

Abstract
We investigated relationships between glucose metabolism, amyloid load and measures of cognitive and functional impairment in Alzheimer’s disease (AD). Patients meeting criteria for probable AD underwent [11C]PIB and [18F]FDG PET imaging and were assessed on a set of clinical measures. PIB Distribution volume ratios and FDG scans were spatially normalized and average PIB counts from regions-of-interest (ROI) were used to compute a measure of global PIB uptake. Separate voxel-wise regressions explored local and global relationships between metabolism, amyloid burden and clinical measures. Regressions reflected cognitive domains assessed by individual measures, with visuospatial tests associated with more posterior metabolism, and language tests associated with metabolism in the left hemisphere. Correlating regional FDG uptake with these measures confirmed these findings. In contrast, no correlations were found between either voxel-wise or regional PIB uptake and any of the clinical measures. Finally, there were no associations between regional PIB and FDG uptake. We conclude that regional and global amyloid burden does not correlate with clinical status or glucose metabolism in AD.
Keywords: amyloid plaques, amyloidosis, Alzheimer’s disease, glucose metabolism, Pittsburgh compound-B, Fluorodeoxyglucose, dementia severity, cognition
1. Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease with a distinct neuropathology involving extracellular neuritic β-amyloid (Aβ) plaques and intraneuronal neurofibrillary tangles (NFT) (Arnold et al., 1991; Braak and Braak, 1991, 1997). Triggered by findings that genetic defects in the amyloid precursor protein (APP) gene lead to overproduction of Aβ and early-onset familial AD (Goate et al., 1991; Naruse et al., 1991), it has been suggested that Aβ is in fact the primary cause of AD (Hardy and Higgins, 1992). According to this “amyloid cascade hypothesis” Aβ causes synaptic dysfunction, synapse loss, and neuronal death resulting in cognitive decline and dementia.
The clinicopathologic evidence supporting this hypothesis remains inconclusive. Several studies suggest a correlation between cognitive decline and Aβ plaque levels in AD (Bussiere et al., 2002; Cummings and Cotman, 1995; Cummings et al., 1996; Haroutunian et al., 1998; Kanne et al., 1998; McLean et al., 1999; Naslund et al., 2000; Parvathy et al., 2001) and in some cases more specifically a regional association between Aβ levels and dementia severity (Bussiere et al., 2002; Cummings et al., 1996; Naslund et al., 2000). However, other studies have challenged this view showing that dementia severity increases as a function of NFT and not plaque density (Arriagada et al., 1992; Berg et al., 1993; Berg et al., 1998; Bierer et al., 1995; Crystal et al., 1988; Duyckaerts et al., 1997; Giannakopoulos et al., 2003; Hof et al., 1992; Price, J. L. et al., 1991). While these discrepancies could arise from technical factors such as sampling methods, (Bussiere et al., 2002), it is possible that differences in amyloid species or mediation of effects by NFT pathology (Bennett et al., 2004; Giannakopoulos et al., 2003) could be responsible.
In vivo uptake of the radiotracer 18F-fluorodeoxyglucose (FDG) is strongly correlated to cerebral synaptic density and activity (Rocher et al., 2003) and has been extensively employed to study metabolic decline in AD. Significant metabolic reductions in association cortex including the precuneus and posterior cingulate have been found to be closely associated with disease severity and to differentiate normal controls (NC) from AD patients (Heiss et al., 1991a; Heiss et al., 1991b; Herholz et al., 2002; Ichimiya et al., 1994; Langbaum et al., 2009; Mielke et al., 1994; Minoshima et al., 1995b). Global and regional effects of Aβ deposition on brain function can now be assessed by combining FDG with the recently developed PET ligand [N-methyl-11C]-2-(4′-methylaminophenyl)-6-hydroxybenzothiazole, more commonly known as Pittsburgh Compound-B (PIB), which specifically binds to fibrillar amyloid both in vitro (Klunk et al., 2003) and in vivo (Bacskai et al., 2003; Klunk et al., 2004). PIB has been used to study AD, normal aging, mild cognitive impairment (MCI), frontotemporal and other amyloid-associated dementias (Aizenstein et al., 2008; Boxer et al., 2007; Engler et al., 2006; Forsberg et al., 2008; Frisoni et al., 2009; Gomperts et al., 2008; Jack et al., 2009; Johnson et al., 2007; Mintun et al., 2006; Mormino et al., 2009; Pike et al., 2007; Rabinovici et al., 2007; Rowe et al., 2007; Sperling et al., 2009). Combining both tracers some authors (Edison et al., 2007; Klunk et al., 2004) have found negative correlations between β-amyloid load and glucose metabolism in temporal and parietal regions but only when either PIB negative healthy controls or PIB negative AD patients were included. Similarly, others (Li et al., 2008) initially found inverse intraregional correlations between FDG and PIB uptake when combining data of NC, MCI and AD patients but none of these correlations survived within the three diagnostic groups. Only a few studies have looked at the relationship between cognition and in vivo amyloid burden as measured by PIB in AD. Three studies reported no significant correlations between PIB uptake and either the Mini-Mental Status Exam (MMSE) (Klunk et al., 2004; Pike et al., 2007; Rowe et al., 2007) or composite episodic memory scores (Pike et al., 2007). One study (Engler et al., 2006) found inverse correlations between regional PIB uptake and MMSE scores at baseline but this finding could not be replicated at the 24 month follow-up. Another (Edison et al., 2007) reported correlations between PIB uptake and scores on the Warrington Recognition Memory Test which were, however, dependent on the inclusion of 2 PIB negative AD patients (i.e. patients with no amyloidosis). A more recent study (Grimmer et al., 2008) found that scores on the Clinical Dementia Rating “sum of boxes” (CDRSB) explained 11–22 % of the regional variance of PIB uptake in frontal cortex, anterior cingulate and nucleus lentiformis but in subsequent voxel-wise regressions these correlations were not reliable once age was included as a nuisance variable.
Taken together these studies suggest that the impact of Aβ on cognition and brain metabolism differs according to diagnosis and in the case of clinical AD pathology it is in fact questionable whether there is any considerable effect in patients with genuine amyloid pathology. Clarity on the latter issue is obviously of crucial importance for the future development and assessment of appropriate disease modifying therapies (Aisen, 2009). The current study was specifically aimed at investigating this relationship in a large cohort of patients diagnosed with probable AD and we hypothesized that FDG would be more strongly associated with cognitive status than PIB.
2. Materials and methods
2.1. Subject selection
Patients were recruited from an AD research cohort followed at the University of California San Francisco Memory and Aging Center (UCSF-MAC). The clinical evaluation included a history and physical examination by a neurologist, a structured caregiver interview administered by a nurse, and a comprehensive battery of neuropsychological tests (Kramer et al., 2003). Clinical diagnoses were assigned by consensus at a multidisciplinary conference using standard research criteria (McKhann et al., 1984). In the current study we used a subset of clinical measures which included the MMSE (Folstein et al., 1975), the CDRSB (Morris, 1993), copy of the modified Rey-Osterrieth figure (ModRey) and a phonemic verbal fluency test (VbFlu) to assess functional status, visuo-spatial memory and executive functions (Kramer et al., 2003). The mean interval between testing and PET was 90 ± 83 days. Although meeting criteria for probable AD one of our patients scored 0 on the CDRSB. However, she has recently declined to a score of 1 which is consistent with the original diagnosis.
