

Summary
The fibroblast growth factor receptor tyrosine kinases (FGFR1, 2, 3, and 4) represent promising therapeutic targets in a number of cancers. We have developed the first potent and selective irreversible inhibitor of FGFR1, 2, 3, and 4 which we named FIIN-1 that forms a covalent bond with cysteine 486 located in the P-loop of the FGFR1 ATP-binding site. We demonstrate that the inhibitor potently inhibits Tel-FGFR1 transformed Ba/F3 cells (EC50 = 14 nM) as well as numerous FGFR-dependent cancer cell lines. A biotin-derivatized version of the inhibitor, FIIN-1-biotin, was shown to covalently label FGFR1 at Cys486. FIIN-1 is a useful probe of FGFR-dependent cellular phenomena and may provide a starting point of the development of therapeutically relevant irreversible inhibitors of wild-type and drug-resistant forms of FGFR kinases.
Introduction
In recent years, targeted therapy has attracted much attention in the field of cancer therapeutics due to the high profile success of inhibitors that target kinases that are aberrantly activated. One validated approach involves targeting protein kinases, particularly receptor tyrosine kinases, which reside at the apex of key signal transduction pathways. There are 518 protein kinase genes encoded in the human genome, many of which have been observed to become constitutively activated by amplification or mutation. Constitutive kinase activation can lead to an oncogene-addicted state that renders cancer cells, but not noncancerous cells, exquisitely sensitive to the inhibitors targeting the activated kinase. This observation has stimulated the development of numerous small molecule kinase inhibitors targeting kinases such as Bcr-Abl, mutant EGFR, V716F Jak-2, FLT3-ITD, c-Kit and PDGFR (Cohen et al., 2002; Ranson, 2002; Savage and Antman, 2002). To date, a dozen small molecule kinase inhibitors have been approved for clinical use and approximately 150 inhibitors are in various stages of clinical development.
Small molecule kinase inhibitors can bind to kinases in a reversible or an irreversible fashion. Reversible kinase inhibitors have been extensively investigated and typically bind to the ATP site with the kinase in an active (type 1) or an inactive (type 2) conformation (Liu and Gray, 2006). Irreversible inhibitors usually possess electrophilic functional groups such as α,β-unsaturated carbonyls and chloro/fluoromethyl carbonyls that react with the nucleophilic sulfhydryl of an active-site cysteine (Zhang et al., 2009). High selectivity of irreversible inhibitors can be achieved by exploiting both the inherent non-covalent selectivity of a given scaffold and the location of a particular cysteine residue within the ATP-site. For example, the most well-characterized, selective irreversible inhibitors of epidermal growth factor receptor (EGFR) such as PD168393 (Fry et al., 1998) were created by appending an acrylamide group to 6-position of 4-anilinoquinazoline scaffold, a pharmacophore known to be EGFR selective, that undergoes Michael reaction with a rare cysteine (Cys773) in the ATP binding site. However potential crossreactivity with other kinases that contain a cysteine at the equivalent position must be considered as recently demonstrated by the cross-reactivity of covalent EGFR inhibitors with Tec-family kinases such as Bmx (Hur et al., 2008). Irreversible inhibitors have been shown to overcome drug-resistance caused by mutation of the ‘gatekeeper’ amino acid, as has been observed for HKI-272, an irreversible EGFR inhibitor, against the T790M EGFR mutant (Carter et al., 2005; Kwak et al.).
The fibroblast growth factor receptor (FGFR) family of receptor tyrosine kinases consists of four family members, FGFR1-4, which bind to 22 different FGF ligands (Koziczak et al., 2004). FGF ligands mediate their pleiotropic actions by binding to FGFRs that have intrinsic intracellular protein tyrosine kinase domain. Upon dimerization, FGFRs can activate an array of downstream signaling pathways, such as MAPK and PKB/Akt pathway. FGF signaling appears to play critical roles not only in normal development and wound healing but also in tumor formation and progression (Powers et al., 2000). Germline activating muations in FGFRs have been found to be associated with the congenital skeletal disorders such as Pfeiffer syndrom, Apert Syndrome, Beare-Stevenson Syndrome, hydrochondroplasia, achondroplasia, and SADDAN Syndrome (Jang et al., 2001; van Rhijn et al., 2001). Somatic mutations of FGFRs that likely result in receptor gain-of-function are present in a variety of cancers such as bladder cancer, gastric cancer, colorectal cancer, endometrial carcinomas, cervical carcinoma, lung squamous cell carcinoma, and hematopoietic diseases (Dutt et al., 2008; Pollock et al., 2007). Interestingly some of the somatic mutations identified in cancers are identical to known germline mutations. These findings have been extended by recent systematic sequencing of cancer genomes that has revealed that the FGF signaling pathway displayed the highest enrichment for kinases carrying non-synonymous mutations among 537 non-redundant pathways that were examined (Greenman et al., 2007). Besides somatic mutations of FGFRs, amplification and overexpression of FGFRs are also present in certain types of human cancers such as breast cancers and prostate cancers and are believed to be involved in tumorigenesis and cancer progression (Devilard et al., 2006; Feng et al., 1997). Recently, two genome-wide association studies identified single nucleotide polymorphisms (SNPs) in FGFR2 as breast cancer susceptibility loci (Hunter et al., 2007), and these SNPs were identified as being linked to upregulated expression of FGFR2 (Meyer et al., 2008). Therefore, FGFR signaling appears to be a plausible target for both genetic diseases and cancers.
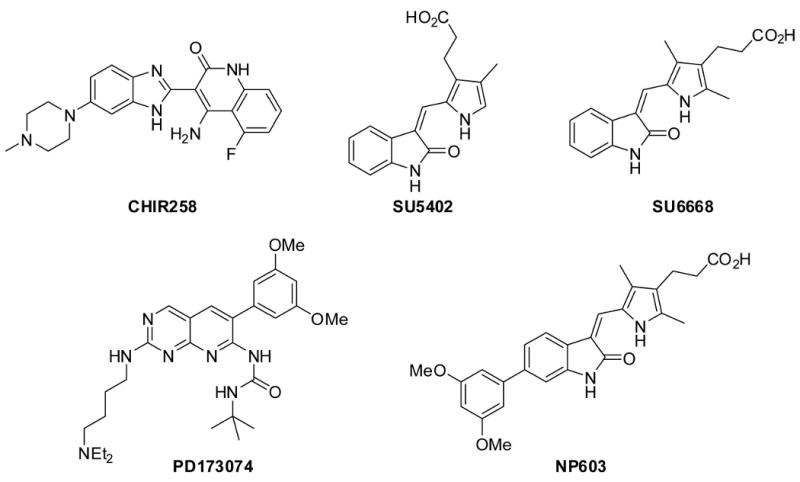
Over the last decade, efforts to discover small molecule FGFR inhibitors have resulted in the discovery of several selective and potent inhibitors that reversibly bind to the FGFR ATP-binding site. For example, the oxindole (SU5402) and the benzimidazole (CHIR258) were reported to be inhibitors of FGFR, VEGFR, and PDGFR (Figure 1) (Mohammadi et al., 1997; Trudel et al., 2005). The inhibitor NP603 was designed as a hybrid of FGFR inhibitors SU6668 and PD173074 and inhibits FGFR1 with IC50 of 0.4 μM (Kammasud et al., 2007). CHIR258 is currently in phase I clinical trials for treatment of AML, multiple myeloma, and malignant melanoma. However, the in vivo efficacy of these reversible FGFR inhibitors is limited by their rapid blood clearance, and therefore there is a compelling need for irreversible FGFR inhibitors with suitable pharmacokinetic properties. To date, no irreversible inhibitors of FGFR kinases have been reported. Here we describe the synthesis and characterization of an irreversible inhibitor of FGFRs which forms a covalent bond with a conserved cysteine (Cys486 of FGFR1) located at the rim of the P-loop.
Figure 1. Small molecule inhibitors of FGFRs.
