resulted in more stable DNA condensation as compared to a dispersed architecture [6]. To maximize the proton sponge effect without compromising the DNA condensing ability, the ratio of R:H in DCE domain is adjusted to 50:50 ratio.
Residue X1was designed to be valine (V) in order to generate RVRR sequences along the DCE unit. This sequence is a substrate for the protease furin which is ubiquitously present in the cell, and specifically abundant inside endosomes [16]. The furin enzyme recognizes the (R-X-R/K-R↓) sequence and cleaves at the ↓. site [17]. This site was engineered in the biopolymer structure to enhance the biodegradability of the DCE unit which could potentially be toxic. Large blocks of cationic amino acids can be toxic to cells as they could bind to various organelles and enzymes inside the cells and interfere with their functions. Degradation of the biopolymer into smaller pieces could reduce the cytotoxicity due to reduction in molecular weight and increase intracellular release of the genetic cargo [18,19].
Residues X2 and X4 are designed to be serine (S) and threonine (T). T and S were selected to increase the solubility of the biopolymer and yield of production while maintaining the balance of R to H (50:50) with no negative impact on DNA condensation. In addition, the terminal hydroxyl groups in S and T could potentially be involved in hydrogen bonding with the DNA backbone enhancing the complex stability.
Residue X3 in the first and second repeating unit is R and in the third repeating unit is H. This is to not only maintain the R to H balance, but incorporate an intrinsic histag into the biopolymer sequence which facilitates its purification via Ni-NTA chromatography.
The second functional domain in the biopolymer structure is a targeting motif that is an affibody with high specificity for biorecognition by model cancer cells over-expressing HER2 [10]. This is to enhance the targetability and internalization of the biopolymer/pDNA complexes via receptor mediated endocytosis. The third functional domain in the biopolymer sequence is an endosme destabilizing motif, namely 5HWYG, which is a synthetic derivative of the influenza virus fusogenic peptide. While influenza virus fusogenic peptide changes conformation at pH 5 to fuse with endosomal membranes, 5HWYG destabilizes endosomal membranes at pH 6.8 (9]. This fusogenic peptide was designed in the biopolymer structure to cooperatively enhance the endosomolytic activity of the histidine residues. The fourth major domain is a M9 nuclear localization signal (M9-NLS) derived from the heterogeneous nuclear ribonucleoprotein (hnRNP Al) [11]. This NLS was deliberately selected because it does not contain clusters of basic amino acid residues minimizing its interaction with pDNA and involvement in the DNA condensation process. Finally, a minor domain encoding cathepsin D substrate (CS) was engineered in between the NLS and the HER2 targeting motif (TM) to facilitate dissociation of the targeting motif from the biopolymer inside the endosomal compartment. This could enhance exposure of NLS to the cell’s importin machinery and facilitate the transport of pDNA across cytoplasm towards nucleus. To correlate structure with function and identify the shortcomings of the biopolymer, several experiments were performed and the functionality of each domain was examined.
The biopolymer sequence was designed, cloned into pET21b expression vector, expressed in E. coli and purified (Fig. 2). The biopolymer was characterized in terms of susceptibility to digestion by furin and cathepsin D enzymes. As mentioned above, three furin substrates (RVRR) were engineered in the DCE structure to facilitate its degradation. It was expected that the furin enzyme digest the biopolymer from these three sites resulting in multiple byproducts. However, Fig. 3a shows that only one site was accessible to the furin enzyme resulting in appearance of one band at ~ 15 kDa and one at below 10 kDa (unresolved). Because furin is a ubiquitous enzyme inside the cells, the degradation of biopolymer at three different pH values was evaluated. As expected maximum degradation of the biopolymer occurred at pH 5.5 which is close to endosomal pH because RVRR is an optimum substrate for endosomal furin [16]. The proteolytic cleavage results by cathepsin D showed that the substrate in the biopolymer structure is accessible and has the potential to be digested by the enzyme (Fig. 3b). As a result, it is expected that the NLS become exposed and play a significant role in gene transfection.