Patients were considered eligible for the study if they had a clinical diagnosis of probable AD and did not have significant co-morbid medical, neurological or psychiatric illness. Patients were recruited between April 2005 and January 2009, and all who consented to participate were enrolled. Two subjects were excluded from the final analysis for technical reasons (motion artifact, incomplete study, etc.). Our final cohort consisted of 39 patients with probable AD.
The study was approved by the Institutional review boards of all participating institutions.
2.2. Image acquisition
11C-PIB was synthesized at the Lawrence Berkeley National Laboratory’s Biomedical Isotope Facility using a previously published protocol (Mathis et al., 2003). 18F-fluorodeoxyglucose was purchased from a commercial vendor (Eastern Isotopes, Sterling, VA). PET scans were performed at Lawrence Berkeley National Laboratory using a Siemens ECAT EXACT HR PET scanner in 3D acquisition mode. All 39 subjects underwent PET imaging with 11C-PIB. Between 10–15 mCi of high specific-activity 11C-PIB was injected as a bolus into an antecubital vein. Dynamic acquisition frames were obtained as follows: 4 × 15 sec, 8 × 30 sec, 9 × 60 sec, 2 × 180 sec, 8 × 300 sec and 3 × 600 sec, for a total of 90 minutes. Thirty-four subjects also underwent PET imaging with 18F-FDG. 18F-FDG imaging began a minimum of two hours following 11C-PIB injection (six Carbon-11 half-lives). Six emission frames of 5 min each were acquired starting 30 minutes after injection of approximately 10 mCi of the tracer, with the patient resting quietly in a well lighted room with minimum ambient noise, and eyes and ears open during tracer uptake. Ten minute transmission scans for attenuation correction were obtained either immediately prior or following each 11C-PIB and 18F-FDG scan. PET data were reconstructed using an ordered subset expectation maximization algorithm with weighted attenuation. Images were smoothed with a 4mm Gaussian kernel with scatter correction. All images were evaluated prior to analysis for patient motion and adequacy of statistical counts.
2.3. Image pre-processing and definition of regions of interest
PET data were pre-processed using SPM2 (http://www.fil.ion.ucl.ac.uk/spm). Origins were manually set to the anterior commissure (AC) in all PET frames. 11C-PIB frames 6-34 were coregistered and resliced with SPM2 PET realignment parameters using a mid-scan frame visually judged to best represent the subject’s anatomy (usually number 17) as the reference frame. Frames 1–5 were coregistered separately to the mean 11C-PIB image obtained from realignment, since these frames typically contain a paucity of anatomical information due to low tracer counts. 18F-FDG scans were realigned with defaults and summed. 11C-PIB and 18F-FDG scans were then separately normalized to the SPM2 PET Montreal Neurological Institute (MNI) space template using the individual patient’s mean 11C-PIB and 18F-FDG scans, respectively. The resulting normalization parameters were then used to perform a reversed (backwards) normalization of two (MNI space) ROI templates that included the cerebellum and the pons. The cerebellum ROI comprised all cerebellar sections from the Automated Anatomic Labeling (AAL) Atlas (Schmahmann et al., 1999; Tzourio-Mazoyer et al., 2002) excluding peduncular white matter. The pons ROI was manually drawn. Reversed normalization warps MNI space ROIs into each subject’s native-space where they were visually inspected and manually edited if needed. It should be noted here that the results presented in this paper were also replicated in homologous analyses involving a subset of thirty patients for which MRs were available at the time in order to rule out any potential bias due to imperfect normalization or masked effects of atrophy. The analysis involved spatial normalzation of PET data based on parameters derived from normalizing the patients’ MRs to the T1 MNI template in SPM5 and subsequent atrophy correction of the PET data. We adopted a two-compartmental partial-volume correction (Meltzer et al., 1990) which involves convolving a brain mask (a sum of the gray and white matter segmented images from the subject’s T1-weighted MRI) with the point-spread function specific to the PET tomograph along all axes which had been previously empirically derived (Klein et al., 1997). This provides a means for estimating the percentage of brain tissue emitting tracer at each voxel. The PET counts for each voxel are then adjusted based on the percentage of estimated brain matter (Meltzer et al., 1999). Since the results from these analyses were essentially the same but slightly attenuated due to the smaller number of cases we present here only the results for the PET based processing stream without MRI data.
2.4. Image analysis
18F-FDG scans were normalized to mean activity in the pons ROI for each subject (Minoshima et al., 1995a). For 11C-PIB, voxel-wise Distribution Volume Ratios (DVRs) were calculated using Logan graphical analysis (Logan et al., 1996), with the cerebellum ROI time-activity curve used as a reference tissue input function (Klunk et al., 2004; Lopresti et al., 2005; Price, J. C. et al., 2005). Kinetic parameters (T = 35–90 min, k2 = 0.15 min−1) were based on previously reported values (Price, J. C. et al., 2005). Finally, all native space DVRs and pons normalized FDG scans were spatially normalized to MNI space using the normalization parameters that had been obtained earlier.
2.5. ROI analysis
We merged and pooled subsets from the original AAL atlas (Tzourio-Mazoyer et al., 2002) to form the following ROIs: FRONTAL = comprised all frontal lobe ROIs except the supplementary motor area (SMA), precentral gyrus and olfactory cortex, ACG = the anterior cingulate, PCG = the posterior cingulate, PARIETAL = all parietal lobe ROIs except the precuneus, PRECUNEUS = the precuneus ROIs only, TEMPORAL = all temporal lobe ROIs including the temporal poles. In order to measure global amyloid burden we also merged a selection of AAL ROIs of brain regions known to be particularly affected by amyloidosis to form a PIB index. These consisted of dorsolateral, medial and orbital superior frontal gyrus, lateral and orbital middle frontal gyrus, anterior cingulate and paracingulate gyri, lateral middle temporal gyrus, angular gyrus, supramarginal gyrus and precuneus.
2.6. Voxel-wise multiple regressions and correlations
Pons normalized FDG scans and DVRs were entered into two separate series of multiple regressions (SPM2) with age, education and gender as nuisance variables to explore the relationship between cognitive/functional measures and either glucose metabolism or amyloid burden. An additional regression model (same nuisance variables) investigated the relationship between global amyloid burden (as measured by the PIB index) and glucose metabolism. All t-maps were thresholded at an uncorrected p < .001 and cluster thresholded at a cluster size of > 100 voxels. Biological parametric mapping (BPM) (Casanova et al., 2007) was also employed to compute voxel-to-voxel correlations between FDG and DVR data. BPM t-maps were thresholded at an uncorrected p < .001.