Results
Design of an irreversible FGFR inhibitor, FIIN-1
We initiated our design efforts using the pyrimidopyridine PD173074 (Figure 1) as a lead structure, because binding assays with 317 kinases (Table S1) demonstrated a selectivity of PD173074 for FGFRs and its co-crystal structure with FGFR1 is available (Mohammadi et al., 1998). Analysis of the crystal structure and comparison to related compounds that inhibit PDGFR, Src, and Abl suggest that the 3,5-dimethoxyphenyl group of PD173074 is essential for FGFR-kinase selectivity. We determined that Cys486 in the P-loop of FGFR1, which was mutated to an alanine in the FGFR1 crystal structure (PDB ID: 2fgi), is located approximately 10 Å away from the pyridine nitrogen of PD173074 (Figure 2B). We decided to attach a phenyl group bearing a meta-acrylamide to the 1-nitrogen of the pyrimido[4,5]pyrimidine, another well-characterized ATP-competitive tyrosine kinase inhibitor template (Figure 2A) (Su et al., 1986). The resulting compound 1 was demonstrated to bind to FGFR and exhibited a good selectivity when tested against a panel of 402 kinases. Unfortunately, the compound was only able to inhibit cellular FGFR1 kinase activity as measured by a Tel-FGFR1 transformed Ba/F3 cell proliferation assay, with a 50% inhibitory concentration (EC50) of 1.5 μM. This potency was insufficient for this compound to be used as a cellular probe of FGFR kinase function. The approximately 300-fold loss of cellular activity of compound 1 relative to PD173074 suggests that replacement of the t-butylurea functionality with the phenylamide moiety of 1 is deleterious to the activity and that a covalent bond with Cys486 was most likely not formed.
Figure 2. Design of irreversible FGFR inhibitor FIIN-1.
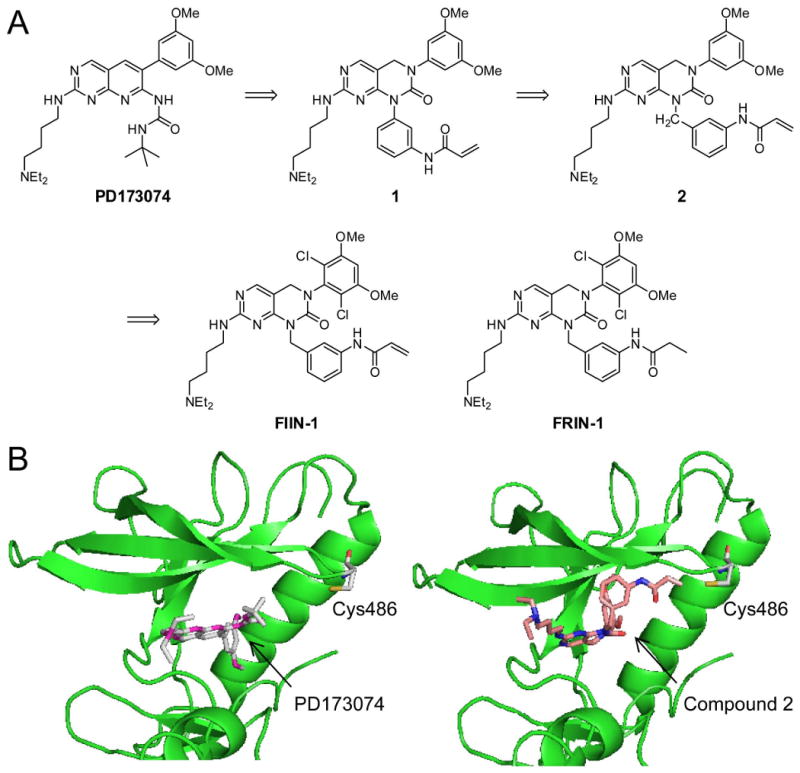
(A) Chemical structure of PD173074 and design of its irreversible analogue FIIN-1. (B) Crystal structure (PDB ID: 2fgi) showing the location of the unique cysteine (Cys486) in FGFR1 relative to the binding site of the PD173074 inhibitor (left). A model of binding mode of the compound 2 within FGFR1 active site (right) demonstrates a proper distance for covalent bond formation between the electrophilic center of the compound and Cys486.
We next introduced a one-carbon spacer to make the corresponding benzylamino analogue 2, which resulted in a compound that possessed an EC50 of 400 nM against cellular Tel-FGFR1 kinase activity. A modeling study indicated that the β-carbon of the acrylamide is positioned 2.9 Å away from Cys486, an ideal distance for covalent bond formation (Figure 2B). We noticed that both 2,6-dichlorophenyl group and 3,5-dimethoxyphenyl groups were utilized as substituents to occupy the hydrophobic region in the ATP binding site (Hamby et al., 1997). We decided to combine the 2,6-dichloro and the 3,5-dimethoxy functionalities in an effort to obtain greater potency and selectivity for FGFR kinases. The resulting compound that we named FIIN-1, FGFR irreversible inhibitor-1 (Figure 2A), blocked proliferation and survival of Ba/F3 cells transformed with Tel-FGFR1 and FGFR3 with EC50's of 14 nM and 10 nM, respectively. To investigate the functional importance of the acrylamide substituent, we synthesized FRIN-1 (FGFR reversible inhibitor-1, Figure 2A) where the acylamide is replaced with a propyl amide which is incapable of forming a covalent bond with Cys486. FRIN-1 is 24-fold less potent against Tel-FGFR1 (EC50 = 340 nM) and 100-fold less potent against Tel-FGFR3 (EC50 = 1040 nM) transformed Ba/F3 cells demonstrating the functional importance of the acrylamide functionality. Assay of recombinant FGFR1 measured in Z′-lyte format (Rodems et al., 2002) demonstrated that FIIN-1 is approximately 2.3 times more potent than FRIN-1 in vitro, supporting that the additional activity is attributed to the irreversible modification (Figure S1).
FIIN-1 is a potent, selective FGFR inhibitor
We next sought to investigate the selectivity of FIIN-1 and FRIN-1 for FGFR family kinases on a kinome-wide level. Both FIIN-1 and FRIN-1 were profiled against a panel of 402 different kinases binding assays using the Ambit KinomeScan technology at a concentration of 10 μM (Karaman et al., 2008). The score indicates the percentage of kinases that retained binding to solid matrix after competition with the inhibitor. Therefore, a lower score for a kinase implies tighter binding of the inhibitor to the kinase. FIIN-1 bound to several kinases including FGFR1-4, Flt1, Flt4, and VEGFR (Table 1 and S1). Kinases that were displaced to greater than 90% of the DMSO control were considered “strong” hits and were further examined in a dose-response format to determine dissociation constants (Kd's). The potent association of FIIN-1 to FGFRs was confirmed with Kd's of 2.8, 6.9, 5.4, 120 nM for FGFR1, 2, 3, and 4, respectively. Only two other kinases associated with FIIN-1 with Kd's below 100 nM were Blk (Kd = 65 nM) and Flt1 (Kd = 32 nM). The biochemical IC50 values of FIIN-1 using the Z′-lyte assays were determined to be 9.2, 6.2, 11.9, and 189 nM against FGFR1, 2, 3, and 4, respectively, and are in an excellent agreement with the measured Kd values. The IC50's for Blk and Flt1 that bound less tightly with FIIN-1 were 381 nM and 661 nM respectively, indicating a moderate inhibition. This good correlation between Kd and IC50 values underscores that the observed binding with kinases translates into the inhibition of kinases. Together, the results from binding and activity assays suggest that FIIN-1 is a selective FGFR inhibitor at a biochemical level and also demonstrate its selectivity over other kinases such as c-Src, TNK1, and YES that have a P-loop cysteine at the same position as the FGFRs (Zhang et al., 2009).
Table 1. Profiling of FGFR inhibitors for binding with a panel of 402 kinases.
Kinases that were displaced by inhibitors to greater than 90% of the DMSO control (Ambit score < 10) from KinomeScan™ platform binding assays for 402 different kinases are listed. Dissociation constants (Kd's) were also measured for selected kinases. See Table S1 for a complete list.
