

Abstract
Energy-dependent protein degradation machines, such as the E. coli protease ClpAP, require regulated interactions between the ATPase component (ClpA) and the protease component (ClpP) for function. Recent studies indicate that the ClpP N-terminus is essential in these interactions, yet the dynamics of this region remain unclear. Here, we use synchrotron hydroxyl radical footprinting and kinetic studies to characterize functionally important conformational changes of the ClpP N-terminus. Footprinting experiments show that the ClpP N-terminus becomes more solvent-exposed upon interaction with ClpA. In the absence of ClpA, deletion of the ClpP N-terminus increases the initial degradation rate of large peptide substrates 5–15 fold. Unlike ClpAP, ClpPΔN exhibits a distinct slow phase of product formation that is eliminated by the addition of hydroxylamine, suggesting that truncation of the N-terminus leads to stabilization of the acyl-enzyme intermediate. These results indicate that 1) the ClpP N-terminus acts as a “gate” controlling substrate access to the active sites, 2) binding of ClpA opens this “gate”, allowing substrate entry and formation of the acyl-enzyme intermediate, and 3) closing of the N-terminal “gate” stimulates acyl-enzyme hydrolysis.
Energy-dependent protein degradation plays a vital role in protein homeostasis in all organisms (1). Examples of proteolytic machines include the eukaryotic 26S proteasome and the bacterial proteases, FtsH, Lon, HslUV, ClpXP, and ClpAP. In these proteolytic complexes, AAA+ ATPases (ATPases associated with various cellular activities) recognize, unfold, and translocate protein substrates into barrel-shaped compartmental proteases where the proteins are hydrolyzed and released as peptide products (2–4). Despite significant sequence divergence, the overall architecture of these complexes is conserved. For example, in ClpAP, ClpXP, HslUV, and the 26S proteasome, the protease subunits (ClpP, HslV, or the 20S peptidase) form either hexameric or heptameric ring structures that stack face to face, resulting in the sequestering of the active sites in a solvent-exposed chamber (5–8). The ATPase subunits (ClpA, ClpX, HslU, or the 19S complex) form hexameric ring-shaped structures that stack co-axially with the ring-shaped proteases, creating a continuous pore through which denatured protein substrates can be passed from the ATPase subunit into the protease compartment (9–13).
Understanding the communication between the ATPase component and the protease component is a key element in understanding the mechanism of protein degradation by these machines. A number of domain interactions have been shown to participate in communication between the protease ClpP and its partner ATPases, ClpX and ClpA. The IGF/L loops of ClpA and ClpX that interact with ClpP are important in the formation of ClpA/XP complexes (14–16), with all six loops being required for complex formation (17). In addition, the N-terminal ClpP axial loops interact with the pore-2 loops of ClpX (17), and mutations and deletions in the ClpP N-terminal region abolish binding to both ClpX and ClpA (18–20).
Although these previous studies revealed important interactions between ClpP and ClpX/ClpA, it remains an open question as to how the IGF/L loops and the N-terminal ClpP loops may change conformation in the course of the catalytic cycle. The sequence of the ClpP N-terminus is highly conserved across many species (18, 19); however, structural studies indicate that that this region of ClpP is flexible (5, 18–23). These observations suggest that this region may play an important dynamic role in the processing of peptide substrates. ClpP structures have been solved from five different organisms, and multiple crystal structures exist for some of these species (5, 18–23) (see Ref. (24) for a review). While the overall structure of ClpP is highly conserved among the structures solved to date, a high degree of variability exists in the conformation of the N-terminal region. In structures for which electron density is observable, the N-terminus crystallizes in one of two ways—in an “up” conformation or in a “down” conformation. In the “up” conformation, which has been observed in Escherichia coli (20, 21), Streptococcus pneumoniae (19), and human (18) enzymes, the N-terminus forms a loop in which the first ~7 residues line the axial pore while residues ~8–16 form a flexible loop extending out of the pore. The “down” conformation (observed in Escherichia coli (20)) is less well-defined. In this form, no residues are visibly protruding from the pore; electron density assigned to the first eleven residues is present in the pore, but is poorly defined. In their 2006 paper, Bewley et al. published a ClpP structure in which 6 of the N-terminal loops on a ClpP heptamer were in the “up” conformation while one was in the “down” conformation (20). They suggested that this arrangement of N-termini may facilitate a local symmetry match between ClpP and ClpX/ClpA, which may play a critical role in complex formation and/or catalysis.
The flexibility of the ClpP N-terminus exhibited in multiple crystal structures, along with the recent finding that it is important in interactions with its partner ATPases, suggests that the ClpP N-terminus could gate access to the ClpP degradation chamber. Such a mechanism is utilized in the proteasome (25–29). The N-termini of the 20S core particle alpha subunits occupy the pore region of this cylindrical protease. Access to the active sites is permitted only by removal of these loops, which is facilitated by the binding of its partner ATPase, the 19S regulatory particle. A similar mechanism may be employed by ClpAP and ClpXP. When exit of peptide products through the equatorial pores of ClpP is blocked, truncation of the N-terminus allows these products to escape the ClpP complex (30), suggesting that occupancy of the N-terminus in the ClpP pore interferes with transport of peptides through the complex.
Here, we describe synchrotron hydroxyl radical footprinting experiments that identify conformational changes that occur in ClpP upon binding to ClpA. This technique permits relative quantification of the solvent accessibility of protein side chains, allowing interaction sites and conformational changes to be mapped to specific areas of the protein. Synchrotron protein footprinting requires the generation of a transient concentration of hydroxyl radicals by direct irradiation of the protein solution for milliseconds with a high-flux x-ray beam. These radicals produce characteristic oxidative modifications on protein side chains (31, 32) in solvent-exposed regions of the protein; areas of the protein not exposed to solvent are protected from these stable oxidations. Protease digestion of the sample followed by liquid-chromatography-coupled mass spectrometry (LCMS) and tandem mass spectrometry (MSMS) allows identification of the oxidized regions and often the specific residue that is modified as well. Binding sites and regions involved in conformational change are then identified by differences in side chain modification rate upon addition of binding partners or small molecule ligands (33, 34).
The synchrotron hydroxyl radical footprinting results show that the ClpP N-terminal residues are shielded from solvent when ClpP is not in complex with ClpA but become significantly more solvent-exposed upon binding of ClpA. To investigate the functional consequences of this conformational change, we also carried out steady-state kinetic characterization of wild-type and N-terminally truncated (19) ClpP. These kinetic studies reveal that removal of the first seven residues of ClpP increases the peptidase initial rate by 5–15 fold. These results provide the first direct evidence that the ClpP N-terminus controls the enzyme's catalytic activity. Moreover, we show here that removal of the N-terminus traps the enzyme in an acyl-enzyme bound form, indicating that an allosteric conformational change involving the N-terminus is necessary to activate the enzyme for acyl-enzyme intermediate breakdown. Together, these results support the hypothesis that motions of the ClpP N-terminus in and out of the ClpP pore regulate both substrate access to the protease active sites and the catalytic efficiency of these active sites for acyl-enzyme hydrolysis.
MATERIALS AND METHODS
Protein purification
ClpP-His6 and ClpA were purified as previously described (35–37). The plasmid encoding ClpPΔN (in which the first seven residues of mature ClpP are removed) (19) was a generous gift from Prof. Walid Houry and Dr. Anna Gribun (University of Toronto). The plasmid was transformed into SG1146GaBL21(DE3) cells (a clpp− strain) and purified in the same way as ClpP-His6.