2.7. Partial correlations
Partial correlations (Statistica 6.0 software, StatSoft Inc., Tulsa, OK) controlling for age, education and gender were performed between regional FDG uptake and cognitive/functional measures, regional and global PIB uptake and cognitive/functional measures, and finally regional FDG and both regional and global PIB uptake.
2.8. PIB positivity
To ensure that our analyses were a true reflection of the relationship between cognition and patients with amyloidosis we determined a cut-off for PIB-positivity using a recently developed iterative method (Aizenstein et al., 2008) that also involved a NC cohort (not presented). Based on this analysis scans with PIB indices > 1.18 were considered PIB positive. This resulted in thirty-five PIB positive and four PIB negative patients. All of the above analyses were then re-run with PIB positive patients only.
3. Results
The patient cohort consisted of thirty-nine AD patients (twenty-four males) with a mean age of 68.3 ± 10.5 years and 16.8 ± 2.8 years of education. Patients’ mean scores for MMSE and CDRSB were 21.8 ± 5.7 and 5.5 ± 3.0, respectively. CDRSB data was only available for thirty-four patients. The cohort had an estimated average disease duration of 5.4 ± 2.8 years (see table 1) and a mean global PIB uptake of 1.67 with a range of 0.92–2.33.
Table 1.
Demographics of AD cohort (N = 39, 24 males)
| Age | Education | MMSE | CDRSB* | DISDUR | |
|---|---|---|---|---|---|
| Mean | 68.3 (10.5) | 16.8 (2.8) | 21.8 (5.7) | 5.5 (3.0) | 5.4 (2.8) |
| Median | 69.5 | 16.0 | 23.0 | 5.0 | 5.0 |
- Data only available for 34 patients
MMSE = mini-mental state exam; CDRSB = clinical dementia rating sum-of-boxes; DISDUR = estimated disease duration (years)
Separate voxel-wise multiple regressions of FDG uptake with each of the cognitive/functional measures controlling for age, education and gender revealed significant effects (uncorrected p < .001 and cluster thresholded to > 100 voxels). MMSE scores were positively correlated with glucose metabolism in bilateral posterior cingulate, inferior parietal lobule, middle temporal, supramarginal, cingulate, middle frontal, superior temporal gyri and left anterior cingulate, middle/inferior orbitofrontal gyrus, insula, hippocampus, parahippocampus, caudate and thalamus. Functional impairment, as measured by CDRSB, was inversely correlated with FDG uptake in bilateral inferior parietal lobule, angular gyrus and left supramarginal gyrus. ModRey scores were positively correlated with metabolism in bilateral precuneus and left superior parietal lobule, middle temporal gyrus, middle/inferior occipital gyrus. VbFlu was positively correlated with left inferior parietal lobule and superior/middle temporal gyrus (see Figure 1).
Figure 1. Multiple regressions between voxel-wise FDG uptake and cognitive/functional measures (controlling for age, gender and education).
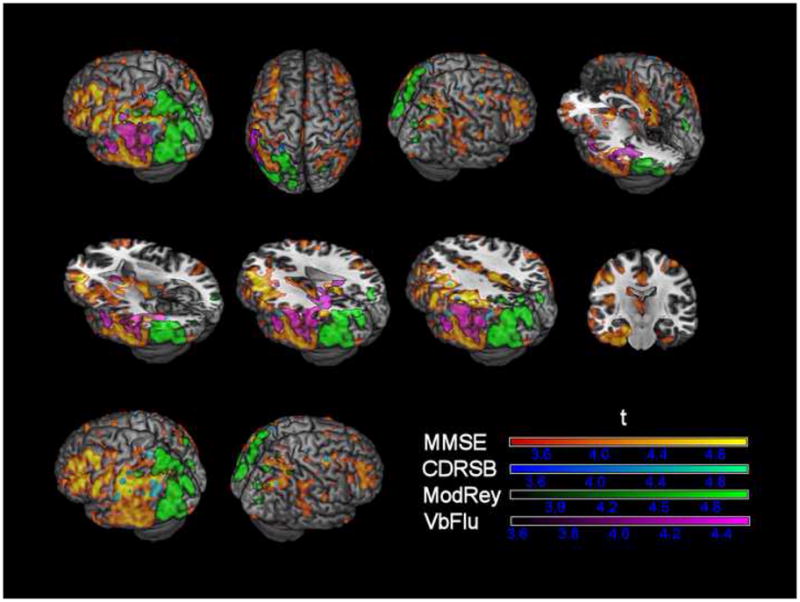
MMSE = mini-mental state exam; CDRSB = clinical dementia rating sum-of-boxes; ModRey = modified Rey test; VbFlu = verbal fluency test; Positive/negative associations between FDG uptake and MMSE, ModRey, VbFlu/CDRSB are simultaneously displayed in the first two rows; since the correlation pattern of CDRSB was spatially coinciding with that of VbFlu the latter was omitted in the last row for better visualization; t-maps are thresholded at p <.001 (uncorrected) and cluster size corrected for clusters > 100 voxels.
The ROI analysis comparing relationships between glucose metabolism and cognitive/functional measures yielded results similar to the voxel-wise analyses (Table 2). Since some of the above analyses were performed including data of one patient that had scored 1 and 17 on MMSE and CDRSB, respectively, we re-ran all relevant analyses excluding this case and confirmed that all effects remained essentially the same (Figures 2a & b).
Table 2.
Partial correlations between regional FDG uptake and cognitive/functional measures controlling for age, gender and education
| FRONTAL | ACG | PCG | PARIETAL | PRECUN | TEMPORAL | |
|---|---|---|---|---|---|---|
| MMSE (N=34) | .4163 | .3557 | .5847 | .5852 | .5346 | .4774 |
| p=.020 | p=.050 | p=.001 | p=.001 | p=.002 | p=.007 | |
| CDRSB (N=30) | −.4602 | −.4640 | −.5645 | −.5416 | −.4781 | −.4801 |
| p=.016 | p=.015 | p=.002 | p=.004 | p=.012 | p=.011 | |
| ModRey (N=21) | .0948 | .0046 | .0239 | .6644 | .3546 | .1235 |
| p=.708 | p=.986 | p=.925 | p=.003 | p=.149 | p=.625 | |
| VbFlu (N=23) | .3807 | .0751 | .5839 | .5526 | .4016 | .5410 |
| p=.098 | p=.753 | p=.007 | p=.012 | p=.079 | p=.014 |
MMSE = mini-mental state exam; CDRSB = clinical dementia rating sum-of-boxes; ModRey = modified Rey test; VbFlu = verbal fluency test; FRONTAL = frontal cortex; ACG = anterior cingulate; PCG = posterior cingulate; PARIETAL = parietal cortex; PRECUN = precuneus; TEMPORAL = temporal cortex
Figure 2.