| FIIN-1 (10 μM) | FRIN-1 (10 μM) | PD173074 (1 μM) | ||||
|---|---|---|---|---|---|---|
| Kinases | Score | Kd (nM) | Score | Kd (nM) | Score | Kd (nM) |
| BLK | 0.5 | 65 | 15 | 2300 | ||
| CAMK1D | 3.4 | 15 | ||||
| CSF1R | 0.2 | 0.25 | ||||
| DDR1 | 0.1 | 0.2 | ||||
| EPHB6 | 1.4 | 0.9 | ||||
| ERK5 | 0.05 | 160 | 0 | 130 | ||
| FGFR1 | 0 | 2.8 | 0 | 3.1 | 0.4 | |
| FGFR2 | 2.1 | 6.9 | 2.4 | 5.6 | 2.9 | |
| FGFR3 | 1.8 | 5.4 | 1.4 | 5.4 | 0.05 | |
| FGFR4 | 0.1 | 120 | 0.25 | 280 | 17 | |
| FLT1 | 0.3 | 32 | 0.55 | 49 | 8 | |
| FLT4 | 0.2 | 120 | 2 | 340 | 6 | |
| FRK | 6 | 9.2 | ||||
| HPK1 | 5.8 | 10 | ||||
| JAK3 | 6.4 | No binding | ||||
| KIT | 1.2 | 420 | 0.55 | 250 | ||
| LCK | 0.7 | 0.6 | ||||
| MAP3K2 | 1.6 | 1.8 | ||||
| MAP3K3 | 0.5 | 0.65 | ||||
| MAP4K3 | 0.85 | 11 | ||||
| MAP4K5 | 0.2 | 2.7 | ||||
| MET | 5.5 | 1000 | 11 | 1400 | ||
| MST1 | 6.6 | 33 | ||||
| MST3 | 4.6 | 4.6 | 7 | |||
| MST4 | 2.6 | 1.6 | ||||
| PDGFRB | 2.4 | 480 | 2.6 | 480 | 38 | |
| TAO1 | 0 | 0 | ||||
| TAOK1 | 0.2 | 0.2 | ||||
| TAOK3 | 0.15 | 0.1 | ||||
| TIE1 | 5.2 | 6.4 | ||||
| TIE2 | 5.6 | 6.8 | ||||
| VEGFR2 | 1.9 | 210 | 5 | 470 | 21 | |
Interestingly, unlike in vitro kinase assay and cellular assay where the irreversible inhibitor FIIN-1 was considerably more potent than the reversible inhibitor FRIN-1, both compounds exhibited similar scores at the 10 μM screening concentration and similar Kd values in the kinase binding assays. It is unlikely that the acrylamide group of FIIN-1 was inactivated by dithiothreitol (DTT, 6 mM) contained in the binding assay buffer, because the potency (IC50) of FIIN-1 and FRIN-1 for FGFR1 was unaffected by 6 mM DTT in vitro (Figure S1). This suggests that the majority of binding energy of FIIN-1 comes from non-covalent binding interactions.
To further examine the selectivity of FIIN-1, we profiled the compound using a panel of various tyrosine kinase-transformed Ba/F3 cells (Melnick et al., 2006). FIIN-1 was not cytotoxic toward wild-type Ba/F3 cells (EC50 > 10 μM) and was barely active against Bcr-Abl (EC50 > 10 μM), NPM-Alk (EC50 > 10 μM), Tpr-Met (EC50 > 10 μM), Tel-Arg (EC50 > 10 μM), Tel-Blk (EC50 = 2 μM), Tel-Bmx (EC50 = 2 μM), Tel-Jak2 (EC50 = 5 μM), Tel-Jak3 (EC50 > 10 μM). The observed low activity against Tel-Blk, Bmx, and Jak3 which contain a reactive cysteine in the ribose binding region further confirms selectivity of FIIN-1.
A biotin-labeled FIIN-1
In order to demonstrate covalent binding of FIIN-1 to FGFR kinases, we synthesized a biotinylated version of FIIN-1 (FIIN-1-biotin, Figure 3A) where a biotin is tethered via a flexible polyethylene glycol linker at the other end of the electrophilic acrylamide group. This design was implemented because molecular modeling suggests that this linker would be directed out of the ATP-binding cleft and toward solvent. As has been demonstrated by the irreversible EGFR inhibitor HKI-272, a dimethylaminomethyl moiety can be appended to the terminal olefin without disrupting the ability of the compound to function as a Michael acceptor (Tsou et al., 2005). The dimethylaminomethyl functionality has also been suggested to enhance reactivity towards the nucleophilic thiol by acting as a general base catalyst. As a control for binding reversibility, we also synthesized a biotinylated FRIN-1 (FRIN-1-biotin, Figure 3A) which lacks the electrophilic center. The cellular EC50 of FIIN-1-biotin against Tel-FGFR1,3 transformed Ba/F3 cells was determined to be 35 nM and 54 nM respectively, which is comparable to the parent compound FIIN-1 and demonstrates that the biotin modification did not significantly affect the potency or cell permeability. To determine whether FIIN-1-biotin could covalently label FGFR1 in cells, HEK293 cells were transiently transfected with a full-length FGFR1 expression vector and were treated with each biotin probe (50 μM) for 2 hours, after which cell lysates were subjected to immunoprecipitation with an anti-FGFR1 antibody. Probing the blot with streptavidin-HRP revealed that FGFR1 was labeled by the FIIN-1-biotin, not by FRIN-1-biotin (Figure 3B).
Figure 3. FIIN-1 irreversibly inhibits FGFR1 and its downstream signaling.
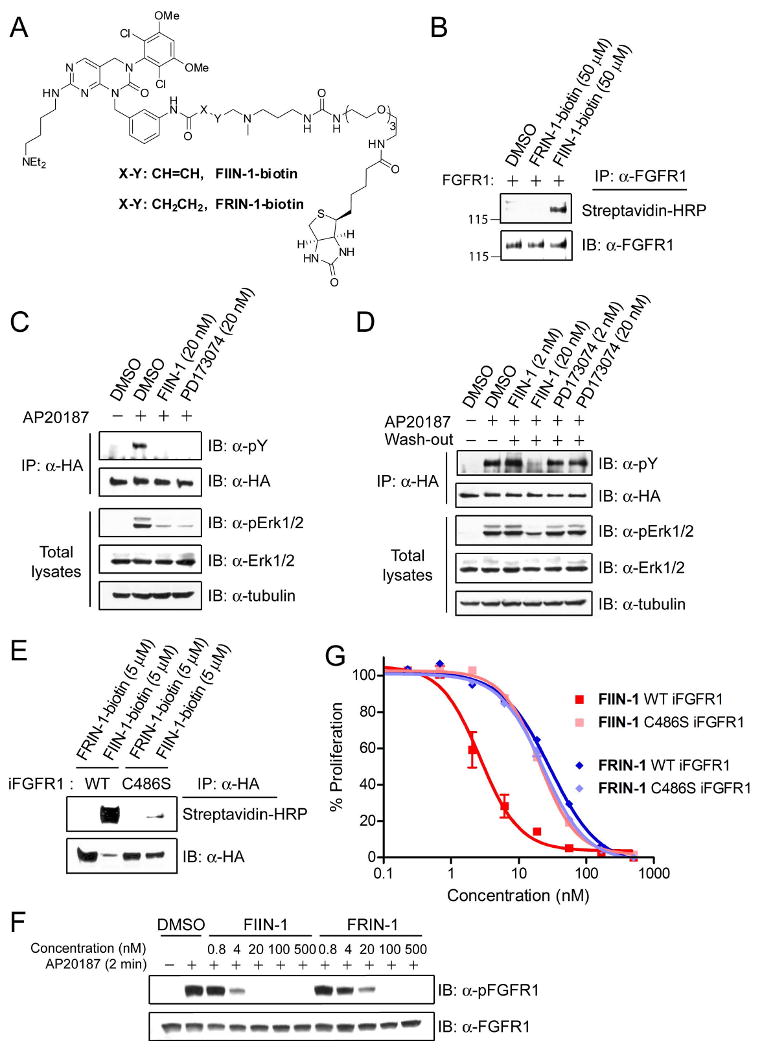
(A) Structures of biotin-tethered inhibitors (FIIN-1-biotin, FRIN-1-biotin). (B) FIIN-1-biotin labeled full-length FGFR1 ectopically expressed in HEK293 cells. (C) MCF10A cells that stably express iFGFR1 were serum-starved for 1 day and were stimulated by AP20187 for 30 min in the presence or absence of 20 nM of the inhibitors. Western blot analysis using immunoprecipated iFGFR1 and total cell lysates showed that both PD173074 (20 nM) and FIIN-1 (20 nM) inhibited iFGFR1 autophosphorylation and its downstream Erk1/2 almost completely. (D) Serum-starved iFGFR1 MCF10A cells were treated with inhibitors for 30 min, extensively washed, and incubated in a serum-free condition for 6 hours prior to iFGFR1 activation. Washout did not affect FIIN-1 (20 nM)'s ability to inhibit FGFR1, but eliminated the inhibitory activity of PD173074 (20 nM). (E) The lysates from MCF10A cells that stably express either WT or C486S iFGFR1 were mixed with the biotin probes (5 μM). Streptavidin-HRP blot of immunoprecipitated iFGFR1 revealed that FIIN-1-biotin labeled WT iFGFR1, but barely labeled C486S iFGFR1. (F) WT or C486S iFGFR1 MCF10A cells were pre-treated with FIIN-1 or FRIN-1 for 1 day in a serum-free condition, and were stimulated with AP20187 for 2 min. FIIN-1 inhibited autophosphorylation of WT iFGFR1 with about 5 times higher potency compared to FRIN-1. (G) Serum-starved MCF10A cells were treated with various doses of inhibitors along with AP20187 for 2 days. Viability of cells was assessed by measuring cellular ATP level. FIIN-1 blocked iFGFR1-dependent proliferation of MCF10A cells with 10-fold higher potency than FRIN-1. Moreover, FIIN-1 and FRIN-1 blocked the proliferation of C486S iFGFR1 MCF10A cells with a similar potency.