Synchrotron hydroxyl radical footprinting
Buffer conditions were optimized to minimize quenching of hydroxyl radicals while maintaining ClpA and ClpP solubility and activity. The optimal buffer conditions were found to be 50 mM sodium cacodylate, pH 7.0, 400 mM KCl, and 20 mM MgCl2 (rxn buffer). ClpP maintains full proteolytic activity in this buffer system (as compared to standard Tris or HEPES buffer conditions), while ClpA maintains 70–80% of its ATPase activity. ClpA and ClpP were purified as described above, except that after purification and before storage, buffer exchange into rxn buffer was performed using a PD10 column (GE Healthcare) according to the manufacturer's instructions. The enzymes were then aliquoted, flash-frozen in liquid nitrogen and shipped overnight in dry ice to Brookhaven National Laboratory (Upton, NY), where they were immediately stored at −80°C. For radiolysis, the proteins were diluted to a concentration of 2 μM, then incubated in 1 mM ATPγS for five minutes prior to exposure to 0–200 ms of mirror-focused (38) polychromatic x-rays at the X28C beamline of the National Synchrotron Light Source using a KinTek quench-flow apparatus (according to published procedures (39)). Methionine amide was added to a final concentration of 10 mM to prevent secondary oxidation of methionine residues (40). Exposed samples were digested overnight at 37°C using sequencing-grade modified trypsin (Promega, Madison, WI) and analyzed via LCMS and MSMS as previously described (33, 41) to determine the extent and location of modification. The rate constant for reduction of the fraction of unmodified peptide with exposure was determined via a chi-squared fit to a single exponential function.
Preparation of ssrA-dabsyl conjugate
The ssrA peptide (AANDENYALAA) was synthesized by the MIT Biopolymers Facility, and a dabsyl group was added to the N-terminus using dabsyl-chloride (Anaspec). The reaction was performed in 66% acetone and 33% bicarbonate buffer (200 mM sodium bicarbonate, pH 9.0). A 3-fold molar excess of dabsyl-chloride was used, and the reaction was allowed to proceed for 30 minutes at 80°C. The conjugate was purified by HPLC using a reverse-phase C-phenyl semi-preparatory column (SB-Phenyl, 5 μm, 9.4 × 250 mm, Agilent). The peptide conjugate was stored dry at −20°C and solubilized in DMSO immediately prior to use.
Peptide degradation assays
All reactions were performed at 37°C in buffer containing 50 mM HEPES, pH 7.5, 100 mM NaCl, 20 mM MgCl2, 1 mM DTT, and 10% glycerol. All assays were performed in triplicate, except for the ssrA-dabsyl experiments, which were performed in duplicate. For insulin chain B degradation, 1 μM tetradecamer was used for reactions with ClpPΔN or wild-type ClpP alone; for reactions with ClpAP, 0.1 μM ClpP14 and 0.2 μM ClpA6 were used, and 2 mM ATPγS was added to the reaction mixture. For both SLY-AMC (N-Succinyl-Leu-Tyr-7-amido-4-methylcoumarin) and ssrA-dabsyl degradation, 0.125 μM ClpP14 or ClpPΔN14 was used; for degradation by ClpAP, 0.25 μM ClpA6 and 2mM ATPγS were added to the reaction. Hydroxylamine (Aldrich) was added to the reaction mixture for trapping experiments. For insulin chain B and ssrA-dabsyl degradation, 7-amino-4-methyl-coumarin (AMC, Sigma) was added as an internal standard (for HPLC analysis). For reactions with SLY-AMC, degradation was monitored as an increase in AMC fluorescence (ex: 345 nm, em: 440 nm) using a microplate spectrofluorimeter (Molecular Devices, Spectramax Gemini XS). Relative fluorescence units were converted into AMC concentration using a standard curve; addition of hydroxylamine did not affect the AMC standard curve (data not shown). For insulin chain B and ssrA-dabsyl degradation, aliquots of the reaction mixture were removed at various time points and quenched by the addition of an equal volume of 7.4 M Gu-HCl. These samples were then analyzed by HPLC using a C18 reverse phase analytical column (Jupiter, 150 × 4.60 mm, 5 μm, Phenomenex). Linear fits of product amplitude vs. time were used to determine degradation rates except where distinct rapid and slow phases were observed. In that case, plots were fitted to the sum of an exponential and a linear function to obtain both an initial rate for the rapid phase and a steady-state rate for the slow phase.
MALDI (Matrix Assisted Laser Desorption/Ionization) mass spectrometry
MALDI spectra were recorded on a PerSeptive Biosystems Voyager-DE STR in positive ion mode using ∝-cyano-4-hydroxycinnamic acid as the matrix. The instrument was calibrated before each use.
RESULTS
Synchrotron hydroxyl radical footprinting
To probe the conformational dynamics of ClpP upon binding of ClpA, we performed synchrotron hydroxyl radical footprinting analysis of the ClpP tetradecamer alone and with ClpA bound. MSMS-verifiable coverage of the ClpP tryptic digest encompassed 49% of the protein (Fig. 1A). Only one tryptic peptide (the N-terminal peptide, residues 1–12) exhibited modification upon x-ray exposure; this low level of observable modification is likely due to the presence of 1 mM ATPγS, a strong hydroxyl radical scavenger that is required for formation of the ClpA hexamer. Another contributing factor is likely to be that a low fraction of the strongly reactive residues in observed ClpP peptides exhibit high solvent accessibility. Solvent accessibility calculations using the ClpP crystal structure (1YG6) predict that the reactive sulfur of methionine 5 is the most exposed in the structure by a significant margin (14 Å2, compared to 6 Å2 for the next most exposed methionine sulfur atom).
FIGURE 1. Synchrotron hydroxyl radical footprinting results.
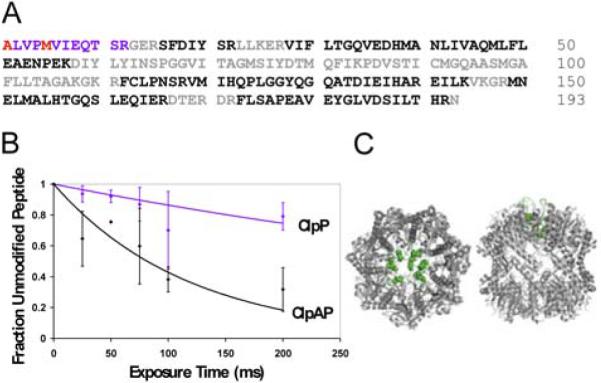
A) Tryptic peptide coverage of ClpP. Color coding of amino acids: Black: MSMS identified peptides, Blue: peptides with modification, Red: specifically identified modified residues, Grey: not identifiable in spectra. B) ClpP peptide 1–12 oxidation curves. blue: ClpP tetradecamer, black: ClpAP complex. C) Left: ClpP top view, Right: ClpP side view. N-terminal residues are highlighted in green. Structure is from Bewley et al. (20), PDB accession code 1YG6.
Nevertheless, the difference in modification rate of this region for the ClpP tetradecamer vs. the ClpAP complex is significant and reveals an important ClpA-induced conformational change in ClpP. The most abundant peptide modification observed was a +16 Da mass shift on methionine 5 (Fig. S1, Supporting Information), accompanied by a substantial decrease in retention time (~2 minutes) during chromatographic separation (Fig. S2, Supporting Information). A +14 Da mass shift on the N-terminal alanine was also observed, but did not contribute significantly to the computed modification rate. The modification rate of this region for ClpP alone is 1–2 sec−1 (the modification rates reported are x-ray dependent rates; the fraction of peptide modified before x-ray exposure is subtracted in the kinetic analysis). Formation of the ClpAP complex might be expected to decrease this rate due to obstruction of free solvent access to the ClpP tetradecamer by the proximity of bound hexameric ClpA. In fact, binding of ATPγS-bound ClpA increases the rate by approximately 6-fold to 7–10 sec−1 (Fig. 1B), indicating that the binding of ClpA causes the ClpP N-termini to assume a more solvent-exposed conformation. The relatively low modification rate observed for ClpP alone suggests that the ClpP N-termini may be primarily in a “closed”, pore-filled conformation when not bound to ClpA; the high modification rate observed for ClpAP may indicate an “opening” of the ClpP pore through removal of the N-termini upon the binding of ClpA.