Figure 2a Partial correlations between MMSE/CDRSB scores and regional FDG uptake controlling for age, gender and education
Figure 2b Partial correlations between MMSE/CDRSB scores and regional FDG uptake controlling for age, gender and education “extreme case” excluded
By contrast, no significant correlations were found between voxel-wise PIB uptake and any of the cognitive/functional measures at an uncorrected p <.001 and even when cluster thresholding was omitted. Consistent with the above we also did not find any partial correlations (r < .35, p > .12) between regional PIB uptake and cognitive/functional measures (Figure 3).
Figure 3.

Partial correlations between MMSE/CDRSB scores and regional PIB uptake controlling for age, gender and education
BPM t-maps correlating PIB and FDG uptake at the voxel level did not show any correlations at an uncorrected p <.001. Similarly, there were no region-to-region and global-to-region associations (r < .18, p > .32) between PIB burden and FDG uptake (Figure 4).
Figure 4.

Partial correlations between regional FDG uptake and global PIB uptake (PIB index) controlling for age, gender and education
Finally, all of the above effects were replicated in additional analyses using thirty-five PIB positive patients only.
4. Discussion
This study investigated in vivo relationships between Aβ plaque load, dementia severity and glucose metabolism in a cohort of 39 patients with probable AD. Consistent with the existing literature, we found cognitive decline to be strongly associated with decreased glucose metabolism in frontal and temporo-parietal regions (Heiss et al., 1991a; Heiss et al., 1991b; Herholz et al., 2002; Langbaum et al., 2009; Mielke et al., 1994; Minoshima et al., 1995b). The observed effects were not driven by extreme cases, differences in age, gender or education and were similar using both voxel-wise and ROI analyses. Patterns of association largely reflected the cognitive domains assessed by the individual tests, with visuospatial tests associated with more posterior regional metabolism, and language tests more associated with metabolism in the left hemisphere. In contrast, we did not find any correlations between global or regional Aβ load and cognitive or functional measures using identical analytic approaches. Furthermore, Aβ burden was not correlated with glucose metabolism in any brain regions.
The failure to find inverse correlations between PIB uptake and either cognitive or functional measures in our patients is unlikely due to lack of power in our analyses, since we did find such relationships for FDG and had an even slightly larger sample for the PIB analyses. Still it is conceivable that a correlation between PIB uptake and cognition might emerge when using larger sample numbers. However, the current data clearly show that even if such an effect existed it would have to be comparably weak and unlikely to be a significant contributor in explaining cognitive decline in AD.
Our results differ from a number of studies that found inverse correlations between PIB uptake and clinical measures (Edison et al., 2007; Grimmer et al., 2008; Klunk et al., 2004). However, some of these studies included AD subjects that did not show elevated PIB uptake. These PIB-negative AD subjects may have biased the results, particularly given the small sample sizes in these studies, since they tended to show high cognitive performance in the face of very low amyloid burden. Notably, the diagnosis of AD was later questioned in a number of PIB-negative AD patients on longitudinal follow-up (Engler et al., 2006). In a separate analysis using a recently developed iterative cut-off technique (Aizenstein et al., 2008) we determined that 4 of our 39 AD patients had global PIB values comparable to those of thirty NC (age = 73.7 ± 6.4). Given the aforementioned studies we looked at PIB uptake as a continuum and even though we did include these subjects no PIB related effects were observed. We also considered the converse argument that inclusion of PIB negative patients might in fact mask the effects related to amyloidosis. However, no such effects were observed when analyzing PIB positive patients only. One study (Grimmer et al., 2008) that did not include PIB negative subjects found that the maximum regional variance of PIB uptake explained by CDRSB scores was in the striatum (22 %) while no effects were observed in several regions that are known to be specifically affected by hypometabolism and amyloidosis, i.e. lateral temporal, parietal cortex, the precuneus and posterior cingulate. Finally, these effects were abolished in a voxel-wise analysis once age was included as a nuisance variable which could explain the discrepancy between their findings and ours.
PIB uptake generally correlates well with post-mortem levels of Aβ deposits (Bacskai et al., 2007; Ikonomovic et al., 2008; Lockhart et al., 2007) and has also been shown to closely reflect Aβ pathology in vivo (Leinonen et al., 2008). However, Rosen et al. (2009) recently reported that one of ten pathologically confirmed cases of AD showed negligible cortical homogenate PIB binding in vitro despite heavy cerebral Aβ-amyloidosis, suggesting that a PIB negative scan is not conclusive evidence for the absence of amyloidosis. While this is important in the context of differential diagnosis (Rabinovici et al., 2007; Rowe et al., 2007) the occurrence of “false negatives” appears to be rare (Rosen et al., 2009) and in our study the majority of PIB scans were positive which is considered to be a truthful indicator of Aβ deposition (i.e. false-positive PIB results compared to pathology have yet to be reported).
Several authors (Prohovnik et al., 2006; Savva et al., 2009) have reported evidence suggesting that in AD patients the association between plaques (and also NFT) and dementia severity diminishes with age and actually disappears by the ninth decade of life. In contrast, in patients under the age of 75 the association has been found to be strong and reliable. The young age of our cohort (mean 68) suggests that the lack of correlations between cognition and PIB uptake in our study is unlikely to be due to age.
There is evidence from mouse models suggesting that Aβ plaques may cause asynchrony of converging neuronal inputs (Stern et al., 2004) and abberant network excitability (Palop et al., 2007). In addition soluble Aβ oligomers have been implicated to play an important role in synaptic loss in AD (Haass and Selkoe, 2007; Walsh and Selkoe, 2007). Supporting this view, oligomers isolated from human AD brains have been shown to decrease dendritic spine density, inhibit long-term potentiation (LTP) and facilitate long-term depression (LTD) in rodent hippocampi (Klyubin et al., 2008; Shankar et al., 2007; Shankar et al., 2008). Furthermore soluble Aβ levels correlate with cognitive status (McLean et al., 1999; Samuels et al., 1999) and some authors have suggested that it is indeed the shift from soluble to insoluble Aβ species that marks the beginning of AD pathology (Wang et al., 1999). Unfortunately, however, given the fact that PIB uptake does not reflect soluble Aβ concentrations we are not able to determine to what extent the neurotoxic effect of soluble species may have contributed to the synapse loss mirrored in the metabolic deficits observed in this study.
Consistent with the literature we replicated the puzzling finding that PIB uptake is high in frontal cortex while this region showed much smaller metabolic decline compared to temporo-parietal areas. Despite a suggestion of a higher proportion of diffuse than neuritic plaques in frontal cortex (McKenzie et al., 1992), neuritic plaque pathology is extensive in this region. Some data suggest that frontal cortex shows lower levels of neuritic plaque pathology than parietal and temporal cortex (Arnold et al., 1991), but neuritic plaques are nevertheless widespread, accompanied by substantial numbers of NFT, and in some studies are as prevalent in frontal cortex as in posterior regions (Tiraboschi et al., 2004).