The biotinylated covalent inhibitor was used to study the kinetics of covalent modification of FGFR1. Recombinant FGFR1 kinase domain was pre-incubated with FIIN-1-biotin (250 nM) for various times (0 – 60 min), and both the in vitro activity and labeling of FGFR1 were measured after the kinase reaction was allowed to proceed for 1 hour (Figure S2A). As expected for a covalent inhibitor, increasing the pre-incubation time with FIIN-1-biotin resulted in increased labeling of FGFR1 and a correlated loss of FGFR1 enzymatic activity consistent with covalent modification being responsible for kinase inactivation. Then we compared the kinetics of FIIN-1 with FRIN-1 (100 nM) using a similar kinetic experiment with FGFR1 (Figure S2B). The result indicates a more rapid inhibition by FIIN-1 than FRIN-1, suggesting that irreversible modification assists a rapid kinase inhibition. In addition, an in vitro time-course study for a longer period (0 - 24 hours) demonstrated that FGFR1 is gradually modified by FIIN-1-biotin reaching a plateau at after approximately 24 hours (Figure S2C).
FIIN-1 irreversibly blocks both activation of FGFR and its downstream signals
To confirm that FIIN-1 is capable of inhibiting FGFR1 signaling, we employed an inducible FGFR1 (iFGFR1) system where oligomerization and activation of FGFR1 are induced upon the treatment of a small molecule AP20187 (Welm et al., 2002). The iFGFR1 construct contains an N-terminal myristylation sequence, an FGFR1 kinase domain, two tandem domains of F36V mutant FKBP12 (FKBPv), and a C-terminal hemagglutinin (HA)-epitope. A stable MCF10A mammary epithelial cell line expressing iFGFR1 construct was made through retroviral transfection and selection with puromycin. The bivalent compound AP20187 exhibits high affinity for the FKBPv domain and induces dimerization and autophosphorylation of the fused FGFR1 kinase domains, which in turn triggers activation of downstream signaling pathways. Serum-starved MCF10A cells that stably express iFGFR1 were treated with 20 nM of either FIIN-1 or PD173074 as a control for 30 min in the presence or absence of AP20187 (100 nM). The cell lysates were immunoprecipiated with anti-HA antibody and the level of iFGFR1 autophosphorylation was examined using anti-phosphotyrosine antibody. Both compounds at a concentration of 20 nM almost completely blocked activation of iFGFR1 and phosphorylation of downstream effectors Erk1/2 (Figure 3C).
We next examined whether FIIN-1 inhibits iFGFR1 activation in an irreversible manner. Serum-starved MCF10A cells were treated with FIIN-1 or PD173074 (2, 20 nM) for 30 min, extensively washed with PBS, and were maintained in serum-free condition for 6 hours prior to activation of iFGFR1 by AP20187. Indeed, FIIN-1 at 20 nM sustained the inhibition of phosphorylation of both iFGFR1 and Erk1/2 even after washout of the drug, while the inhibitory activity of the reversible inhibitor PD173074 at 20 nM was almost completely eliminated by the washout procedure (Figure 3D). This result further supports that FIIN-1 inhibits FGFR1 in an irreversible manner. Likewise, a similar wash-out experiment with FRIN-1 demonstrated that the FGFR1 and downstream Erk1/2 inhibition occurred in a reversible fashion (Figure S3).
In order to examine if Cys486 of FGFR1 is labeled by FIIN-1 in cells, we generated a C486S mutant iFGFR1 construct and produced a stable MCF10A cell line expressing C486S iFGFR1 using retroviral transduction. MCF10A cells that stably express either WT or C486S iFGFR1 were lysed, incubated with the biotin probes (5 μM each) for 1 hour, and immunoprecipitated with an anti-HA antibody. The blot with streptavidin-HRP showed that FIIN-1-biotin strongly labeled WT iFGFR1, but barely labeled C486S iFGFR1 (Figure 3E). This result confirms that Cys486 is the primary labeling site of FIIN-1.
We next investigated the functional implication of covalent modification of Cys486 on FGFR1 inhibition. Serum-starved iFGFR1 MCF10A cells were treated with increasing doses of the inhibitors before stimulus with AP20187 and examination of FGFR1 autophosphorylation (Y653/Y654) status. Intriguingly, when the inhibitors were pre-treated for 10 or 30 min, FIIN-1 and FRIN-1 inhibited autophosphorylation with almost equivalent potency (data not shown). However, when inhibitors were pre-treated for a long term (24 hours), which the in vitro studies suggest is sufficient time to lead to almost complete labeling (Figure S2B), FIIN-1 exhibited an approximately 5-fold greater potency compared to FRIN-1 (Figure 3F). We then compared activity of the inhibitors for iFGFR1-dependent cellular proliferation. Wild-type and C486S iFGFR1 MCF10A cells were serum-starved and treated with AP20187 and FIIN-1/FRIN-1 for 2 days in a serum-free condition, and cell proliferation was measured (Figure 3G). Indeed, FIIN-1 inhibited the proliferation of WT cells (EC50 = 2.7 nM) 10 times more potently than FRIN-1 (EC50 = 29 nM). As expected the C486S iFGFR1 cells which are resistant to covalent labeling, were inhibited by FIIN-1 (EC50 = 20 nM) and FRIN-1 (EC50 = 23 nM) with equivalent potency. These biochemical and cellular experiments demonstrate that the ability to covalently modify Cys486 of FGFR1 contributes to the improved potency of FIIN-1 relative to FRIN-1 despite both compounds being potent FGFR1 inhibitors without covalent bond formation.
FIIN-1 abolishes iFGFR1-mediated mammary epithelial cell transformation in 3D culture
MCF10A cells grown in three dimensions (3D) recapitulate several features of mammary epithelium in vivo (Schmeichel and Bissell, 2003; Shaw et al., 2004), and have been used as a unique system to study the signaling pathways involved in the early progression of breast cancer (Debnath et al., 2003b; Muthuswamy et al., 2001; Radisky et al., 2001). Wild-type MCF10A cells form spherical structures that consist of an outer layer of polarized, growth-arrested epithelial cells surrounding a hollow lumen. Activation of iFGFR1 in MCF10A cells resulted in cellular transformation and formation of the large and disorganized structures in Matrigel, similar to that observed previously in the iFGFR1-activated mouse mammary HC11 cell line (Xian et al., 2007). To investigate the effects of FIIN-1 on FGFR1-mediated transformation of mammary epithelial cells in a more physiologically relevant context, we first examined the morphology of iFGFR1-activated cells in 3D culture in the absence or presence of FIIN-1 and PD173074 (Figure 4A). The abnormal morphogenesis induced by iFGFR1 activation was prevented by the treatment of either FIIN-1 (20 nM) or PD173074 (1 μM). Moreover, iFGFR1 activation-induced Akt phosphorylation and luminal cell survival were also abolished by either FIIN-1 or PD173074, as assessed by immunostaining with antibodies against phospho-Akt (Figure 4B) and activated caspase 3 (Figure 4C). Importantly, neither FIIN-1 nor PD173074 affected the growth of wild-type MCF10A cells, suggesting that FGF signaling is dispensable to normal mammary epithelial cells and inhibition of FGF pathway in cancer might be effective and selective to inhibit cancer cells growth with low toxicity to non-cancerous cells.
Figure 4. FIIN-1 abrogates the effects of iFGFR1 activation in iFGFR1-transformed MCF10A cells in 3D culture.