The ClpPΔN mutation accelerates degradation of insulin chain B and induces distinct rapid and slow phases of product formation
Our hydroxyl radical footprinting data suggested an “opening” of the ClpP N-termini upon ClpA binding; in order to test this hypothesis, we turned to kinetic studies. To mimic the ClpP open conformation, we expressed and purified a truncation mutant (ClpPΔN) in which the first seven residues of the mature N-terminus are removed (19). Previous studies (18, 19) have shown that this deletion greatly diminishes binding to both ClpX and ClpA. Therefore, all studies with this mutant were performed in the absence of either ATPase.
Previous work has demonstrated that wild-type ClpP is inefficient at degrading large (~30 amino acid) peptides such as oxidized insulin chain B and glucagon. However, this work also showed that the addition of ClpA and either ATP or the nonhydrolyzable analog ATPγS greatly increased the efficiency of degradation (42, 43). Based on our hydroxyl radical footprinting data, we hypothesized that the increase in degradation efficiency induced by the ClpA hexamer was due to opening of the ClpP pore by removal of the N-terminus. This hypothesis predicts that the ClpPΔN mutant will mimic ClpAP in the processing of large substrates. To test this hypothesis, we compared the insulin chain B degradation kinetics of ClpPΔN to those of ClpAP and wild-type ClpP.
Degradation of insulin chain B yields four major products and several less-abundant products (see Supporting Information Fig. S3 for a representative HPLC trace). ClpPΔN, ClpAP, and wild-type ClpP produces the same four major products, as judged by HPLC retention time. The formation rates of all four products were measured for each of the three enzyme conditions, and the steady-state kinetic parameters were determined. Plots of the formation rate of the most abundant product as a function of concentration are shown in Fig. 2A. Wild-type ClpP, ClpAP, and ClpPΔN each exhibit a Km for insulin chain B of ~350 μM. However, ClpAP and ClpPΔN exhibit significantly higher maximal formation rates for each of the major peptide products compared to wild-type ClpP. The apparent kcats for each of the four major peptides produced by ClpAP are 8–20 fold higher than those measured using wild-type ClpP, while the maximal formation rates with ClpPΔN are 5–15 fold higher than wild-type ClpP (Fig. 2A).
FIGURE 2. Insulin chain B degradation.

A) Michaelis-Menten plots with ClpAP, wild-type ClpP, and ClpPΔN. ClpPΔN rapid phase rates shown in red and slow phase rates shown in orange. Values for Km are 350±60 μM, 340±80 μM, and 330±90 μM for wild-type ClpP, ClpPΔN, and ClpAP, respectively. Values for apparent kcat are 2.4±0.2 min−1, 13±1 min−1, and 19±2 min−1 for wild-type ClpP, ClpPΔN, and ClpAP, respectively. B) ClpAP, wild-type ClpP, and ClpPΔN degradation of 10 μM insulin chain B with 1 μM enzyme. Only ClpPΔN shows a rapid phase. C) ClpPΔN degradation of insulin chain B at various concentrations of substrate. Only low substrate concentrations exhibit a distinct rapid phase. Error bars represent standard deviation of three trials.
Unexpectedly, and unlike wild-type ClpP and ClpAP, at low concentrations of insulin chain B (≤150 μM) the rate of product formation by ClpPΔN exhibits a rapid phase followed by a slow phase in which product formation was essentially undetectable (Fig. 2B). The amplitude of this rapid phase depends on the substrate concentration, with higher amplitudes observed at higher substrate concentrations (Fig. 2C). At insulin chain B concentrations of 250 μM and higher, only the rapid phase is evident (Fig. 2C). Furthermore, the largest rapid phase amplitude observed (at 150 μM insulin chain B) is approximately stoichiometric with the active site concentration (i.e., 1 equivalent of product was generated for each equivalent of protease active site in the ClpP tetradecameric complex), indicating that the rapid phase represents at least one complete substrate turnover. The observation of a distinct slow phase in ClpPΔN kinetics suggests that a step that is rapid for the wild-type enzyme becomes rate-limiting in the absence of the ClpP N-terminus; one possibility (precedented in serine proteases, especially with ester substrates (44)) is that acyl-enzyme breakdown is rate-limiting. The data also indicate that the substrate concentration affects the ability of the enzyme to process and/or bypass the long-lived species that predominates in the slow phase of product formation.
Kinetic contributions of substrate entry, acyl-enzyme formation, and acyl-enzyme breakdown
Comparing the kinetics of large and small peptide substrates is one way to determine the relative contributions of substrate entry and subsequent covalent chemical steps to the rapid and slow phases of product formation. Previous work has demonstrated that addition of ClpA and ATP or ATPγS slightly decreases the degradation rate of the small peptide substrate, N-succinyl-Leu-Tyr-7-amido-4-methyl-coumarin (SLY-AMC) compared to the rate seen with ClpP alone (~3-fold decrease in rate at a concentration of 1 mM) (42). Previous results have also shown that truncation of the ClpP N-terminus has a relatively modest effect on the degradation rate of SLY-AMC (~2-fold increase in the steady-state rate at a concentration of 0.5 mM) (19), consistent with the hypothesis that this substrate can enter either through the axial or equatorial pores of ClpP. These results suggest that access to the ClpP degradation chamber is not rate-limiting for small substrates such as SLY-AMC. If the rapid phase of the ClpPΔN-catalyzed reaction corresponds to substrate entry and acyl-enzyme formation, while the slow phase corresponds to the breakdown of a stabilized acyl-enzyme, rapid and slow phases will be observed for ClpPΔN-catalyzed proteolysis of the small substrate. However, if access to the ClpP active sites is not rate-limiting for small substrates, removal of the N terminus will not accelerate the rapid phase compared to the wild-type rate. In order to test these hypotheses, we measured the degradation kinetics of SLY-AMC with wild-type ClpP, ClpPΔN, and ClpAP.
The previously reported Km of SLY-AMC with wild-type ClpP is 1 mM (37). However, we observe an apparent decrease in the steady-state rate of degradation at SLY-AMC concentrations greater than 250 μM. We suspect that this decrease in apparent rate is due to the inner filter effect (absorption of emitted photons by dye molecules in highly absorbing solutions (45)), and therefore our rates above 250 μM SLY-AMC are not accurate. Nevertheless, we were able to compare the degradation rates of SLY-AMC with wild-type ClpP, ClpAP, and ClpPΔN at various concentrations of substrate. At all concentrations used, the initial degradation rate is the same within error for wild-type ClpP, ClpAP, and ClpPΔN (see Fig. 3A).2 The lack of rate enhancement seen with ClpPΔN as compared to wild-type ClpP supports the hypothesis that the rapid kinetic phase corresponds to substrate entry and/or acyl-enzyme formation. It also suggests that, in contrast to the observation for large substrates, removal of the N-terminal region of ClpP does not accelerate substrate entry/acyl-enzyme formation for small substrates.
FIGURE 3. Degradation of SLY-AMC.

A) ClpAP, wild-type ClpP, and ClpPΔN degradation rate constants as a function of SLY-AMC concentration. B) Kinetics of 50 μM SLY-AMC degradation with ClpAP, wild-type ClpP, and ClpPΔN (using 0.125 μM ClpP14). Only ClpPΔN exhibits a distinct rapid phase. C) ClpPΔN degradation of SLY-AMC at various concentrations of substrate. A rapid phase is present at concentrations ≤250 μM. The apparent change in rate seen with 500 μM and 1 mM is also present with wild-type ClpP and ClpAP and is due to saturation of fluorescence signal; it does not represent a distinct rapid phase. Traces shown in (B) and (C) are the average of 3 trials.