The possibility that Aβ accumulates later in the frontal lobes than in posterior areas, and therefore is exposed for a shorter durations to the toxic effects of Aβ does not seem likely in view of contrary histological and imaging data (Mintun et al., 2006; Thal et al., 2002). Similarly, duration of exposure to NFT does not adequately explain the anatomic discrepancies in glucose metabolism since frontal and posterior regions show NFT at similar disease stages (Braak and Braak, 1995).
A potential explanation for the differential impact of Aβ burden on synaptic density/activity as measured by FDG (Rocher et al., 2003) comes from studies comparing dendritic variation in different brain regions. Jacobs and colleagues found that basilar dendrites and associated spines were significantly more extensive in the anterior portion of the superior frontal cortex (BA 10) than in cuneus, calcarine and superior occipital lobes (BA 18), which they attributed to broader vs. discrete sampling of afferent information (Jacobs et al., 1997). A similar caudal-rostral progression of dendritic size and spine number has also been reported in monkeys (Elston et al., 1996; Elston and Rosa, 1997, 1998a, 1998b). The higher dendritic complexity observed by Jacobs and colleagues, which involved measures such as total dendritic length, spine number & density could suggest a type of neuronal reserve due to higher redundancy of synapses within the frontal lobes. This could explain why the impact of neurotoxicity appears to be reduced in frontal/anterior compared to posterior regions. It is possible that due to the a priori higher synaptic/dendritic density in frontal areas the detrimental effect of Aβ and/or NFT is delayed compared to other regions in the brain.
To date our study represents the largest AD cohort investigation of the impact of amyloid load on cognitive status and metabolism. The results are clear-cut and show that once the clinical diagnosis of AD has been reached, amyloid burden is no longer associated with dementia severity or hypometabolism. This finding is consistent with previous papers (Edison et al., 2007; Engler et al., 2006; Grimmer et al., 2008; Klunk et al., 2004; Pike et al., 2007; Rowe et al., 2007) and it is unlikely that the relationship between Aβ and dementia severity changes over the course of the disease since there is neither an appreciable change of PIB binding over time nor any correlation between plaque burden and disease duration (Edison et al., 2007; Engler et al., 2006; Ingelsson et al., 2004; Jack et al., 2009).
A recent longitudinal study of MRI and PIB changes across the diagnostic spectrum showed that in contrast to brain atrophy, which accelerated in correlation with clinical status, longitudinal changes in PIB uptake were low, did not reliably correlate with dementia severity and did not differ between healthy controls, MCI and AD patients (Jack et al., 2009). This has lead to the suggestion that the lifetime progression of plaque burden in AD is either marked by a sharp prodromal increase followed by a plateau throughout MCI and AD (Ingelsson et al., 2004) or by a slow but constant increase over an individual’s lifespan (Jack et al., 2009). Importantly, both models share the assumption that neurodegeneration and with it cognitive decline are no longer associated with Aβ burden once the AD stage is reached. Instead further functional decline in AD may be driven by increases in NFT burden (Ingelsson et al., 2004) or other pathogenic processes downstream from Aβ accumulation. Consistent with this notion, there is evidence from mouse models suggesting that Aβ neurotoxicity may indeed depend on the simultaneous expression of tau (Rapoport et al., 2002; Roberson et al., 2007).
The data presented here have important implications for the future development and evaluation of disease modifying therapies (Aisen, 2009) in clinical AD. It appears that in contrast to NC and MCI where there may be relationships between Aβ levels and both cognition and brain atrophy (Mormino et al., 2009; Pike et al., 2007), lowering Aβ levels in clinical AD may not be the most promising approach to either halt or reverse disease progression, as cognitive decline and neurodegeneration appear to proceed largely independent of plaque load at this point. Similarly, while PIB remains important in the context of differential diagnosis (Rabinovici et al., 2007; Rabinovici et al., 2008; Rowe et al., 2007), one of its most promising future applications lies in the detection of individuals harboring amyloid deposits prior to the onset of symptoms (Reiman et al., 2009) as potential candidates for preventive anti-amyloid interventions.
The mere existence of Aβ plaques in the aging brain is not sufficient to cause dementia (Aizenstein et al., 2008; Jack et al., 2008; Mintun et al., 2006; Mormino et al., 2009). However, Aβ appears to be a conditio sine qua non for the emergence of AD. Whether its role lies in the interaction with NFT pathology (Bennett et al., 2004; Rapoport et al., 2002; Roberson et al., 2007), brain injury (Brody et al., 2008), soluble oligomers (Walsh and Selkoe, 2007) or functional network deficits (Buckner et al., 2005; Sperling et al., 2009) remains to be explored.
Acknowledgments
This work was supported by National Institute on Aging grants NIA K23-AG031861 (GDR), NIA AG027859 (WJJ), NIA P01-AG1972403 and P50-AG023501 (BLM), Alzheimer’s Association NIRG-07-59422 (GDR) and ZEN-08-87090 (WJJ), John Douglas French Alzheimer’s Foundation (GDR).
Footnotes
Disclosure statement
The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aisen PS. Alzheimer’s disease therapeutic research: the path forward. Alzheimers Res Ther. 2009;1:2. doi: 10.1186/alzrt2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, Ziolko SK, James JA, Snitz BE, Houck PR, Bi W, Cohen AD, Lopresti BJ, DeKosky ST, Halligan EM, Klunk WE. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65:1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex. 1991;1:103–116. doi: 10.1093/cercor/1.1.103. [DOI] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Hickey GA, Skoch J, Kajdasz ST, Wang Y, Huang GF, Mathis CA, Klunk WE, Hyman BT. Four-dimensional multiphoton imaging of brain entry, amyloid binding, and clearance of an amyloid-beta ligand in transgenic mice. Proc Natl Acad Sci U S A. 2003;100:12462–12467. doi: 10.1073/pnas.2034101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacskai BJ, Frosch MP, Freeman SH, Raymond SB, Augustinack JC, Johnson KA, Irizarry MC, Klunk WE, Mathis CA, Dekosky ST, Greenberg SM, Hyman BT, Growdon JH. Molecular imaging with Pittsburgh Compound B confirmed at autopsy: a case report. Arch Neurol. 2007;64:431–434. doi: 10.1001/archneur.64.3.431. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol. 2004;61:378–384. doi: 10.1001/archneur.61.3.378. [DOI] [PubMed] [Google Scholar]