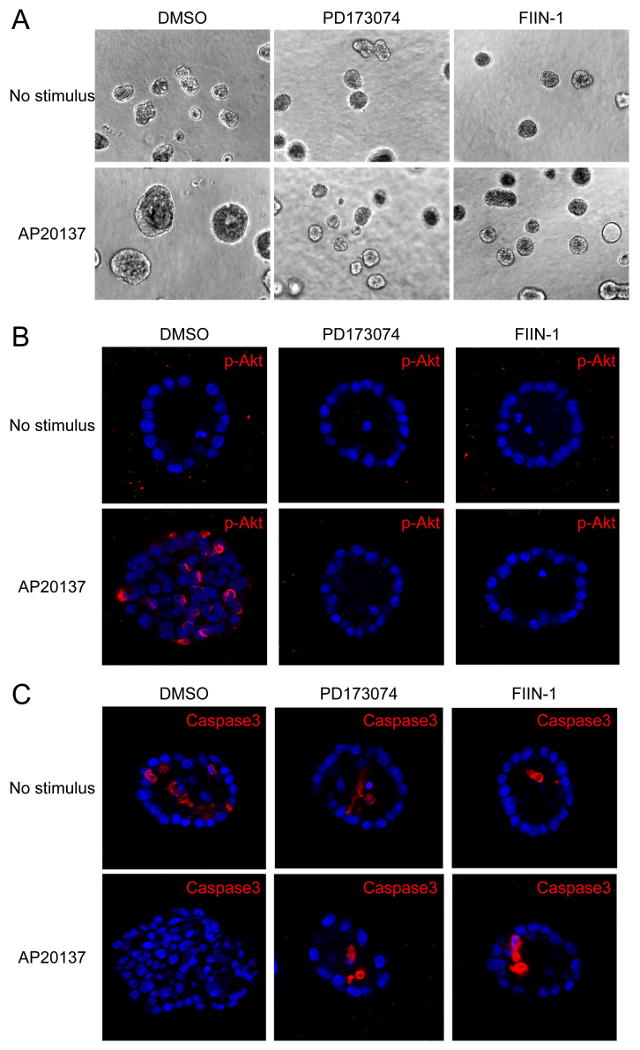
(A) PD173074 (1 μM) and FIIN-1 (20 nM) inhibited abnormal morphogenesis induced by iFGFR1 activation. (B) Both inhibitors abolished iFGFR1-mediated Akt phosphorylation (Ser473) and (C) iFGFR1-mediated cell growth and luminal cell survival in 3D culture. Neither drug was toxic to the out layer of cells in 3D culture.
FIIN-1 inhibits proliferation of FGF signaling-sensitive cancer cell lines
We screened 687 human cancer cell lines and identified the cancer cell lines that were uniquely sensitive to PD173074 (Table S2) and were therefore presumed to be dependent on FGFR signaling for their survival (McDermott et al., 2007). We re-tested a subset of the sensitive and resistant cell lines with a range of concentrations of PD173074 and FIIN-1, by measuring cell viability after 72 hours. As expected, FIIN-1 was capable of inhibiting the viability of cell lines that had been shown to be sensitive to PD173074 (Table 2). In general, FIIN-1 inhibited proliferation of these cell lines at lower concentrations than PD173074. These results suggest that FIIN-1 will be a valuable probe to identify cancer cell lines that are sensitive to the inhibition of FGFR kinase activity.
Table 2. Antiproliferative activity of FIIN-1 against various cancer cell lines.
Cell viability was measured using Syto-60 staining method following treatment of inhibitors for 72 hours. Cells are ranked according to their EC50 for PD173074. Copy numbers for FGFR1, 2, and 3 genes in each cell line are also shown. Copy number changes were derived from a 500K SNP array (diploid = 2). NA: not available
| Cell line | Organ | 500K SNP array copy number | EC50 (μM) | |||
|---|---|---|---|---|---|---|
| FGFR1 | FGFR2 | FGFR3 | PD173074 | FIIN-1 | ||
| KATO III | Stomach | 2.18 | 14.95 | 1.76 | 0.022 | 0.014 |
| RT4 | Bladder | 2.1 | 1.71 | 1.73 | 0.05 | 0.07 |
| SNU-16 | Stomach | 1.71 | 15.14 | 1.25 | 0.12 | 0.03 |
| STS-0421 | Muscle | NA | NA | NA | 0.12 | NA |
| G-401 | Kidney | 1.94 | 1.95 | 1.93 | 0.21 | 0.14 |
| SBC-3 | Lung | 2.44 | 1.81 | 1.88 | 0.32 | 0.08 |
| A2.1 | Pancreas | NA | NA | NA | 0.34 | 0.23 |
| A2780 | Ovary | 1.94 | 2 | 2.14 | 0.82 | 0.22 |
| FU97 | Stomach | 1.89 | 1.72 | 1.75 | 1.16 | 0.65 |
| G-402 | Kidney | 1.89 | 1.91 | 1.91 | 2.48 | 1.65 |
| R082-W-1 | Thyroid | 2.46 | 1.68 | 1.87 | 2.5 | >5 |
| LU99A | Lung | 2.07 | 2.08 | 1.62 | 3 | >5 |
| RD-ES | Bone | NA | NA | NA | >5 | 2.3 |
| PA-1 | Ovary | NA | NA | NA | >5 | 4.6 |
| JAR | Choriocarcinoma | NA | NA | NA | >5 | >5 |
| H520 | Lung | NA | NA | NA | >5 | 4.5 |
| VM-CUB1 | Bladder | 2.01 | 2.05 | 1.7 | >5 | >5 |
| NCI-H1703 | Lung | 4.19 | 1.73 | 1.8 | >5 | >5 |
| JHH-7 | Liver | 1.33 | 1.84 | 1.85 | >5 | >5 |
| TOV-112D | Ovary | NA | NA | NA | >5 | >5 |
We examined the effect of FIIN-1 on cell survival and downstream pro-survival signaling pathways in the KATO-III and SNU-16 gastric cancer cell lines (Figure 5A). These two cell lines harbor FGFR2 gene amplification and have previously been shown to be dependent of FGFR2 activation for their survival (Kunii et al., 2008; Takeda et al., 2007). FIIN-1 potently suppressed the pro-survival Akt and Erk1/2 pathways in both of these cell lines. FGFR2 gene amplification in these cancer cell lines resulted in an oncogene-addicted state that rendered cell proliferation hypersensitive to the FGFR inhibitors (Table 2). Interestingly, FIIN-1 also inhibited cell survival and survival pathways in other cell lines such as A2780 and SBC-3 cells that have not previously been shown to be dependent on the FGFR family for survival (Figure 5A). This may indicate a role for targets of FIIN-1 other than FGFRs. We performed a streptavidin pulldown experiment using the extracts of MCF10A cells that express iFGFR1, and identified a number of intracellular proteins bound to FIIN-1-biotin using mass spectrometry analysis (Figure S4). A subset of the identified proteins (Table S3) could be related to FGFR-independent pro-survival pathways, but more in-depth study on the drug-sensitivity of these particular cell lines is currently under investigation.
Figure 5. FIIN-1 inhibits FGFR2 and pro-survival signaling pathways, and inhibits FGFR2-dependent cell growth.
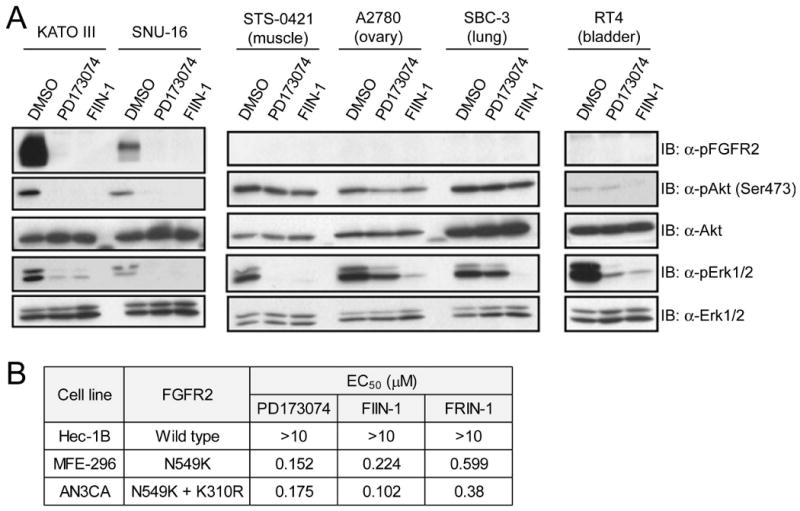
(A) FIIN-1 inhibited pro-survival signaling pathways in the FGFR2-amplified gastric cancer cell lines including KATO III and SNU-16 and in the other cancer cell lines that were previously shown to be independent of FGFR signaling for survival. Cells were treated for 6 hours with 200 nM of each compound. (B) Proliferation of the endometrial carcinoma cell lines (MFE-296, AN3CA) that express activating FGFR2 mutants were abrogated by FGFR inhibitors, while Hec-1B cells that express WT FGFR2 showed no sensitivity to the inhibitors.