In order to identify the minimum required quantity of biopolymer needed to effectively neutralize the negative charges in pEGFP and minimize possible toxicity, a DNA neutralization study was performed. Various amounts of biopolymer were added to pEGFP at two different pH values (i.e., 7.4 and 5.5) to neutralize the negative charges. At pH 7.4, histidine residues are partially charged and do not make significant contribution to pDNA condensation. At pH 5.5 which is below the pKa of histidine (i.e., 6.0), histidine residues are protonated creating a continuous block of positive charge in the DCE segment with significant contribution to pDNA condensation. Thereby, at the lower pH, significantly less biopolymer is required to neutralize and condense pDNA (Fig. 4). Based on this observation, all the biopolymer/pDNA complexes were formed at pH 5.5.
In the next step, the ability of the biopolymer to efficiently condense pDNA into nanosize carriers suitable for receptor binding and cellular uptake was examined. Particle size studies demonstrated that at N:P ratios greater than 5 particles less than 100 nm in size can be obtained (Fig. 5a). This size range (< 150 nm) is optimal for targeted vectors as they can easily fit into clathrin-coated vesicles and internalize via receptor mediated endocytosis [20,21]. The surface charge studies showed that the zeta potential of the nanoparticles remained slightly positive even at high N:P ratios. This could be an indication that the affibody molecules are exposed at the surface imparting slight positive charge. It is noteworthy that the pi value for the affibody is 9.4 which make it positively charged at physiologic pH or lower. The exposure of the affibody on the surface of the nanoparticles was further investigated using an inhibition assay which will be discussed later.
The stability of nanoparticles in the presence of serum proteins and protection from serum nucleases is also of paramount importance. The serum stability was demonstrated by incubating the biopolymer/DNA complexes with serum. It was observed that the serum proteins were not able to dissociate the complexes and the biopolymer effectively protected the pDNA from serum endonucleases (Fig. 5b). Thus far, we have shown that the biopolymer is able to condense pDNA into stable nanosize particles in a size range that is suitable for cellular uptake. To examine the ability of these nanoparticles to enter the cells and mediate gene transfer, a cell transfection study was performed.
The optimum ratio of biopolymer to pEGFP for maximum gene transfer efficiency was determined by transfecting SKOV-3 cells with biopolymer/pEGFP complexes at various N:P ratios. The highest rate of gene transfection efficiency was observed at N:P 14 (Fig. 6a) and used in subsequent studies. To examine whether biopolymer related toxicity had any impact on determination of gene transfection efficiency, a cell toxicity assay was performed. The results of the cell toxicity assay indicated that the biopolymer did not have any significant effect on the viability of SKOV-3 cells in the range tested and as a result, did not alter the absolute values of transfection efficiency (Fig. 6b).
It was mentioned that an affibody was engineered in the biopolymer structure to target HER2 positive cells and facilitate internalization of nanocomplexes via receptor-mediated endocytosis. To demonstrate the functionality of the targeting moiety in the biopolymer structure, methods similar to those used for immunolipoplexes and targeted biopolymers were performed [7,22,23]. The cell targetability was demonstrated by transfecting HER2 positive and HER2 negative cell lines with biopolymer/pEGFP complexes. SKOV-3 was selected because the ability of the HER2 affibody to target this cell line has previously been illustrated [10]. Well characterized MDA-MB-231 breast cancer and PC-3 prostate cancer cell lines with low levels of HER2 expression were used as controls [24]. The results of this study show that the biopolymer/pEGFP complexes could selectively internalize into HER2 positive cells but not HER2 negative cells (Fig. 7a). Internalization of nanoparticles into SKOV-3 cells specifically via HER2-mediated endocytosis was illustrated by performing an inhibition assay (Fig. 7b). As result of these studies, it was concluded that the affibody in the biopolymer structure was functional and exposed on the surface of the nanoparticles.