- Berg L, McKeel DW, Jr, Miller JP, Baty J, Morris JC. Neuropathological indexes of Alzheimer’s disease in demented and nondemented persons aged 80 years and older. Arch Neurol. 1993;50:349–358. doi: 10.1001/archneur.1993.00540040011008. [DOI] [PubMed] [Google Scholar]
- Berg L, McKeel DW, Jr, Miller JP, Storandt M, Rubin EH, Morris JC, Baty J, Coats M, Norton J, Goate AM, Price JL, Gearing M, Mirra SS, Saunders AM. Clinicopathologic studies in cognitively healthy aging and Alzheimer’s disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol. 1998;55:326–335. doi: 10.1001/archneur.55.3.326. [DOI] [PubMed] [Google Scholar]
- Bierer LM, Hof PR, Purohit DP, Carlin L, Schmeidler J, Davis KL, Perl DP. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch Neurol. 1995;52:81–88. doi: 10.1001/archneur.1995.00540250089017. [DOI] [PubMed] [Google Scholar]
- Boxer AL, Rabinovici GD, Kepe V, Goldman J, Furst AJ, Huang SC, Baker SL, O’Neil JP, Chui H, Geschwind MD, Small GW, Barrio JR, Jagust W, Miller BL. Amyloid imaging in distinguishing atypical prion disease from Alzheimer disease. Neurology. 2007;69:283–290. doi: 10.1212/01.wnl.0000265815.38958.b6. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271–278. doi: 10.1016/0197-4580(95)00021-6. discussion 278–284. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Diagnostic criteria for neuropathologic assessment of Alzheimer’s disease. Neurobiol Aging. 1997;18:S85–88. doi: 10.1016/s0197-4580(97)00062-6. [DOI] [PubMed] [Google Scholar]
- Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, Zipfel GJ, Holtzman DM. Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science. 2008;321:1221–1224. doi: 10.1126/science.1161591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis CA, Morris JC, Mintun MA. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussiere T, Friend PD, Sadeghi N, Wicinski B, Lin GI, Bouras C, Giannakopoulos P, Robakis NK, Morrison JH, Perl DP, Hof PR. Stereologic assessment of the total cortical volume occupied by amyloid deposits and its relationship with cognitive status in aging and Alzheimer’s disease. Neuroscience. 2002;112:75–91. doi: 10.1016/s0306-4522(02)00056-8. [DOI] [PubMed] [Google Scholar]
- Casanova R, Srikanth R, Baer A, Laurienti PJ, Burdette JH, Hayasaka S, Flowers L, Wood F, Maldjian JA. Biological parametric mapping: A statistical toolbox for multimodality brain image analysis. Neuroimage. 2007;34:137–143. doi: 10.1016/j.neuroimage.2006.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crystal H, Dickson D, Fuld P, Masur D, Scott R, Mehler M, Masdeu J, Kawas C, Aronson M, Wolfson L. Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer’s disease. Neurology. 1988;38:1682–1687. doi: 10.1212/wnl.38.11.1682. [DOI] [PubMed] [Google Scholar]
- Cummings BJ, Cotman CW. Image analysis of beta-amyloid load in Alzheimer’s disease and relation to dementia severity. Lancet. 1995;346:1524–1528. doi: 10.1016/s0140-6736(95)92053-6. [DOI] [PubMed] [Google Scholar]
- Cummings BJ, Pike CJ, Shankle R, Cotman CW. Beta-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer’s disease. Neurobiol Aging. 1996;17:921–933. doi: 10.1016/s0197-4580(96)00170-4. [DOI] [PubMed] [Google Scholar]
- Duyckaerts C, Bennecib M, Grignon Y, Uchihara T, He Y, Piette F, Hauw JJ. Modeling the relation between neurofibrillary tangles and intellectual status. Neurobiol Aging. 1997;18:267–273. doi: 10.1016/s0197-4580(97)80306-5. [DOI] [PubMed] [Google Scholar]
- Edison P, Archer HA, Hinz R, Hammers A, Pavese N, Tai YF, Hotton G, Cutler D, Fox N, Kennedy A, Rossor M, Brooks DJ. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology. 2007;68:501–508. doi: 10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- Elston GN, Rosa MG, Calford MB. Comparison of dendritic fields of layer III pyramidal neurons in striate and extrastriate visual areas of the marmoset: a Lucifer yellow intracellular injection. Cereb Cortex. 1996;6:807–813. doi: 10.1093/cercor/6.6.807. [DOI] [PubMed] [Google Scholar]
- Elston GN, Rosa MG. The occipitoparietal pathway of the macaque monkey: comparison of pyramidal cell morphology in layer III of functionally related cortical visual areas. Cereb Cortex. 1997;7:432–452. doi: 10.1093/cercor/7.5.432. [DOI] [PubMed] [Google Scholar]
- Elston GN, Rosa MG. Morphological variation of layer III pyramidal neurones in the occipitotemporal pathway of the macaque monkey visual cortex. Cereb Cortex. 1998a;8:278–294. doi: 10.1093/cercor/8.3.278. [DOI] [PubMed] [Google Scholar]
- Elston GN, Rosa MG. Complex dendritic fields of pyramidal cells in the frontal eye field of the macaque monkey: comparison with parietal areas 7a and LIP. Neuroreport. 1998b;9:127–131. doi: 10.1097/00001756-199801050-00025. [DOI] [PubMed] [Google Scholar]
- Engler H, Forsberg A, Almkvist O, Blomquist G, Larsson E, Savitcheva I, Wall A, Ringheim A, Langstrom B, Nordberg A. Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain. 2006;129:2856–2866. doi: 10.1093/brain/awl178. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Forsberg A, Engler H, Almkvist O, Blomquist G, Hagman G, Wall A, Ringheim A, Langstrom B, Nordberg A. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol Aging. 2008;29:1456–1465. doi: 10.1016/j.neurobiolaging.2007.03.029. [DOI] [PubMed] [Google Scholar]
- Frisoni GB, Lorenzi M, Caroli A, Kemppainen N, Nagren K, Rinne JO. In vivo mapping of amyloid toxicity in Alzheimer disease. Neurology. 2009;72:1504–1511. doi: 10.1212/WNL.0b013e3181a2e896. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, Morrison JH, Gold G, Hof PR. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Gomperts SN, Rentz DM, Moran E, Becker JA, Locascio JJ, Klunk WE, Mathis CA, Elmaleh DR, Shoup T, Fischman AJ, Hyman BT, Growdon JH, Johnson KA. Imaging amyloid deposition in Lewy body diseases. Neurology. 2008;71:903–910. doi: 10.1212/01.wnl.0000326146.60732.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmer T, Henriksen G, Wester HJ, Forstl H, Klunk WE, Mathis CA, Kurz A, Drzezga A. Clinical severity of Alzheimer’s disease is associated with PIB uptake in PET. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.01.016. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Haroutunian V, Perl DP, Purohit DP, Marin D, Khan K, Lantz M, Davis KL, Mohs RC. Regional distribution of neuritic plaques in the nondemented elderly and subjects with very mild Alzheimer disease. Arch Neurol. 1998;55:1185–1191. doi: 10.1001/archneur.55.9.1185. [DOI] [PubMed] [Google Scholar]