As observed with insulin chain B, the kinetics of SLY-AMC degradation by ClpPΔN exhibit a rapid phase comprising multiple turnovers followed by a slow phase, whereas no slow phase is seen with wild-type ClpP or ClpAP (Fig. 3B). Consistent with the insulin chain B results, distinct rapid and slow phases are only observed at low (≤250 μM) concentrations of SLY-AMC, and the amplitude of the ClpPΔN rapid phase increases with increasing substrate concentration (Fig. 3C). In this case, the largest rapid phase amplitude observed (at a SLY-AMC concentration of 250 μM) is greater than stoichiometric: 5 equivalents of product are generated per equivalent of protease active site in the complex, indicating that the rapid phase represents multiple substrate turnovers. These observations show that increased substrate concentrations help the enzyme achieve catalytic turnover rather than becoming trapped in a stabilized intermediate state.
Hydroxylamine eliminates the ClpPΔN slow phase
The existence of a slow phase in the ClpPΔN degradation kinetics suggests that a step following acyl-enzyme formation is rate-limiting for catalysis at low substrate concentrations. One possibility is that breakdown of a stabilized acyl-enzyme intermediate is the rate-limiting step. In order to test this hypothesis, we measured the kinetics of SLY-AMC degradation in increasing concentrations of the strong nucleophile hydroxylamine (46–48) (Fig. 4A). We found that the slow-phase rate of ClpPΔN degradation increases with increasing concentrations of hydroxylamine, and at 2.4 M NH2OH the slow phase is not observed (Fig. 4A and 4B). These data are consistent with hydroxylamine increasing the ClpPΔN slow-phase rate of degradation by accelerating the rate of acyl-enzyme breakdown.
FIGURE 4. The effects of hydroxylamine on the degradation of SLY-AMC.
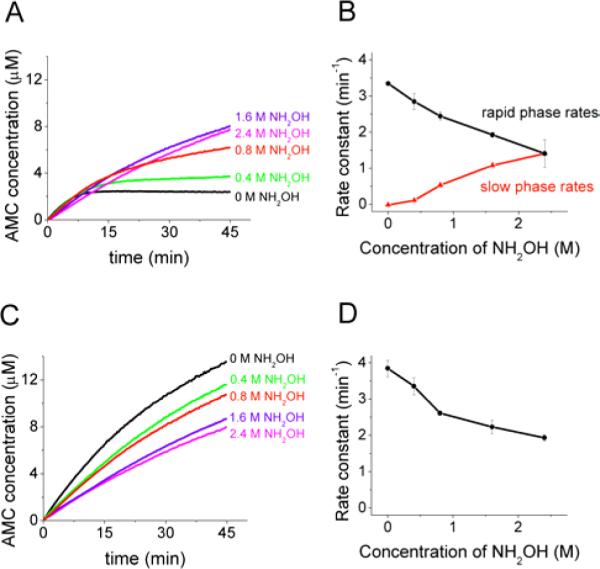
A) Time course of ClpPΔN degradation of SLY-AMC. B) Rate constant of SLY-AMC degradation by ClpPΔN as a function of hydroxylamine concentration. C) Time course of wild-type ClpP degradation of SLY-AMC. D) Rate constant of SLY-AMC degradation by wild-type ClpP as a function of hydroxylamine concentration. Reaction conditions: 50 μM SLY-AMC with 0.125 μM ClpP tetradecamer. Traces shown are the average of three trials, and error bars represent the standard deviation of three trials.
In contrast, the rate of degradation by wild-type ClpP decreases with increasing concentrations of hydroxylamine, as does the rapid phase rate of degradation with ClpPΔN (Fig. 4B,C,D). This inhibitory effect of high (~molar) concentrations of hydroxylamine on the rapid phase of turnover does not have an obvious specific molecular mechanism. However, the effects of hydroxylamine on the rapid phase are similar for wild-type ClpP and ClpPΔN and were therefore not interpreted further in terms of possible functions for the N-terminal domain. The possibility that hydroxylamine has non-specific effects on the rapid kinetic phase nonetheless highlighted the importance of additional experiments to determine whether its effects on the slow phase are due to acceleration of acyl-enzyme breakdown.
Hydroxylamine traps a stabilized acyl-enzyme intermediate
In order to more rigorously test the mechanism of ClpPΔN slow-phase rate acceleration by hydroxylamine, we used MALDI-MS analysis to test for the presence of hydroxamic acid products following addition of hydroxylamine. However, the low molecular weight of the peptide products of SLY-AMC degradation precludes their observation using MALDI-MS. The complex product mixture obtained upon degradation of insulin chain B (Fig. S3, Supporting Information) also complicates interpretation of the MALDI spectrum. To simplify the analysis, we used as a substrate the ssrA degradation tag (AANDENYALAA), derivatized with a dabsyl group at the N-terminus to facilitate detection by UV absorbance. Previous studies in our lab indicated that degradation of this substrate yields two primary products (AANDENYA + LAA) (data not shown).
First, we characterized the degradation of ssrA-dabsyl with wild-type ClpP, ClpAP/ATPγS, and ClpPΔN. At 10 μM ssrA-dabsyl, we found that the initial rate of degradation of this intermediate-sized substrate is 3-fold faster with ClpAP as compared to wild-type Clp and 5-fold faster with ClpPΔN as compared to wild-type ClpP (Fig. 5). This is consistent with the insulin chain B data, and indicates that removal of the N-terminus, either through truncation or by complex with ClpA, increases the rate of acyl-enzyme formation for this 11-amino acid long substrate. As expected, we also observed distinct rapid and slow phases in the kinetics of ClpPΔN degradation (Fig. 5).
FIGURE 5. Degradation of ssrA-dabsyl.
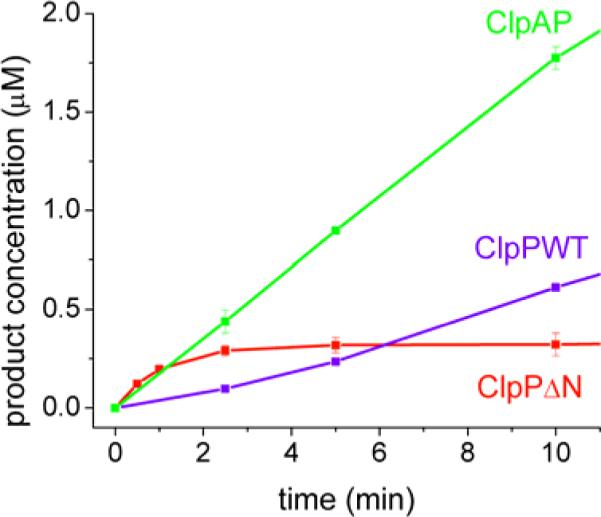
Conditions: 10 μM ssrA-dabsyl with 0.125 μM enzyme. Rate constants are 0.53 min−1, 1.42 min−1, 2.51 min−1, and 0.02 min−1 for wild-type ClpP, ClpAP/ATPγS, ClpPΔN rapid phase rate and ClpPΔN slow phase rate, respectively. Error bars represent the standard deviation of two trials.