Thus far, we have examined the functionality of DCE and TM segments in the biopolymer. The next motif in the biopolymer structure that was characterized was the pH responsive fusogenic peptide. The role of this motif is to assist in escape of cargo from the endosomes into cytosol. Fusogenic peptide 5HWYG assumes an alpha helical structure at pH 6.8 which helps the biopolymer to fuse with the endosome membranes and destabilize its integrity. The results of hemolysis assay show that lysis occurred only in high concentrations (20 μg) and at low pH (6.0) indicating the potential for endosomolytic activity (Fig. 8a). Under these circumstances, the possibility of cell damage by the biopolymer while in the circulating blood stream is minimal, as circulation will dilute the biopolymer concentration and the pH will remain at physiological levels until taken into endosomal compartments. Therefore, the position of the FP at the N-terminus of the biopolymer with the specified sequence preserves its pH-responsive functionality. The ability of the biopolymer to effectively disrupt endosome membranes resulting in the escape of the genetic material into cytosol was examined by transfecting the SKOV-3 cells in the presence and absence of chloroquine and bafilomycin. Chloroquine is a buffering agent with the capacity to burst endosomes and release the trapped complexes (if any) into cytosol [25]. Transfection in the presence chloroquine revealed no significant change in gene expression in comparison to cells transfected in its absence (Fig. 8b). This suggests that the biopolymer/pEGFP nanocarriers that were internalized via receptor mediated endocytosis, could effectively escape from the endosomes and did not remain trapped. Moreover, treatment with bafilomycin showed that a low pH was necessary for endosomal escape as a significant decrease in gene expression was observed when cells were treated with baflomycin at the time of transfection. This is due to the fact that baflomycin is an inhibitor of the vacuolar ATPase endosomal protein pump [26]. Inhibition of this pump reduced the release of the complexes into the cytosol and subsequent gene transfection efficiency due to the loss of proton sponge effect as well as conformational change of FP. Interestingly, the results of cell transfection with biopolymer without fusogenic peptide revealed the significant impact of this motif on efficient escape of nanocarriers from endosomes. Even though significant number of histidine residues were present in the biopolymer sequence, it was not sufficient to disrupt endosomes efficiently without the help of fusogenic peptide. This could be one important reason behind the fact that viruses never evolved to utilize proton sponge effect as a means to lyse endosomes.
It is well understood that the escape of the pDNA from the endosomes into cytosol is not sufficient to mediate gene expression as the target site for gene expression is the cell nucleus. To provide the means of active transport towards nucleus for pDNA, a M9-NLS was engineered in the biopolymer structure. The effect of M9-NLS in the biopolymer structure on gene transfer efficiency was evaluated by transfecting SKOV-3 cells with the biopolymer and biopolymer without NLS. The results showed significant decrease in transfection efficiency when cells were transfected with biopolymer without NLS (Fig. 9). This was expected as Gustin et al. (2001), have shown that when the M9-NLS is fused to GFP, the distribution of the resulting protein (GFP-M9NLS) was restricted to the nucleus [27,28]. Using the SWISS-MODEL program, the tertiary structure of M9-NLS was predicted (Fig. 10). Interestingly, the M9-NLS appears to be an unstructured peptide with no apparent α-helical or β-sheet structure. Lack of secondary structure and the fact that M9-NLS does not possess the ability to condense DNA could perhaps make it easier for the cell’s importin machinery to recognize its binding site and assist in gene transfer process to the nucleus and subsequent transfection. Although our speculation on the mechanism of transport is probable, more in depth studies on this subject are required to unravel the details of the intracellular trafficking processes involved. The effect of microtubules on the transport of the nanoparticles was also examined by transfecting cells in the presence of nocodazole. Nocodazole (microtubule depolymerizer) is a drug that is normally used to demonstrate the effect microtubules on the trafficking of non-viral vectors towards nucleus [14]. This drug has also been used to demonstrate the effect of NLS on microtubule-mediated transport of the viruses towards nucleus [29]. Pre-treatment of cells with nocodazole causes collapse of the shuttling system that interacts with the NLS sequence [30]. Consequently, it was expected that a significant reduction in gene transfection efficiency in the absence of microtubule network would be observed which was confirmed in SKOV-3 transfection studies. The observed results were another indication that the NLS in the biopolymer structure utilized microtubules to facilitate translocation of the nanoparticles towards nucleus (Fig. 9). Although this shuttling system does not guarantee nuclear entry; it does provide an opportunity based on proximity for the nanoparticles to enter the cells during the mitosis phase of the cell cycle when the nuclear membrane dissolves.