- Heiss WD, Kessler J, Szelies B, Grond M, Fink G, Herholz K. Positron emission tomography in the differential diagnosis of organic dementias. J Neural Transm Suppl. 1991a;33:13–19. doi: 10.1007/978-3-7091-9135-4_3. [DOI] [PubMed] [Google Scholar]
- Heiss WD, Szelies B, Kessler J, Herholz K. Abnormalities of energy metabolism in Alzheimer’s disease studied with PET. Ann N Y Acad Sci. 1991b;640:65–71. doi: 10.1111/j.1749-6632.1991.tb00192.x. [DOI] [PubMed] [Google Scholar]
- Herholz K, Salmon E, Perani D, Baron JC, Holthoff V, Frolich L, Schonknecht P, Ito K, Mielke R, Kalbe E, Zundorf G, Delbeuck X, Pelati O, Anchisi D, Fazio F, Kerrouche N, Desgranges B, Eustache F, Beuthien-Baumann B, Menzel C, Schroder J, Kato T, Arahata Y, Henze M, Heiss WD. Discrimination between Alzheimer dementia and controls by automated analysis of multicenter FDG PET. Neuroimage. 2002;17:302–316. doi: 10.1006/nimg.2002.1208. [DOI] [PubMed] [Google Scholar]
- Hof PR, Bierer LM, Perl DP, Delacourte A, Buee L, Bouras C, Morrison JH. Evidence for early vulnerability of the medial and inferior aspects of the temporal lobe in an 82-year-old patient with preclinical signs of dementia. Regional and laminar distribution of neurofibrillary tangles and senile plaques. Arch Neurol. 1992;49:946–953. doi: 10.1001/archneur.1992.00530330070019. [DOI] [PubMed] [Google Scholar]
- Ichimiya A, Herholz K, Mielke R, Kessler J, Slansky I, Heiss WD. Difference of regional cerebral metabolic pattern between presenile and senile dementia of the Alzheimer type: a factor analytic study. J Neurol Sci. 1994;123:11–17. doi: 10.1016/0022-510x(94)90197-x. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, Lopresti BJ, Ziolko S, Bi W, Paljug WR, Debnath ML, Hope CE, Isanski BA, Hamilton RL, DeKosky ST. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131:1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, Knopman DS, Boeve BF, Klunk WE, Mathis CA, Petersen RC. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer’s disease and amnestic mild cognitive impairment. Brain. 2008;131:665–680. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Shiung MM, Gunter JL, Boeve BF, Kemp BJ, Weiner M, Petersen RC. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain. 2009 doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs B, Driscoll L, Schall M. Life-span dendritic and spine changes in areas 10 and 18 of human cortex: a quantitative Golgi study. J Comp Neurol. 1997;386:661–680. [PubMed] [Google Scholar]
- Johnson KA, Gregas M, Becker JA, Kinnecom C, Salat DH, Moran EK, Smith EE, Rosand J, Rentz DM, Klunk WE, Mathis CA, Price JC, Dekosky ST, Fischman AJ, Greenberg SM. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol. 2007;62:229–234. doi: 10.1002/ana.21164. [DOI] [PubMed] [Google Scholar]
- Kanne SM, Balota DA, Storandt M, McKeel DW, Jr, Morris JC. Relating anatomy to function in Alzheimer’s disease: neuropsychological profiles predict regional neuropathology 5 years later. Neurology. 1998;50:979–985. doi: 10.1212/wnl.50.4.979. [DOI] [PubMed] [Google Scholar]
- Klein GJ, Teng X, Jagust WJ, Eberling JL, Acharya A, Reutter BW, Huesman RH. A methodology for specifying PET VOI’s using multimodality techniques. IEEE Trans Med Imaging. 1997;16:405–415. doi: 10.1109/42.611350. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Shao L, Hamilton RL, Ikonomovic MD, DeKosky ST, Mathis CA. The binding of 2-(4′-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci. 2003;23:2086–2092. doi: 10.1523/JNEUROSCI.23-06-02086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28:4231–4237. doi: 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer JH, Jurik J, Sha SJ, Rankin KP, Rosen HJ, Johnson JK, Miller BL. Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn Behav Neurol. 2003;16:211–218. doi: 10.1097/00146965-200312000-00002. [DOI] [PubMed] [Google Scholar]
- Langbaum JB, Chen K, Lee W, Reschke C, Bandy D, Fleisher AS, Alexander GE, Foster NL, Weiner MW, Koeppe RA, Jagust WJ, Reiman EM. Categorical and correlational analyses of baseline fluorodeoxyglucose positron emission tomography images from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) Neuroimage. 2009;45:1107–1116. doi: 10.1016/j.neuroimage.2008.12.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen V, Alafuzoff I, Aalto S, Suotunen T, Savolainen S, Nagren K, Tapiola T, Pirttila T, Rinne J, Jaaskelainen JE, Soininen H, Rinne JO. Assessment of beta-amyloid in a frontal cortical brain biopsy specimen and by positron emission tomography with carbon 11-labeled Pittsburgh Compound B. Arch Neurol. 2008;65:1304–1309. doi: 10.1001/archneur.65.10.noc80013. [DOI] [PubMed] [Google Scholar]
- Li Y, Rinne JO, Mosconi L, Pirraglia E, Rusinek H, DeSanti S, Kemppainen N, Nagren K, Kim BC, Tsui W, de Leon MJ. Regional analysis of FDG and PIB-PET images in normal aging, mild cognitive impairment, and Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2008;35:2169–2181. doi: 10.1007/s00259-008-0833-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart A, Lamb JR, Osredkar T, Sue LI, Joyce JN, Ye L, Libri V, Leppert D, Beach TG. PIB is a non-specific imaging marker of amyloid-beta (Abeta) peptide-related cerebral amyloidosis. Brain. 2007;130:2607–2615. doi: 10.1093/brain/awm191. [DOI] [PubMed] [Google Scholar]
- Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab. 1996;16:834–840. doi: 10.1097/00004647-199609000-00008. [DOI] [PubMed] [Google Scholar]
- Lopresti BJ, Klunk WE, Mathis CA, Hoge JA, Ziolko SK, Lu X, Meltzer CC, Schimmel K, Tsopelas ND, DeKosky ST, Price JC. Simplified quantification of Pittsburgh Compound B amyloid imaging PET studies: a comparative analysis. J Nucl Med. 2005;46:1959–1972. [PubMed] [Google Scholar]
- Mathis CA, Wang Y, Holt DP, Huang GF, Debnath ML, Klunk WE. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem. 2003;46:2740–2754. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- McKenzie JE, Gentleman SM, Royston MC, Edwards RJ, Roberts GW. Quantification of plaque types in sulci and gyri of the medial frontal lobe in patients with Alzheimer’s disease. Neurosci Lett. 1992;143:23–26. doi: 10.1016/0304-3940(92)90224-u. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Meltzer CC, Leal JP, Mayberg HS, Wagner HN, Jr, Frost JJ. Correction of PET data for partial volume effects in human cerebral cortex by MR imaging. J Comput Assist Tomogr. 1990;14:561–570. doi: 10.1097/00004728-199007000-00011. [DOI] [PubMed] [Google Scholar]