Upon addition of hydroxylamine, we observed the formation of a new peak in the HPLC trace (Fig. 6A). MALDI analysis confirmed that this new peak is the hydroxamic acid peptide product (Fig. S4, Supporting Information). Kinetic analysis revealed that, as observed with SLY-AMC degradation, the addition of increasing concentrations of hydroxylamine accelerates the slow-phase rate of ClpPΔN degradation (Fig. 7A and 7B). This is achieved largely through formation of the hydroxamic acid product, although we did see a modest increase in the production of the carboxylic acid product. (At 1.6 M NH2OH the concentration of the carboxylic acid product increased ~2-fold, Fig. 6A.) As a control, we measured the kinetics of degradation with wild-type ClpP at increasing hydroxylamine concentrations. We found that while addition of hydroxylamine does cause limited formation of the hydroxamic acid product, the rates of formation are significantly slower than the rates of formation with ClpPΔN. For example, the rate of hydroxamic acid production is 15-fold slower at 1.6 M NH2OH with wild-type ClpP as compared to ClpPΔN (Fig. 6B). As observed with SLY-AMC, the wild-type ClpP degradation rate decreases with increasing concentrations of hydroxylamine, as does the rapid phase rate of degradation by ClpPΔN (see Figure 7B,C,D). These data show that hydroxylamine increases the ClpPΔN rate of acyl-enzyme breakdown through formation of the hydroxamic acid product, consistent with the trapping of an acyl-enzyme intermediate.
FIGURE 6. Degradation of ssrA-dabsyl with hydroxylamine produces the hydroxamic acid product.
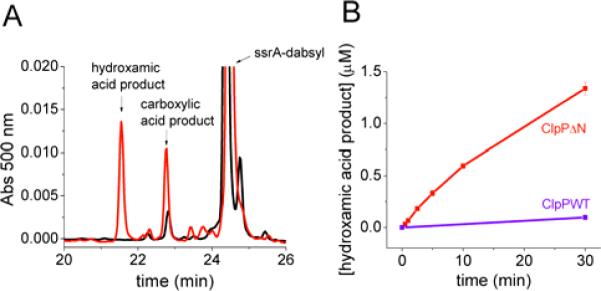
A) ClpPΔN degradation of ssrA-dabsyl (0.125 μM ClpΔN14, 10 μM ssrA-dabsyl). Shown in black is the reverse-phase chromatogram of the reaction mixture after 30 min. in the absence of NH2OH. Shown in red is the chromatogram of the reaction mixture after 30 min. in the presence of 1.6 M NH2OH. B) Concentration of the hydroxamic acid product over time with 1.6 M NH2OH. (conditions: 0.125 μM ClpP tetradecamer, 10 μM ssrA-dabsyl). Error bars represent standard deviation of two trials.
FIGURE 7. The effects of hydroxylamine on the kinetics ssrA-dabsyl degradation.
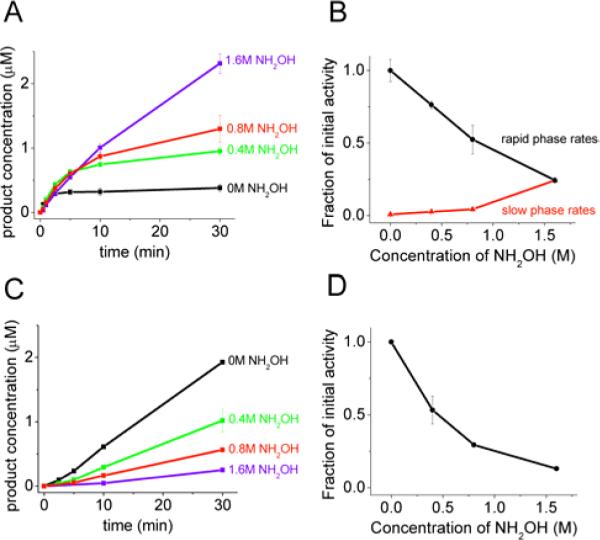
A) Time course of ClpPΔN degradation of ssrA-dabsyl. B) Rate constant of ssrA-dabsyl degradation by ClpPΔN as a function of hydroxylamine concentration. C) Time course of wild-type ClpP degradation of ssrA-dabsyl. D) Rate constant of ssrA-dabsyl degradation by wild-type ClpP as a function of hydroxylamine concentration. Reaction conditions: 10 μM ssrA-dabsyl with 0.125 μM ClpP tetradecamer. Traces shown are the average of two trials, and error bars represent the standard deviation of two trials. For (C) and (D) rates are shown as a fraction of the initial rate at 0 M NH2OH.
DISCUSSION
Recent biochemical studies have shown that the N-terminus of ClpP is an important component in the interaction with its partner ATPases ClpX and ClpA (17–20). Furthermore, the conformational flexibility of the N-terminus has been highlighted in a series of ClpP structures showing the N-terminus in both an “up” conformation in which the N-terminal residues form a well-ordered β-sheet outside the central pore and a “down” conformation in which the first several N-terminal residues are disordered and appear to occupy the central pore (18–21). This conformational flexibility suggests that while the N-terminus is important for interaction with ClpX and ClpA, it may also be playing a more dynamic role in the overall processing of peptide and protein substrates.
Synchrotron hydroxyl radical footprinting was used to probe the dynamics of the ClpP N-terminal region. These experiments show that the first few residues of the ClpP N-terminus become more solvent-exposed upon interaction with ATPγS-bound ClpA. This observation is consistent with ClpP changing from a “closed” conformation to an “open” conformation upon binding to ClpA. Structurally, this may correspond to a change from the “down” conformation to the “up” conformation (20). The previous observations that the ClpP N-terminus is required for ClpA/ClpP interaction (19, 20) and that the N-terminus can functionally occlude the axial pore of the complex (30) suggested that the N-terminus exits the ClpP pore and resides at the ClpA/ClpP interface. The footprinting results provide direct structural evidence for this hypothesis in the intact ClpAP complex.
The relatively low coverage of ClpP (49%) precludes a definitive statement that domain motions outside of the N-terminus do not occur. However, while the coverage is incomplete, it spans most of the ClpP sequence; the only large regions of the protein that are not covered at all are sheets 3–6 and helix C (as defined in ref. 5). It is therefore unlikely that a global conformational change involving large changes in solvent exposure has been missed due to limited peptide coverage.
Complementary kinetic experiments provide evidence that this “opening” of the N-terminus is functionally important in peptide processing. Previous studies have reported that the processing of long (~30 amino acid) peptides by ClpP is significantly accelerated by the addition of ClpA and ATPγS. The current experiments indicate that an N-terminal ClpP truncation mutant mimics the ClpAP state by accelerating the degradation rate of insulin chain B 5–15-fold above wild-type ClpP levels. This rate acceleration was also observed for an 11-amino-acid-long peptide (ssrA-dabsyl), but not for the short peptide SLY-AMC. These results are consistent with a model in which the ClpP serine protease active sites are accessible to large peptide substrates only when the N-terminus is in the “open” conformation, but small peptides may enter the ClpP chamber through the equatorial pores. The “open” conformation can be mimicked by an elimination of the first seven N-terminal residues (ClpPΔN) or by removal of the ClpP N-terminal residues from the pore of ClpP upon interaction with ATP-bound ClpA.
The unexpected observation of distinct rapid and slow phases for the ClpPΔN mutant kinetics supports a novel role for the ClpP N-terminus in regulating the reactivity of the acyl-enzyme intermediate. Studies with three peptides ranging in size from 2 amino acids to 30 amino acids showed distinct rapid and slow phases in product formation kinetics with ClpPΔN, but not with ClpAP or wild-type ClpP. The N-terminal truncation induces a slow phase without decreasing the rate of the rapid phase, indicating that this structural perturbation has a selective effect on a subset of microscopic reaction steps rather than acting simply to disrupt the enzyme's structure. The slow phase is eliminated at high concentrations of the strong nucleophile hydroxylamine with concomitant formation of the hydroxamic acid product, consistent with the ClpPΔN mutant trapping the active-site serine in a slowly hydrolyzed acyl-enzyme form that is broken down by hydroxylaminolysis. The slow phase for ClpPΔN can be interpreted as breakdown of the acyl-enzyme intermediate, which appears to be highly stabilized in the absence of the ClpP N-terminus.