Fig. 10.
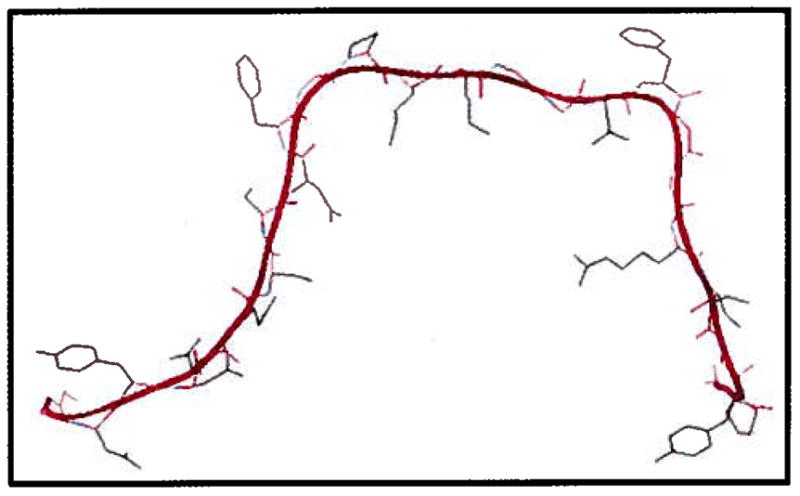
The 3D structure of the M9-NLS predicted by SWISS-MODEL program.
5. Future direction
We demonstrated that a biopolymer with well defined architecture can be engineered that is customizable, easy to engineer, nontoxic, and able to perform multiple tasks. The biopolymer in this study can condense pDNA into stable nanosize particles suitable for cellular uptake, target cancer cells, effectively disrupt endosomes and enhance translocation of the genetic material towards nucleus. However, the transfection efficiency may still have room for improvement. This opens the door for more studies aimed at optimizing the biopolymer architecture in order to achieve higher rates of gene transfer. One area that is of particular interest is the optimization of ligand density on the surface of the nanoparticles which could significantly impact particle internalization [31]. This can be studied by varying the number of repeating units (n) in the biopolymer structure to create biopolymers with different molecular weights. Furthermore, by changing the X amino acid residues in the biopolymer structure, more basic or lipophilic constructs can be obtained which could in turn impact gene transfer efficiency. Such constructs can be used as a basis to generate a library of biopolymers with various gene transfer capabilities. For example, substitution of the HER2 targeting motif with other targeting peptides could help to target and transfect different subpopulation of cancer cells. A series of studies are in progress to evaluate the in vivo gene transfer efficiency as well as potential immunogenicity of this biopolymeric gene delivery system.
Acknowledgments
This work was supported by the startup funds from the Washington State University to Hatefi and NIH biotechnology training fellowship (T-32 GM008336) to Canine.