- Meltzer CC, Kinahan PE, Greer PJ, Nichols TE, Comtat C, Cantwell MN, Lin MP, Price JC. Comparative evaluation of MR-based partial-volume correction schemes for PET. J Nucl Med. 1999;40:2053–2065. [PubMed] [Google Scholar]
- Mielke R, Herholz K, Grond M, Kessler J, Heiss WD. Clinical deterioration in probable Alzheimer’s disease correlates with progressive metabolic impairment of association areas. Dementia. 1994;5:36–41. doi: 10.1159/000106692. [DOI] [PubMed] [Google Scholar]
- Minoshima S, Frey KA, Foster NL, Kuhl DE. Preserved pontine glucose metabolism in Alzheimer disease: a reference region for functional brain image (PET) analysis. J Comput Assist Tomogr. 1995a;19:541–547. doi: 10.1097/00004728-199507000-00006. [DOI] [PubMed] [Google Scholar]
- Minoshima S, Frey KA, Koeppe RA, Foster NL, Kuhl DE. A diagnostic approach in Alzheimer’s disease using three-dimensional stereotactic surface projections of fluorine-18-FDG PET. J Nucl Med. 1995b;36:1238–1248. [PubMed] [Google Scholar]
- Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, Klunk WE, Mathis CA, DeKosky ST, Morris JC. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- Mormino EC, Kluth JT, Madison CM, Rabinovici GD, Baker SL, Miller BL, Koeppe RA, Mathis CA, Weiner MW, Jagust WJ. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain. 2009;132:1310–1323. doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- Naruse S, Igarashi S, Kobayashi H, Aoki K, Inuzuka T, Kaneko K, Shimizu T, Iihara K, Kojima T, Miyatake T, et al. Mis-sense mutation Val----Ile in exon 17 of amyloid precursor protein gene in Japanese familial Alzheimer’s disease. Lancet. 1991;337:978–979. doi: 10.1016/0140-6736(91)91612-x. [DOI] [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. Jama. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvathy S, Davies P, Haroutunian V, Purohit DP, Davis KL, Mohs RC, Park H, Moran TM, Chan JY, Buxbaum JD. Correlation between Abetax-40-, Abetax-42-, and Abetax-43-containing amyloid plaques and cognitive decline. Arch Neurol. 2001;58:2025–2032. doi: 10.1001/archneur.58.12.2025. [DOI] [PubMed] [Google Scholar]
- Pike KE, Savage G, Villemagne VL, Ng S, Moss SA, Maruff P, Mathis CA, Klunk WE, Masters CL, Rowe CC. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain. 2007;130:2837–2844. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- Price JC, Klunk WE, Lopresti BJ, Lu X, Hoge JA, Ziolko SK, Holt DP, Meltzer CC, DeKosky ST, Mathis CA. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. J Cereb Blood Flow Metab. 2005;25:1528–1547. doi: 10.1038/sj.jcbfm.9600146. [DOI] [PubMed] [Google Scholar]
- Price JL, Davis PB, Morris JC, White DL. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer’s disease. Neurobiol Aging. 1991;12:295–312. doi: 10.1016/0197-4580(91)90006-6. [DOI] [PubMed] [Google Scholar]
- Prohovnik I, Perl DP, Davis KL, Libow L, Lesser G, Haroutunian V. Dissociation of neuropathology from severity of dementia in late-onset Alzheimer disease. Neurology. 2006;66:49–55. doi: 10.1212/01.wnl.0000191298.68045.50. [DOI] [PubMed] [Google Scholar]
- Rabinovici GD, Furst AJ, O’Neil JP, Racine CA, Mormino EC, Baker SL, Chetty S, Patel P, Pagliaro TA, Klunk WE, Mathis CA, Rosen HJ, Miller BL, Jagust WJ. 11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration. Neurology. 2007;68:1205–1212. doi: 10.1212/01.wnl.0000259035.98480.ed. [DOI] [PubMed] [Google Scholar]
- Rabinovici GD, Jagust WJ, Furst AJ, Ogar JM, Racine CA, Mormino EC, O’Neil JP, Lal RA, Dronkers NF, Miller BL, Gorno-Tempini ML. Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann Neurol. 2008;64:388–401. doi: 10.1002/ana.21451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99:6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, Ayutyanont N, Keppler J, Reeder SA, Langbaum JB, Alexander GE, Klunk WE, Mathis CA, Price JC, Aizenstein HJ, DeKosky ST, Caselli RJ. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106:6820–6825. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Rocher AB, Chapon F, Blaizot X, Baron JC, Chavoix C. Resting-state brain glucose utilization as measured by PET is directly related to regional synaptophysin levels: a study in baboons. Neuroimage. 2003;20:1894–1898. doi: 10.1016/j.neuroimage.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Rosen RF, Ciliax BJ, Wingo TS, Gearing M, Dooyema J, Lah JJ, Ghiso JA, Levine H, 3rd, Walker LC. Deficient high-affinity binding of Pittsburgh compound B in a case of Alzheimer’s disease. Acta Neuropathol. 2009 doi: 10.1007/s00401-009-0583-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, Cowie TF, Dickinson KL, Maruff P, Darby D, Smith C, Woodward M, Merory J, Tochon-Danguy H, O’Keefe G, Klunk WE, Mathis CA, Price JC, Masters CL, Villemagne VL. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- Samuels SC, Silverman JM, Marin DB, Peskind ER, Younki SG, Greenberg DA, Schnur E, Santoro J, Davis KL. CSF beta-amyloid, cognition, and APOE genotype in Alzheimer’s disease. Neurology. 1999;52:547–551. doi: 10.1212/wnl.52.3.547. [DOI] [PubMed] [Google Scholar]
- Savva GM, Wharton SB, Ince PG, Forster G, Matthews FE, Brayne C. Age, neuropathology, and dementia. N Engl J Med. 2009;360:2302–2309. doi: 10.1056/NEJMoa0806142. [DOI] [PubMed] [Google Scholar]
- Schmahmann JD, Doyon J, McDonald D, Holmes C, Lavoie K, Hurwitz AS, Kabani N, Toga A, Evans A, Petrides M. Three-dimensional MRI atlas of the human cerebellum in proportional stereotaxic space. Neuroimage. 1999;10:233–260. doi: 10.1006/nimg.1999.0459. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Laviolette PS, O’Keefe K, O’Brien J, Rentz DM, Pihlajamaki M, Marshall G, Hyman BT, Selkoe DJ, Hedden T, Buckner RL, Becker JA, Johnson KA. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–188. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern EA, Bacskai BJ, Hickey GA, Attenello FJ, Lombardo JA, Hyman BT. Cortical synaptic integration in vivo is disrupted by amyloid-beta plaques. J Neurosci. 2004;24:4535–4540. doi: 10.1523/JNEUROSCI.0462-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology. 2004;62:1984–1989. doi: 10.1212/01.wnl.0000129697.01779.0a. [DOI] [PubMed] [Google Scholar]
- Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, Mazoyer B, Joliot M. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage. 2002;15:273–289. doi: 10.1006/nimg.2001.0978. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Wang J, Dickson DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer’s disease from normal and pathologic aging. Exp Neurol. 1999;158:328–337. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]