Cancer-associated FGFR2 mutations have been identified in endometrial carcinoma by high-throughput DNA sequencing (Dutt et al., 2008). We investigated whether cell lines bearing these mutations were sensitive to growth inhibition by FIIN-1. Indeed, MFE-296 and AN3CA endometrial carcinoma cell lines harboring activating FGFR2 mutations (N549K, N549K/K310R) presented sensitivity (sub-micromolar EC50 values) to FIIN-1, FRIN-1, and PD173074 in cell proliferation assays, whereas Hec-1B cells that express wild type FGFR2 were not sensitive to all three FGFR inhibitors (Figure 5B). These results indicate that FIIN-1 is also an effective agents against cancer cells that are ‘oncogene-addicted’ based upon FGFR2 mutations.
FIIN-1 weakly inhibits FGFR1 gatekeeper mutant V561M
Many patients being treated with small molecule kinase inhibitors develop resistance to the therapy primarily as a result of mutations that prevent efficient kinase inhibition. For example, resistance to reversible EGFR inhibitors gefitinib and erlotinib in treatment of non-small cell lung cancers can be acquired as a result of mutations in EGFR kinase domain, particularly by the gatekeeper mutation T790M (Pao et al., 2005). The gatekeeper position also appears to be a hotspot for mutation in Bcr-Abl, c-Kit, and PDGFR (Branford et al., 2002). Mutation of the gatekeeper position can severely interfere with inhibitors binding in the ATP site of kinase while still maintaining normal or enhanced kinase function. It has been reported that irreversible EGFR inhibitors such at HKI-272 that target Cys773 can inhibit the T790M gatekeeper mutant (Carter et al., 2005; Kwak et al., 2005; Yun et al., 2008). We therefore investigated whether FIIN-1 inhibits the V561M gatekeeper mutant of FGFR1 which has been reported to induce resistance to PD173074 (Figure 6A,B) (Blencke et al., 2004). HEK293 cells were transiently transfected with full-length of either WT FGFR1 or V561M FGFR1, and autophosphorylation of both receptors were examined following 1 hour drug treatment. Interestingly, the V561M FGFR1 mutation results in a higher level of FGFR1 autophosphorylation relative to wild-type FGFR1. The activating nature of the gatekeeper mutation has recently been documented for several other kinases including c-Src, PDGFR, and c-Abl (Azam et al., 2008). Concentrations up to 10 μM of PD173074 were incapable of inhibiting autophosphorylation of FGFR1-V561M in agreement with published results. This resistance likely results from a steric clash between the dimethoxylphenyl ring of PD173074 and the gatekeeper position (Figure 6C,D). In contrast, FIIN-1 effectively blocked autophosphorylation of FGFR1-V561M at a concentration of 10 μM. This result demonstrates that covalent inhibition of FGFR1 potentially overcomes the V561M mutation which is expected to arise when reversible FGFR inhibitors enter clinical development. But the relatively low potency on V561M also argues that FIIN-1 needs to be further optimized to become a potent V561M FGFR1 inhibitor.
Figure 6. FIIN-1 moderately inhibits V561M mutant of FGFR1.
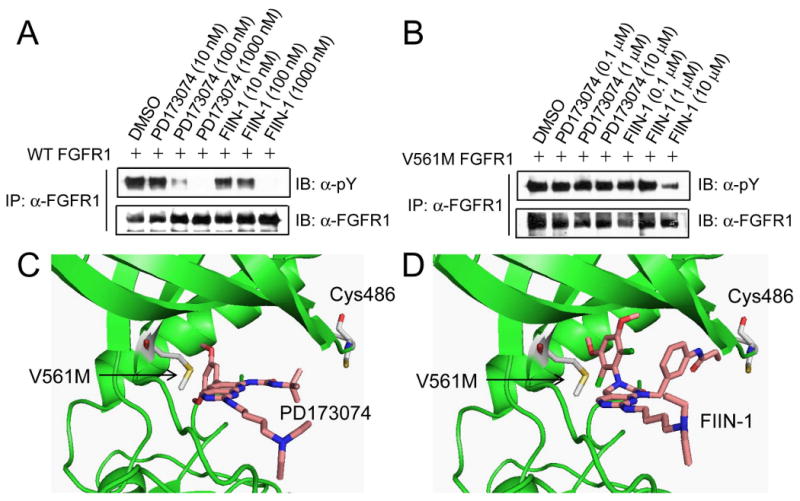
Dose-response inhibition of autophosphorylation of (A) WT and (B) V561M mutant of full-length FGFR1 by PD173074 and FIIN-1. FIIN-1 (10 μM) inhibited V561M FGFR1, whereas PD173074 (10 μM) that was nearly equipotent with FIIN-1 against WT FGFR1 was inactive to the mutant. (C) Binding mode of PD173074 within V561M FGFR1. (D) Molecular model of FIIN-1 bound to V561M FGFR1.
Discussion
Using a structure-guided approach, we have developed a highly potent and selective irreversible inhibitor of FGFRs, FIIN-1, that is capable of forming a covalent bond with a conserved cysteine located in the P-loop. FIIN-1 exhibited a high degree of selectivity when tested against a panel of 402 kinases with sub-500 nM dissociation constants only observed for FGFR1-4, Flt1, Flt4, and VEGFR kinase. FIIN-1 blocked autophosphorylation of FGFR1 and the phosphorylation of FGFR's downstream effectors Erk1/2 in MCF-10A cells that stably express iFGFR1, and repressed the proliferation of FGFR1,3-transformed Ba/F3 cells with an EC50 of 14 and 10 nM, respectively. Four lines of evidence were provided to support the claim that FIIN-1 forms a covalent bond with Cys486 of FGFR1. First, the corresponding reversible inhibitor FRIN-1 lost 24-fold and 100-fold activity against cellular Tel-FGFR1 and 3 kinase activity, respectively. Second, the biotin modified analog FIIN-1-biotin but not its reversible analog FRIN-1-biotin is capable of labeling FGFR1 from cell lysates. Third, FIIN-1 maintains inhibition of FGFR1 kinase autophosphorylation and downstream signaling following washout of the drug whereas the reversible inhibitor FRIN-1 does not. Fourth, the C486S mutant of iFGFR1 is not covalently labeled FIIN-1-biotin and cells bearing this mutation are more weakly inhibited by FIIN-1 relative to wild-type iFGFR1-transformed cells.
Recent findings have highlighted the role of activating mutations, amplifications, and over-expression of FGFRs in the pathogenesis of a variety of human tumors including multiple myeloma, breast, prostate, colon, bladder, and endometrial cancers (Dutt et al., 2008; Hunter et al., 2007; Jang et al., 2001; Pollock et al., 2007; Powers et al., 2000; van Rhijn et al., 2001). These findings have stimulated the development of reversible FGFR inhibitors as potential therapeutics as exemplified by CHIR258, XL228, and XL999 that are currently in clinical trials for several cancers. Although no kinase inhibitor-resistant mutations have been reported for FGFRs from clinical samples, analogy to other kinases suggests that mutations at the gatekeeper position can be anticipated. Our results suggest that irreversible FGFR inhibitors could overcome gatekeeper mutations such as V561M similar to what has been observed for irreversible inhibitors of the T790M EGFR gatekeeper mutant. However further optimization of FRIN-1 will be required to obtain an inhibitor with a useful level of potency against the V561M mutant.
The methods utilized to develop irreversible FGFR inhibitors are likely to be generally applicable to many kinases with cysteine located in the ATP binding site. The approach relies on (i) non-covalent recognition of an active site to obtain selectivity between all kinases that have a cysteine at a given position and (ii) covalent bond formation to get specificity relative to the rest of the kinome. By appending the electrophiles that target particular cysteines, a remarkable selectivity can be also achieved. A similar approach has been previously used to make selective inhibitors of EGFR (Fry et al., 1998), Btk (Pan et al., 2007), Rsk1, 2 (Cohen et al., 2005), and the T790 mutant of EGFR (Zhou et al., 2009). More than 200 different kinases which represent 35% of human kinome have at least one cysteine residue in the ATP–binding site (Zhang et al., 2009), suggesting the broad utility of this approach for developing selective irreversible kinase inhibitors.