References
- 1.Lv H, Zhang S, Wang B, Cui S, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release. 2006;114(1):100–109. doi: 10.1016/j.jconrel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 2.Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat Rev Drug Discov. 2005;4(7):581–593. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 3.Cappello J, Crissman J, Dorman M, Mikolajczak M, Textor G, Marquet M, Ferrari F. Genetic engineering of structural protein polymers. Biotechnol Prog. 1990;6(3):198–202. doi: 10.1021/bp00003a006. [DOI] [PubMed] [Google Scholar]
- 4.Urry DW. Physical chemistry of biological free energy transduction as demonstrated by elastic protein-based polymers. J Phys Chem, B. 1997;101(51):11007–11028. [Google Scholar]
- 5.Cappello J. Synthetically designed protein-polymer biomaterials. In: Park K, editor. Controlled drug delivery: The Next generation. American Chemical Socity; Washington, D.C: 1997. pp. 439–467. [Google Scholar]
- 6.Canine BF, Wang Y, Hatefi A. Evaluation of the effect of vector architecture on DNA condensation and gene transfer efficiency. J Control Release. 2008;129(2):117–123. doi: 10.1016/j.jconrel.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hatefi A, Megeed Z, Ghandehari H. Recombinant polymer-protein fusion: a promising approach towards efficient and targeted gene delivery. J Gene Med. 2006;8(4):468–476. doi: 10.1002/jgm.872. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Mangipudi SS, Canine BF, Hatefi A. A designer biomimetic vector with a chimeric architecture for targeted gene transfer. J Control Release. 2009 Mar 17; doi: 10.1016/j.jconrel.2009.03.005. ( http://dx.doi.org/10.1016/j.jconrel.2009.03.005) [DOI] [PMC free article] [PubMed]
- 9.Midoux P, Kichler A, Boutin V, Maurizot JC, Monsigny M. Membrane permeabilization and efficient gene transfer by a peptide containing several histidines. Bioconjug Chem. 1998;9(2):260–267. doi: 10.1021/bc9701611. [DOI] [PubMed] [Google Scholar]
- 10.Orlova A, Magnusson M, Eriksson TL, Nilsson M, Larsson B, Hoiden-Guthenberg I, Widstrom C, Carlsson J, Tolmachev V, Stahl S, Nilsson FY. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res. 2006;66(8):4339–4348. doi: 10.1158/0008-5472.CAN-05-3521. [DOI] [PubMed] [Google Scholar]
- 11.Siomi H, Dreyfuss G. A nuclear localization domain in the hnRNP A1 protein. J Cell Biol. 1995;129(3):551–560. doi: 10.1083/jcb.129.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haines AM, Irvine AS, Mountain A, Charlesworth J, Farrow NA, Husain RD, Hyde H, Ketteringham H, McDermott RH, Mulcahy AF, Mustoe TL, Reid SC, Rouquette M, Shaw JC, Thatcher DR, Welsh JH, Williams DE, Zauner W, Phillips RO. CL22 – a novel cationic peptide for efficient transfection of mammalian cells. Gene Then. 2001;8(2):99–110. doi: 10.1038/sj.gt.3301314. [DOI] [PubMed] [Google Scholar]
- 13.Lah T, Turk V. Autolysis studies of cathepsin D, Hoppe Seylers. Z Physiol Chem. 1982;363(3):247–254. doi: 10.1515/bchm2.1982.363.1.247. [DOI] [PubMed] [Google Scholar]
- 14.Sun J, Wirtz D, Hanes J. Efficient active transport of gene nanocarriers to the cell nucleus. Proc Natl Acad Sci U S A. 2003;100(7):3878–3882. doi: 10.1073/pnas.0636277100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Behr JP. The proton sponge: a trick to enter cells the viruses did not exploit. Chimica. 1997;51:34–36. [Google Scholar]
- 16.Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev, Mol Cell Biol. 2002;3(10):753–766. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bravo DA, Gleason JB, Sanchez RI, Roth RA, Fuller RS. Accurate and efficient cleavage of the human insulin proreceptor by the human proprotein-processing protease furin. Characterization and kinetic parameters using the purified, secreted soluble protease expressed by a recombinant baculovirus. J Biol Chem. 1994;269(41):25830–25837. [PubMed] [Google Scholar]