The rapid phase in ClpPΔN kinetics is similar to the well-characterized burst phase kinetics of other serine proteases in the sense that formation of the putative acyl-enzyme intermediate occurs during this phase. However, the observation that multiple turnovers can take place during the rapid phase is unusual. If the enzyme were in an active form (corresponding to the purple enzyme forms in Figure 8A) throughout the catalytic cycle, only one equivalent of product per equivalent of active site would be expected in the burst phase. The production of multiple equivalents of product followed by a nearly complete loss of activity suggests that active ClpP can undergo a global shift to an inactive conformation that does not hydrolyze the acyl-enzyme intermediate. In that case, the number of turnovers before inactivation will depend on the relative rates of acyl-enzyme hydrolysis and entry into the inactive conformation (Fig. 8A and Supporting Information text). The observation that the inactive conformation is only present for the ClpPΔN mutant indicates that the N-terminus regulates acyl-enzyme intermediate reactivity by regulating the ability of ClpP to maintain an active conformation. One possibility is that a conformational change of the N-terminus initiates an Monod-Wyman-Changeux -type allosteric conformational change: occupation of the central ClpP pore by the N-terminus may stabilize the conformation of the active site that can hydrolyze the acyl-enzyme intermediate, while removal of the N-terminus may stabilize the inactive form of the enzyme.
FIGURE 8. Schematic model showing the gating function of the ClpP N-terminus.

A) Degradation by ClpPΔN. After formation of the acyl-enzyme intermediate, the enzyme partitions between an active state that is competent for acyl-enzyme breakdown and an long-lived inactive state (shown in green) in which the acyl-enzyme intermediate is stabilized. Binding of a second substrate to the putative N-terminal binding site accelerates the rate of entry into the active (hydrolysis-competent) state. B) Degradation by ClpAP. ClpA is shown in red, and ClpP is shown in blue. Binding of ClpA causes removal of the ClpP N-terminus from the pore of ClpP, allowing substrates to enter the protease chamber and form the acyl-enzyme intermediate. Re-entry of the N-terminal loops into the ClpP pore causes acyl-enzyme breakdown, and prevents the complex from assuming the inactive conformation.
Motion of the N-terminus from the “up” to the “down” conformation may provide the structural trigger for the proposed allosteric conformational change, allowing efficient acyl-enzyme hydrolysis in the wild-type enzyme. Szyk et al. have recently reported the crystal structure of wild-type ClpP with an inhibitor peptide covalently bound to the active site, mimicking the tetrahedral intermediate (21). The structure shows the N-termini of ClpP in the “up” conformation and suggests that the acyl-enzyme intemediate may be stabilized when the ClpP N-termini adopt this conformation. This may also explain why ClpPΔN, which functionally mimics the “open” or “up” conformation, stabilizes the acyl-enzyme intermediate. Isolation of the intact acyl-enzyme intermediate itself would open up many new avenues for understanding its structure and reactivity, especially its remarkable stability at low substrate concentrations; the kinetics of the slow phase suggest that this intermediate will be stable enough to isolate, and efforts in our laboratories to do so are ongoing.
The observation that high substrate concentrations increase the amplitude of the ClpPΔN rapid kinetic phase is also consistent with the idea that peptide occupancy in ClpP helps regulate the reactivity of the acyl-enzyme intermediate. One possibility is that occupancy of the axial pores by peptides triggers the shift to an active conformation. Because ClpP has a large (50 Å) central cavity, substrate peptides or a translocating polypeptide might simultaneously occupy the axial pore and the central cavity. If, for the wild-type enzyme, an N-terminal conformational change from the “up” to the “down” conformation (ie, the “closed”, pore-filled conformation) causes the active site to assume a conformation that favors acyl-enzyme hydrolysis, a substrate peptide at high concentration might fill the pore in place of the N-terminus for the ClpPΔN enzyme, therefore facilitating the conformational change to this hydrolysis-active state. In this ClpPΔN mechanistic model (Fig. 8A), increasing the substrate concentration increases the probability that the enzyme hydrolyzes the acyl-enzyme intermediate rather than assuming the hydrolysis-incompetent form. This model thus explains the observation that increasing the substrate concentration increases the rapid phase amplitude and accounts for a rapid phase composed of multiple turnovers.
Our data are most consistent with a mechanism in which increased substrate binding at the putative N-terminal peptide binding sites within the central ClpP pore causes accelerated acyl-enzyme breakdown. Nevertheless, at this point we cannot rule out the possibility that acylation of the active site serine also helps to shift the conformational equilibrium that triggers acyl-enzyme hydrolysis. In that case, high substrate concentrations might favor acyl-enzyme hydrolysis by increasing the steady-state concentration of the acyl-enzyme intermediate. However, if acylation is the allosteric trigger for acyl-enzyme breakdown, one would expect the wild-type enzyme to also exhibit biphasic kinetics at subsaturating concentrations of substrate; such kinetics are not observed.
Regardless of the exact mechanism(s) at work, the change from active to inactive states appears to be cooperative and to affect the entire ClpP complex. The substrate concentration dependence of the rapid phase amplitude is steep, consistent with a cooperative allosteric transition induced by the binding of substrate peptides (see Supporting Information text and Figures S5 and S6 for details). Other ring-shaped enzymes, such as the chaperonin GroEL ATPase, couple enzymatic activity to a cooperative allosteric conformational change (49), and the catalytic cycle of ClpP may be analogous.
Taken together, the footprinting and kinetic data suggest a new model for proteolysis by wild-type ClpAP (Fig. 8B). In this model, an interaction with the ATP-bound form of ClpA would cause the ClpP N-terminus to assume the “up” conformation. This conformational change would open the ClpP axial pore, providing substrate access to the ClpP serine protease active sites and enabling formation of the acyl-enzyme intermediate. Re-entry of the ClpP N-terminus into the axial pore (ie, the “down” conformation) would lead to efficient hydrolysis of the acyl-enzyme intermediate and escape of the second peptide product via the equatorial pores. One important unresolved question is exactly how ATP-driven conformational changes of ClpA are coupled to the conformational changes of ClpP that regulate acyl-enzyme reactivity. It may be that motions of the ClpA D2 loop, previously proposed to drive substrate translocation (50), also drive ClpP conformational changes. Further footprinting and kinetic studies are expected to help resolve this question.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Janna Kiselar for assistance with MS and Dr. Sayan Gupta for footprinting advice; Mike Sullivan, John Toomey and Don Abel for expert technical support for the X28C beamline; Prof. Walid Houry and Dr. Anna Gribun for the plasmid encoding ClpPΔN and for ClpPΔN purification advice; and Profs. Bob Sauer and Tania Baker for the ClpA expression plasmid.
Work in the Licht laboratory is supported in part by a Beckman Young Investigator Award. Work in the Center for Synchrotron Biosciences, Center for Proteomics, Case Western Reserve University, is supported by the National Institute for Biomedical Imaging and Bioengineering under P41-EB-01979 and through a Ruth L. Kirschstein National Service Award (NIH T32 HL007887). The National Synchrotron Light Source at Brookhaven National Laboratory is supported by the Department of Energy under contract DE-AC02-98CH10886.