Significance
Gain of function mutations in FGF receptors caused by chromosomal translocation, gene fusion, and gene amplification have been identified in a variety of human cancers including myeloid tumors and breast cancers. Several FGFR inhibitors have been developed as potential therapeutic agents and some are being evaluated in clinical trials for cancer treatment. So far, only reversible inhibitors of FGFRs have been developed. This article reports the first irreversible inhibitor of FGFRs, namely FIIN-1, with nanomolar IC50's. Based on the structural information of PD173074 bound to FGFR1, the irreversible analogue FIIN-1 was created by incorporating an acrylamide moiety that forms a covalent bond with the Cys486 positioned in the P-loop within the active site. FIIN-1 exhibited nanomolar inhibition of FGFRs, and surprisingly, showed moderate or nearly no affinity to c-Fgr, LIMK1, c-Src, TNK1, and YES that bear the identical cysteine in the P-loop. This remarkable selectivity toward FGFRs indicates that the selectivity and potency of FIIN-1 was determined primarily by noncovalent FGFR-drug interaction. The covalent linkage resulted in the irreversible blockage of FGFR1 activation and its downstream signals, which could potentially be advantageous in achieving efficacious in vivo action. But more notably, the ability of FIIN-1 to form a covalent bond resulted in a moderate potency against the gatekeeper mutant of FGFR1 that is resistant to the reversible inhibitor PD173074. This result suggests that irreversible FGFR inhibitors such as FIIN-1 should be considered promising leads to target FGFR mutants that may emerge as mutants resistant to the first-generation reversible inhibitors.
Experimental Procedures
In vitro FGFR1 activity assay
Inhibitors were assayed against recombinant kinase domain FGFR1 using the Z′-LYTE™ Enzymatic Kinase Assay format (Invitrogen Corporation). Assays were carried out using 8 ng of FGFR1, 2 μM peptide substrate, and 40 μM ATP for 1 hour reaction. The detailed procedure is described in the SelectScreen™ Customer Protocol located at http://www.invitrogen.com/kinaseprofiling.
Washout experiment
MCF10A cells expressing iFGFR1 were cultured in 100 mm dishes, and at about 90% confluence, cells were serum-starved for 1 day. Cells were then incubated with FGFR inhibitors for 30 min, washed with PBS three times, and were maintained in serum-free condition for 6 hours. After which, cells were treated with AP20187 (100 nM) in a serum-free media for 30 min, and harvested. The resulting lysates were analyzed for iFGFR1 autophosphorylation using immunoblot and immunoprecipitation.
Cell viability assays
Cell viability of Tel-FGFR1 or 3-transformed Ba/F3 cells was determined using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) (Promega) assay based on manufacturer's procedure. Cells (40,000 cells, 100 μL/well) were seeded in media in a 96-well plate and treated with 1 μL of each pre-diluted compound (3-fold, 12-point dilutions from 1 mM DMSO stock) for 48 hours. After addition of MTT reagents, absorbance was measured by using SpectraMax M5 (Molecular Devices). Each test was duplicated. Cell viability was calculated by Atreatment/ADMSO × 100% (A represents the absorbance recorded at 570 nm).
All other Ba/F3 cells including WT Ba/F3 cells were engineered to express luciferase, and therefore their viability was measured using luciferase reporter gene assay. Each Ba/F3 cell line (4,000 cells in 50 μL) were plated to white 384 well microtiter plates (Corning), and the preplated compounds were transferred (300 nL) to the assay plates using a 384-pin transfer device. Each data point was triplicated. After incubation for 48 hours in cell incubator, 25 μL of BrightGlo (Promega) was added and luminescence was quantified by using Envision multilabel plate reader (PerkinElmer).
Proliferation of iFGFR1 expressing MCF10A cells was assessed by measuring cellular ATP. Cells were serum-starved for 1 day and seeded to 96 well plates (100 μL, 20,000 cells per well). In the serum-free condition, cells were treated with AP20187 and increasing concentrations of inhibitors for 2 days. Then an equal volume of CellTiter-Glo Luminescent Cell Viability Assay (Promega) reagent was added, and luminescence signal was measured after 10 min.
MFE-296, AN3CA, Hec-1B cells were treated with inhibitors on the second day after plating 1,000 cells. Cell survival was assessed 4 days later using the WST-1 assay (Roche). Each data point represents the median of six replicate wells for each tumor cell line and inhibitor concentration.
For all other cancer cell lines, Syto-60 staining method was used. Cells (3000-4000 cells) were seeded on 96-well plates and incubated overnight. Following drug addition, the plates were returned to the incubator for an additional 72 hours. Each plate was fixed in 4% formaldehyde, washed with distilled water, and 100 μL of 1:5000 Syto-60 fluorescent dye (Invitrogen) was added for an additional 1 hour. Following a final wash with distilled water, each plate was read on a SpectraMax M5 plate reader, and all values were expressed as a fraction of the untreated control wells. EC50 values for all viability assays were calculated using GraphPad Prism 4 (GraphPad Software).
Supplementary Material
Table S1, related to Table 1. Ambit KinomeScan profiling of FIIN-1 (10 μM), FRIN-1 (10 μM), and PD173074 (1 μM) for a panel of 402 kinases. Listed are the kinases that show a strong affinity with each inhibitor (Ambit score < 35).
Table S2, related to Table 2 and Figure 5. Antiproliferative activity of PD173074 against 687 cancer cell lines. Cell viability was screened with three concentrations of PD173074 (0.02, 0.2, and 2 μM) for 72 hours. Values are the ratio of cells relative to the control wells.
Table S3, related to Figure S4 Proteins identified from FIIN-1-biotin pulldown. The streptavidin-enriched proteins (Figure S4) were processed for on-beads tryptic digestion and subsequent MS analysis. The unique proteins identified from FIIN-1-biotin are listed.
Acknowledgments
This work was supported by grants from the Damon Runyon Cancer Foundation, the Mary Kay Ash Foundation, and the National Science Foundation. We acknowledge generous assistance from Ambit Biosciences for performing KinomeScan profiling and Invitrogen for performing the Z′-lyte kinase assays.
Footnotes
Supplemental Data
Supplemental data contains four supplementary figures, three supplementary tables, and supplementary procedures that describe chemical synthesis and other protocols, and can be found with this article online at http://www.cell.com/chemistry-biology/supplemental/.
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol. 2008;15:1109–1118. doi: 10.1038/nsmb.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blencke S, Zech B, Engkvist O, Greff Z, Orfi L, Horvath Z, Keri G, Ullrich A, Daub H. Characterization of a conserved structural determinant controlling protein kinase sensitivity to selective inhibitors. Chem Biol. 2004;11:691–701. doi: 10.1016/j.chembiol.2004.02.029. [DOI] [PubMed] [Google Scholar]
- Branford S, Rudzki Z, Walsh S, Grigg A, Arthur C, Taylor K, Herrmann R, Lynch KP, Hughes TP. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002;99:3472–3475. doi: 10.1182/blood.v99.9.3472. [DOI] [PubMed] [Google Scholar]
- Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK, Milanov ZV, Atteridge CE, Biggs WH, 3rd, Edeen PT, et al. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci USA. 2005;102:11011–11016. doi: 10.1073/pnas.0504952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MH, Williams G, Johnson JR, Duan J, Gobburu J, Rahman A, Benson K, Leighton J, Kim SK, Wood R, et al. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin Cancer Res. 2002;8:935–942. [PubMed] [Google Scholar]
- Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308:1318–1321. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, Walker SJ, Brugge JS. Akt activation disrupts mammary acinar architecture and enhances proliferation in an mTOR-dependent manner. J Cell Biol. 2003;163:315–326. doi: 10.1083/jcb.200304159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devilard E, Bladou F, Ramuz O, Karsenty G, Dales JP, Gravis G, Nguyen C, Bertucci F, Xerri L, Birnbaum D. FGFR1 and WT1 are markers of human prostate cancer progression. BMC Cancer. 2006;6:272. doi: 10.1186/1471-2407-6-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutt A, Salvesen HB, Chen TH, Ramos AH, Onofrio RC, Hatton C, Nicoletti R, Winckler W, Grewal R, Hanna M, et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci USA. 2008;105:8713–8717. doi: 10.1073/pnas.0803379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Wang F, Matsubara A, Kan M, McKeehan WL. Fibroblast growth factor receptor 2 limits and receptor 1 accelerates tumorigenicity of prostate epithelial cells. Cancer Res. 1997;57:5369–5378. [PubMed] [Google Scholar]