- 18.Oupicky D, Parker AL, Seymour LW. Laterally stabilized complexes of DNA with linear reducible polycations: strategy for triggered intracellular activation of DNA delivery vectors. J Am Chem Soc. 2002;124(1):8–9. doi: 10.1021/ja016440n. [DOI] [PubMed] [Google Scholar]
- 19.Read ML, Bremner KH, Oupicky D, Green NK, Searle PF, Seymour LW. Vectors based on reducible polycations facilitate intracellular release of nucleic acids. J Gene Med. 2003;5(3):232–245. doi: 10.1002/jgm.331. [DOI] [PubMed] [Google Scholar]
- 20.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377(Pt 1):159–169. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Putnam D, Gentry CA, Pack DW, Langer R. Polymer-based gene delivery with low cytotoxicity by a unique balance of side-chain termini. Proc Natl Acad Sci U S A. 2001;98(3):1200–1205. doi: 10.1073/pnas.031577698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hayes ME, Drummond DC, Hong K, Zheng WW, Khorosheva VA, Cohen JA, Park ONtCJW, Marks JD, Benz CC, Kirpotin DB. Increased target specificity of anti-HER2 genospheres by modification of surface charge and degree of PEGylation. Mol Pharmacol. 2006;3(6):726–736. doi: 10.1021/mp060040v. [DOI] [PubMed] [Google Scholar]
- 23.Yang T, Choi MK, Cui FD, Lee SJ, Chung SJ, Shim CK, Kim DD. Antitumor effect of paclitaxel-loaded PEGylated immunoliposomes against human breast cancer cells. Pharm Res. 2007;24(12):2402–2411. doi: 10.1007/s11095-007-9425-y. [DOI] [PubMed] [Google Scholar]
- 24.Rusnak DW, Alligood KJ, Mullin RJ, Spehar GM, Arenas-Elliott C, Martin AM, Degenhardt Y, Rudolph SK, Haws TFJ, Hudson-Curtis BL, Gilmer TM. Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb, GW572016) in an expanded panel of human normal and tumour cell lines. Cell Prolif. 2007;40(4):580–594. doi: 10.1111/j.1365-2184.2007.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xavier J, Singh S, Dean DA, Rao NM, Gopal V. Designed multi-domain protein as a carrier of nucleic acids into cells. J Control Release. 2009;133(2):154–160. doi: 10.1016/j.jconrel.2008.09.090. [DOI] [PubMed] [Google Scholar]
- 26.Bowman EJ, Siebers A, Altendorf K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci U S A. 1988;85(21):7972–7976. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gustin KE, Sarnow P. Effects of poliovirus infection on nucleo-cytoplasmic trafficking and nuclear pore complex composition. EMBO J. 2001;20(1–2):240–249. doi: 10.1093/emboj/20.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gustin KE, Sarnow P. Inhibition of nuclear import and alteration of nuclear pore complex composition by rhinovirus. J Virol. 2002;76(17):8787–8796. doi: 10.1128/JVI.76.17.8787-8796.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suh J, Wirtz D, Hanes J. Real-time intracellular transport of gene nanocarriers studied by multiple particle tracking. Biotechnol Prog. 2004;20(2):598–602. doi: 10.1021/bp034251y. [DOI] [PubMed] [Google Scholar]
- 30.Salman H, Abu-Arish A, Oliel S, Loyter A, Klafter J, Granek R, Elbaum M. Nuclear localization signal peptides induce molecular delivery along microtubules. Biophys J. 2005;89(3):2134–2145. doi: 10.1529/biophysj.105.060160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee Y, Sampson NS. Romping the cellular landscape: linear scaffolds for molecular recognition. Curr Opin Struct Biol. 2006;16(4):544–550. doi: 10.1016/j.sbi.2006.05.015. [DOI] [PubMed] [Google Scholar]