Abbreviations
- AAA+
ATPases associated with various cellular activities
- LCMS
liquid-chromatography-coupled mass spectrometry
- MSMS
tandem mass spectrometry
- ATPγS
adenosine 5′-(3-thiotriphosphate)
- HPLC
high-performance liquid chromatography
- SLY-AMC
N-Succinyl-Leu-Tyr-7-amido-4-methylcoumarin
- MALDI
matrix-assisted laser desorption/ionization
- ssrA
peptide of sequence AANDENYALAA
- DMSO
dimethyl sulfoxide
- HEPES
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)
- DTT
dithiothreitol
- PDB
protein data bank
- AMC
7-amino-4-methylcoumarin
Footnotes
Previous studies have reported that ClpPΔN hydrolyzes SLY-AMC 2-fold more rapidly than the wild-type enzyme, based on rates at a single substrate concentration (0.5 mM), Ref. 19. We also observe somewhat more rapid rates for ClpPΔN at high substrate concentrations (Fig. 3A). However, at these concentrations, the apparent rate decreases with increasing substrate concentration due to substrate inhibition and/or inner-filter effects. We have therefore chosen to use the maximal hydrolytic rate constant in interpreting this kinetic data.
SUPPORTING INFORMATION AVAILABLE Synchrotron footprinting tandem MS spectra and chromatographic traces, a representative HPLC trace of insulin chain B degradation products, MALDI spectra of ssrA-dabsyl carboxylic acid and hydroxamic acid products, and a detailed quantitative model of ClpPΔN-catalyzed degradation. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Gottesman S, Maurizi MR, Wickner S. Regulatory subunits of energy-dependent proteases. Cell. 1997;91:435–438. doi: 10.1016/s0092-8674(00)80428-6. [DOI] [PubMed] [Google Scholar]
- 2.Ogura T, Wilkinson AJ. AAA+ superfamily ATPases: common structure--diverse function. Genes Cells. 2001;6:575–597. doi: 10.1046/j.1365-2443.2001.00447.x. [DOI] [PubMed] [Google Scholar]
- 3.Gottesman S. Proteolysis in bacterial regulatory circuits. Annu. Rev. Cell Dev. Biol. 2003;19:565–587. doi: 10.1146/annurev.cellbio.19.110701.153228. [DOI] [PubMed] [Google Scholar]
- 4.Sauer RT, Bolon DN, Burton BM, Burton RE, Flynn JM, Grant RA, Hersch GL, Joshi SA, Kenniston JA, Levchenko I, Neher SB, Oakes ESC, Siddiqui SM, Wah DA, Baker TA. Sculpting the proteome with AAA+ proteases and disassembly machines. Cell. 2004;119:9–18. doi: 10.1016/j.cell.2004.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang JM, Hartling JA, Flanagan JM. The structure of ClpP at 2.3 Å resolution suggests a model for ATP-dependent proteolysis. Cell. 1997;91:447–456. doi: 10.1016/s0092-8674(00)80431-6. [DOI] [PubMed] [Google Scholar]
- 6.Bochtler M, Ditzel L, Groll M, Huber R. Crystal structure of heat shock locus V (HslV) from Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1997;94:6070–6074. doi: 10.1073/pnas.94.12.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 8.Lowe J, Stock D, Jap R, Zwickl P, Baumeister W, Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 Å resolution. Science. 1995;268:533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 9.Bochtler M, Hartmann C, Song HK, Bourenkov GP, Bartunik HD, Huber R. The structures of HslU and ATP-dependent protease HslU-HslV. Nature. 2000;403:800–805. doi: 10.1038/35001629. [DOI] [PubMed] [Google Scholar]
- 10.Kessel M, Maurizi MR, Kim B, Kocsis E, Trus BL, Singh SK, Steven AC. Homology in structural organization between Escherichia coli ClpAP protease and the eukaryotic 26S proteasome. J. Mol. Biol. 1995;250:587–594. doi: 10.1006/jmbi.1995.0400. [DOI] [PubMed] [Google Scholar]
- 11.Grimaud R, Kessel M, Beuron F, Steven AC, Maurizi MR. Enzymatic and structural similarities between the Escherichia coli ATP-dependent proteases, ClpXP and ClpAP. J. Biol. Chem. 1998;273:12476–12481. doi: 10.1074/jbc.273.20.12476. [DOI] [PubMed] [Google Scholar]
- 12.Guo F, Maurizi MR, Esser L, Xia D. Crystal structure of ClpA, an Hsp100 chaperone and regulator of ClpAP protease. J. Biol. Chem. 2002;277:46743–46752. doi: 10.1074/jbc.M207796200. [DOI] [PubMed] [Google Scholar]
- 13.Kim DY, Kim KK. Crystal structure of ClpX molecular chaperone from Helicobacter pylori. J. Biol. Chem. 2003;278:50664–50670. doi: 10.1074/jbc.M305882200. [DOI] [PubMed] [Google Scholar]
- 14.Kim YI, Levchenko I, Fraczkowska K, Woodruff RV, Sauer RT, Baker TA. Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat. Struct. Biol. 2001;8:230–233. doi: 10.1038/84967. [DOI] [PubMed] [Google Scholar]
- 15.Singh SK, Rozycki J, Ortega J, Ishikawa T, Lo J, Steven AC, Maurizi MR. Functional domains of the ClpA and ClpX molecular chaperones identified by limited proteolysis and deletion analysis. J. Biol. Chem. 2001;276:29420–29429. doi: 10.1074/jbc.M103489200. [DOI] [PubMed] [Google Scholar]
- 16.Joshi SA, Hersch GL, Baker TA, Sauer RT. Communication between ClpX and ClpP during substrate processing and degradation. Nat. Struct. Mol. Biol. 2004;11:404–411. doi: 10.1038/nsmb752. [DOI] [PubMed] [Google Scholar]
- 17.Martin A, Baker TA, Sauer RT. Distinct static and dynamic interactions control ATPase-peptidase communication in a AAA+ protease. Mol. Cell. 2007;27:41–52. doi: 10.1016/j.molcel.2007.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang SG, Maurizi MR, Thompson M, Mueser T, Ahvazi B. Crystallography and mutagenesis point to an essential role for the N-terminus of human mitochondrial ClpP. J. Struct. Biol. 2004;148:338–352. doi: 10.1016/j.jsb.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 19.Gribun A, Kimber MS, Ching R, Sprangers R, Fiebig KM, Houry WA. The ClpP double ring tetradecameric protease exhibits plastic ring-ring interactions, and the N termini of its subunits form flexible loops that are essential for ClpXP and ClpAP complex formation. J. Biol. Chem. 2005;280:16185–16196. doi: 10.1074/jbc.M414124200. [DOI] [PubMed] [Google Scholar]
- 20.Bewley MC, Graziano V, Griffin K, Flanagan JM. The asymmetry in the mature amino-terminus of ClpP facilitates a local symmetry match in ClpAP and ClpXP complexes. J. Struct. Biol. 2006;153:113–128. doi: 10.1016/j.jsb.2005.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szyk A, Maurizi MR. Crystal structure at 1.9 Å of E. coli ClpP with a peptide covalently bound at the active site. J. Struct. Biol. 2006;156:165–174. doi: 10.1016/j.jsb.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 22.Ingvarsson H, Mate MJ, Hogbom M, Portnoi D, Benaroudj N, Alzari PM, Ortiz-Lombardia M, Unge T. Insights into the inter-ring plasticity of caseinolytic from the X-ray structure of Mycobacterium tuberculosis ClpP1. Acta Crystallogr. D Biol. Crystallogr. 2007;63:249–259. doi: 10.1107/S0907444906050530. [DOI] [PubMed] [Google Scholar]