- Fry DW, Bridges AJ, Denny WA, Doherty A, Greis KD, Hicks JL, Hook KE, Keller PR, Leopold WR, Loo JA, et al. Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. Proc Natl Acad Sci USA. 1998;95:12022–12027. doi: 10.1073/pnas.95.20.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamby JM, Connolly CJ, Schroeder MC, Winters RT, Showalter HD, Panek RL, Major TC, Olsewski B, Ryan MJ, Dahring T, et al. Structure-activity relationships for a novel series of pyrido[2,3-d]pyrimidine tyrosine kinase inhibitors. J Med Chem. 1997;40:2296–2303. doi: 10.1021/jm970367n. [DOI] [PubMed] [Google Scholar]
- Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39:870–874. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur W, Velentza A, Kim S, Flatauer L, Jiang X, Valente D, Mason DE, Suzuki M, Larson B, Zhang J, et al. Clinical stage EGFR inhibitors irreversibly alkylate Bmx kinase. Bioorg Med Chem Lett. 2008;18:5916–5919. doi: 10.1016/j.bmcl.2008.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang JH, Shin KH, Park JG. Mutations in fibroblast growth factor receptor 2 and fibroblast growth factor receptor 3 genes associated with human gastric and colorectal cancers. Cancer Res. 2001;61:3541–3543. [PubMed] [Google Scholar]
- Kammasud N, Boonyarat C, Tsunoda S, Sakurai H, Saiki I, Grierson DS, Vajragupta O. Novel inhibitor for fibroblast growth factor receptor tyrosine kinase. Bioorg Med Chem Lett. 2007;17:4812–4818. doi: 10.1016/j.bmcl.2007.06.058. [DOI] [PubMed] [Google Scholar]
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Koziczak M, Holbro T, Hynes NE. Blocking of FGFR signaling inhibits breast cancer cell proliferation through downregulation of D-type cyclins. Oncogene. 2004;23:3501–3508. doi: 10.1038/sj.onc.1207331. [DOI] [PubMed] [Google Scholar]
- Kunii K, Davis L, Gorenstein J, Hatch H, Yashiro M, Di Bacco A, Elbi C, Lutterbach B. FGFR2-amplified gastric cancer cell lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer Res. 2008;68:2340–2348. doi: 10.1158/0008-5472.CAN-07-5229. [DOI] [PubMed] [Google Scholar]
- Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA. 2005;102:7665–7670. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2:358–364. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- McDermott U, Sharma SV, Dowell L, Greninger P, Montagut C, Lamb J, Archibald H, Raudales R, Tam A, Lee D, et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci USA. 2007;104:19936–19941. doi: 10.1073/pnas.0707498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick JS, Janes J, Kim S, Chang JY, Sipes DG, Gunderson D, Jarnes L, Matzen JT, Garcia ME, Hood TL, et al. An efficient rapid system for profiling the cellular activities of molecular libraries. Proc Natl Acad Sci USA. 2006;103:3153–3158. doi: 10.1073/pnas.0511292103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KB, Maia AT, O'Reilly M, Teschendorff AE, Chin SF, Caldas C, Ponder BA. Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol. 2008;6:e108. doi: 10.1371/journal.pbio.0060108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M, Froum S, Hamby JM, Schroeder MC, Panek RL, Lu GH, Eliseenkova AV, Green D, Schlessinger J, Hubbard SR. Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain. EMBO J. 1998;17:5896–5904. doi: 10.1093/emboj/17.20.5896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M, McMahon G, Sun L, Tang C, Hirth P, Yeh BK, Hubbard SR, Schlessinger J. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science. 1997;276:955–960. doi: 10.1126/science.276.5314.955. [DOI] [PubMed] [Google Scholar]
- Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001;3:785–792. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ory DS, Neugeboren BA, Mulligan RC. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc Natl Acad Sci USA. 1996;93:11400–11406. doi: 10.1073/pnas.93.21.11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, Mendonca RV, Sweeney MD, Scott KC, Grothaus PG, et al. Discovery of Selective Irreversible Inhibitors for Bruton's Tyrosine Kinase. ChemMedChem. 2007;2:58–61. doi: 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock PM, Gartside MG, Dejeza LC, Powell MA, Mallon MA, Davies H, Mohammadi M, Futreal PA, Stratton MR, Trent JM, et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene. 2007;26:7158–7162. doi: 10.1038/sj.onc.1210529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer. 2000;7:165–197. doi: 10.1677/erc.0.0070165. [DOI] [PubMed] [Google Scholar]
- Radisky D, Hagios C, Bissell MJ. Tumors are unique organs defined by abnormal signaling and context. Semin Cancer Biol. 2001;11:87–95. doi: 10.1006/scbi.2000.0360. [DOI] [PubMed] [Google Scholar]
- Ranson M. ZD1839 (Iressa): for more than just non-small cell lung cancer. Oncologist. 2002;7 4:16–24. doi: 10.1634/theoncologist.7-suppl_4-16. [DOI] [PubMed] [Google Scholar]
- Rodems SM, Hamman BD, Lin C, Zhao J, Shah S, Heidary D, Makings L, Stack JH, Pollok BA. A FRET-based assay platform for ultra-high density drug screening of protein kinases and phosphatases. Assay Drug Dev Technol. 2002;1:9–19. doi: 10.1089/154065802761001266. [DOI] [PubMed] [Google Scholar]
- Savage DG, Antman KH. Imatinib mesylate-a new oral targeted therapy. N Engl J Med. 2002;346:683–693. doi: 10.1056/NEJMra013339. [DOI] [PubMed] [Google Scholar]
- Schmeichel KL, Bissell MJ. Modeling tissue-specific signaling and organ function in three dimensions. J Cell Sci. 2003;116:2377–2388. doi: 10.1242/jcs.00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw KR, Wrobel CN, Brugge JS. Use of three-dimensional basement membrane cultures to model oncogene-induced changes in mammary epithelial morphogenesis. J Mammary Gland Biol Neoplasia. 2004;9:297–310. doi: 10.1007/s10911-004-1402-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su TL, Huang JT, Burchenal JH, Watanabe KA, Fox JJ. Synthesis and biological activities of 5-deaza analogues of aminopterin and folic acid. J Med Chem. 1986;29:709–715. doi: 10.1021/jm00155a021. [DOI] [PubMed] [Google Scholar]
- Takeda M, Arao T, Yokote H, Komatsu T, Yanagihara K, Sasaki H, Yamada Y, Tamura T, Fukuoka K, Kimura H, et al. AZD2171 shows potent antitumor activity against gastric cancer over-expressing fibroblast growth factor receptor 2/keratinocyte growth factor receptor. Clin Cancer Res. 2007;13:3051–3057. doi: 10.1158/1078-0432.CCR-06-2743. [DOI] [PubMed] [Google Scholar]
- Trudel S, Li ZH, Wei E, Wiesmann M, Chang H, Chen C, Reece D, Heise C, Stewart AK. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood. 2005;105:2941–2948. doi: 10.1182/blood-2004-10-3913. [DOI] [PubMed] [Google Scholar]
- Tsou HR, Overbeek-Klumpers EG, Hallett WA, Reich MF, Floyd MB, Johnson BD, Michalak RS, Nilakantan R, Discafani C, Golas J, et al. Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J Med Chem. 2005;48:1107–1131. doi: 10.1021/jm040159c. [DOI] [PubMed] [Google Scholar]
- van Rhijn BW, Lurkin I, Radvanyi F, Kirkels WJ, van der Kwast TH, Zwarthoff EC. The fibroblast growth factor receptor 3 (FGFR3) mutation is a strong indicator of superficial bladder cancer with low recurrence rate. Cancer Res. 2001;61:1265–1268. [PubMed] [Google Scholar]
- Welm BE, Freeman KW, Chen M, Contreras A, Spencer DM, Rosen JM. Inducible dimerization of FGFR1: development of a mouse model to analyze progressive transformation of the mammary gland. J Cell Biol. 2002;157:703–714. doi: 10.1083/jcb.200107119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xian W, Schwertfeger KL, Rosen JM. Distinct roles of fibroblast growth factor receptor 1 and 2 in regulating cell survival and epithelial-mesenchymal transition. Mol Endocrinol. 2007;21:987–1000. doi: 10.1210/me.2006-0518. [DOI] [PubMed] [Google Scholar]
- Xian W, Schwertfeger KL, Vargo-Gogola T, Rosen JM. Pleiotropic effects of FGFR1 on cell proliferation, survival, and migration in a 3D mammary epithelial cell model. J Cell Biol. 2005;171:663–673. doi: 10.1083/jcb.200505098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, related to Table 1. Ambit KinomeScan profiling of FIIN-1 (10 μM), FRIN-1 (10 μM), and PD173074 (1 μM) for a panel of 402 kinases. Listed are the kinases that show a strong affinity with each inhibitor (Ambit score < 35).
Table S2, related to Table 2 and Figure 5. Antiproliferative activity of PD173074 against 687 cancer cell lines. Cell viability was screened with three concentrations of PD173074 (0.02, 0.2, and 2 μM) for 72 hours. Values are the ratio of cells relative to the control wells.
Table S3, related to Figure S4 Proteins identified from FIIN-1-biotin pulldown. The streptavidin-enriched proteins (Figure S4) were processed for on-beads tryptic digestion and subsequent MS analysis. The unique proteins identified from FIIN-1-biotin are listed.