- 23.Vedadi M, Lew J, Artz J, Amani M, Zhao Y, Dong AP, Wasney GA, Gao M, Hills T, Brokx S, Qiu W, Sharma S, Diassiti A, Alam Z, Melone M, Mulichak A, Wernimont A, Bray J, Loppnau P, Plotnikova O, Newberry K, Sundararajan E, Houston S, Walker J, Tempel W, Bochkarev A, Kozieradzki L, Edwards A, Arrowsmith C, Roos D, Kain K, Hui R. Genome-scale protein expression and structural biology of Plasmodium falciparum and related Apicomplexan organisms. Mol. Biochem. Parasit. 2007;151:100–110. doi: 10.1016/j.molbiopara.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 24.Yu AYH, Houry WA. ClpP: A distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett. 2007;581:3749–3757. doi: 10.1016/j.febslet.2007.04.076. [DOI] [PubMed] [Google Scholar]
- 25.Groll M, Bajorek M, Kohler A, Moroder L, Rubin DM, Huber R, Glickman MH, Finley D. A gated channel into the proteasome core particle. Nat. Struct. Biol. 2000;7:1062–1067. doi: 10.1038/80992. [DOI] [PubMed] [Google Scholar]
- 26.Kohler A, Cascio P, Leggett DS, Woo KM, Goldberg AL, Finley D. The axial channel of the proteasome core particle is gated by the Rpt2 ATPase and controls both substrate entry and product release. Mol. Cell. 2001;7:1143–1152. doi: 10.1016/s1097-2765(01)00274-x. [DOI] [PubMed] [Google Scholar]
- 27.Smith DM, Kafri G, Cheng YF, Ng D, Walz T, Goldberg AL. ATP binding to PAN or the 26S ATPases causes association with the 20S proteasome, gate opening, and translocation of unfolded proteins. Mol. Cell. 2005;20:687–698. doi: 10.1016/j.molcel.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 28.Smith DM, Chang SC, Park S, Finley D, Cheng Y, Goldberg AL. Docking of the proteasomal ATPases' carboxyl termini in the 20S proteasome's alpha ring opens the gate for substrate entry. Mol. Cell. 2007;27:731–744. doi: 10.1016/j.molcel.2007.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rabl J, Smith DM, Yu Y, Chang SC, Goldberg AL, Cheng Y. Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol. Cell. 2008;30:360–368. doi: 10.1016/j.molcel.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sprangers R, Gribun A, Hwang PM, Houry WA, Kay LE. Quantitative NMR spectroscopy of supramolecular complexes: dynamic side pores in ClpP are important for product release. Proc. Natl. Acad. Sci. U.S.A. 2005;102:16678–16683. doi: 10.1073/pnas.0507370102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu GZ, Chance MR. Radiolytic modification and reactivity of amino acid residues serving as structural probes for protein footprinting. Anal. Chem. 2005;77:4549–4555. doi: 10.1021/ac050299+. [DOI] [PubMed] [Google Scholar]
- 32.Xu G, Chance MR. Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem. Rev. 2007;107:3514–3543. doi: 10.1021/cr0682047. [DOI] [PubMed] [Google Scholar]
- 33.Kiselar JG, Maleknia SD, Sullivan M, Downard KM, Chance MR. Hydroxyl radical probe of protein surfaces using synchrotron X-ray radiolysis and mass spectrometry. Int. J. Radiat. Biol. 2002;78:101–114. doi: 10.1080/09553000110094805. [DOI] [PubMed] [Google Scholar]
- 34.Takamoto K, Chance MR. Radiolytic protein footprinting with mass Spectrometry to probe the structure of macromolecular complexes. Annu. Rev. Biophys. Biomol. Struct. 2006;35:251–276. doi: 10.1146/annurev.biophys.35.040405.102050. [DOI] [PubMed] [Google Scholar]
- 35.Choi KH, Licht S. Control of peptide product sizes by the energy-dependent protease ClpAP. Biochemistry. 2005;44:13921–13931. doi: 10.1021/bi0505060. [DOI] [PubMed] [Google Scholar]
- 36.Kim YI, Burton RE, Burton BM, Sauer RT, Baker TA. Dynamics of substrate denaturation and translocation by the ClpXP degradation machine. Mol. Cell. 2000;5:639–648. doi: 10.1016/s1097-2765(00)80243-9. [DOI] [PubMed] [Google Scholar]
- 37.Maurizi MR, Thompson MW, Singh SK, Kim SH. Endopeptidase Clp: ATP-dependent Clp protease from Escherichia coli. Methods Enzymol. 1994;244:314–331. doi: 10.1016/0076-6879(94)44025-5. [DOI] [PubMed] [Google Scholar]
- 38.Sullivan MR, Rekhi S, Bohon J, Gupta S, Abel D, Toomey J, Chance MR. Installation and testing of a focusing mirror at beamline X28C for high flux x-ray radiolysis of biological macromolecules. Rev. Sci. Instrum. 2008;79:025101–025108. doi: 10.1063/1.2839027. [DOI] [PubMed] [Google Scholar]
- 39.Gupta S, Sullivan M, Toomey J, Kiselar J, Chance MR. The Beamline X28C of the Center for Synchrotron Biosciences: a national resource for biomolecular structure and dynamics experiments using synchrotron footprinting. J. Synchrotron Radiat. 2007;14:233–43. doi: 10.1107/S0909049507013118. [DOI] [PubMed] [Google Scholar]
- 40.Xu G, Kiselar J, He Q, Chance MR. Secondary reactions and strategies to improve quantitative protein footprinting. Anal. Chem. 2005;77:3029–3037. doi: 10.1021/ac048282z. [DOI] [PubMed] [Google Scholar]
- 41.Guan JQ, Vorobiev S, Almo SC, Chance MR. Mapping the G-actin binding surface of cofilin using synchrotron protein footprinting. Biochemistry. 2002;41:5765–5775. doi: 10.1021/bi0121104. [DOI] [PubMed] [Google Scholar]
- 42.Thompson MW, Singh SK, Maurizi MR. Processive degradation of proteins by the ATP-dependent Clp protease from Escherichia coli. Requirement for the multiple array of active sites in ClpP but not ATP hydrolysis. J. Biol. Chem. 1994;269:18209–18215. [PubMed] [Google Scholar]
- 43.Thompson MW, Maurizi MR. Activity and specificity of Escherichia coli ClpAP protease in cleaving model peptide substrates. J. Biol. Chem. 1994;269:18201–18208. [PubMed] [Google Scholar]
- 44.Hedstrom L. Serine protease mechanism and specificity. Chem. Rev. 2002;102:4501–4524. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- 45.Liu Y, Kati W, Chen CM, Tripathi R, Molla A, Kohlbrenner W. Use of a fluorescence plate reader for measuring kinetic parameters with inner filter effect correction. Anal. Biochem. 1999;267:331–335. doi: 10.1006/abio.1998.3014. [DOI] [PubMed] [Google Scholar]
- 46.Caplow M, Jencks WP. The apparent absence of an acyl-enzyme intermediate in the chymotrypsin-catalyzed hydrolysis of acetyl-L-tyrosine hydroxamic acid. J. Biol. Chem. 1963;238:1907–1909. [PubMed] [Google Scholar]
- 47.Kezdy FJ, Clement GE, Bender ML. The alpha-chymotrypsin-catalyzed hydrolysis And hydroxylaminolysis of N-acetyl-L-tyrosine ethyl ester. J. Biol. Chem. 1963;238:3141–3143. [PubMed] [Google Scholar]
- 48.Epand RM, Wilson IB. On the question of an acyl-enzyme intermediate in the chymotrypsin-catalyzed hydrolysis of hydroxamic acids. J. Biol. Chem. 1963;238:3138–3140. [PubMed] [Google Scholar]
- 49.Horovitz A, Willison KR. Allosteric regulation of chaperonins. Curr. Opin. Struct. Biol. 2005;15:646–651. doi: 10.1016/j.sbi.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 50.Hinnerwisch J, Fenton WA, Furtak KJ, Farr GW, Horwich AL. Loops in the central channel of ClpA chaperone mediate protein binding, unfolding, and translocation. Cell. 2005;121:1029–1041. doi: 10.1016/j.cell.2005.04.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.