

Abstract
Alzheimer disease (AD) is an age-related neurodegenerative disorder, characterized histopathologically by the presence of senile plaques (SP), neurofibrillary tangles and synapse loss in selected brain regions. Positron emission tomography (PET) studies of glucose metabolism revealed decreased energetics in brain of subjects with AD and arguably its earliest form, mild cognitive impairment (MCI), and this decrease correlated with brain structural studies using MRI. The main component of senile plaques is amyloid beta-peptide (Aβ), a 40–42 amino acid peptide that as oligomers is capable of inducing oxidative stress under both in vitro and in vivo conditions and is neurotoxic. In the mitochondria isolated from AD brain, Aβ oligomers that correlated with the reported increased oxidative stress markers in AD have been reported. The markers of oxidative stress have been localized in the brain regions of AD and MCI that show pathological hallmarks of this disease, suggesting the possible role of Aβ in the initiation of the free-radical mediated process and consequently to the build up oxidative stress and AD pathogenesis. Using redox proteomics our laboratory found a number of oxidatively modified brain proteins that are directly in or are associated with the mitochondrial proteome, consistent with a possible involvement of the mitochondrial targeted oxidatively modified proteins in AD progression or pathogenesis. The precise mechanistic link between mitochondrial oxidative damage and role of oligomeric Aβ has not been explicated. In this review, we discuss the role of the oxidation of mitochondria-relevant brain proteins to the pathogenesis and progression of AD.
Keywords: Oxidative stress, Alzheimer’s disease, Mitochondria, MCI
Introduction
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder, characterized clinically by the impairment of cognitive functions and behavioral alterations (Katzman and Saitoh 1991; Salmon et al. 2002). AD is histopathologically characterized by the presence of senile plaques (SP), neurofibrillary tangles and synapse loss in selected brain regions that are responsible for learning, memory processing, and executive functioning. The main component of senile plaques is amyloid beta-peptide (Aβ), a 40–42 amino acid peptide that is derived by proteolytic cleavage of amyloid precursor protein (APP) by the action of beta- and gamma- secretases, and SP are extra cellular in localization (Zhang and Xu 2007).
Role of Aβ(1-42) in oxidative stress in brain of subject with AD and MCI
A number of in vitro and in vivo studies showed that Aβ(1-42) is a neurotoxic peptide that exists in various forms, e.g., soluble (monomers, oligomers, and protofibrils) and insoluble (fibrils) forms. Recent studies suggest that the small oligomers of Aβ are the actual toxic species of this peptide rather than Aβ fibrils (Drake et al. 2003; Lambert et al. 2001; Oda et al. 1995; Walsh et al. 1999). The most concrete evidence suggesting the role of Aβ in AD pathogenesis comes from the genetic observations from familial Alzheimer disease [FAD] (Hardy and Selkoe 2002) that showed that mutations of the genes of amyloid precursor protein (Goate et al. 1991), presenilin-1 and presenilin-2 (Cruts et al. 1998) leads to increased accumulation of fibrillar Aβ in the brain (Hardy and Selkoe 2002; Saido 2003) of FAD subjects.
Oxidative stress in AD brain is manifested by elevated lipid peroxidation (Butterfield et al. 2001; Butterfield and Lauderback 2002; Montine et al. 2002; Sayre et al. 1997), protein oxidation (Butterfield and Stadtman 1997; Good et al. 1996; Hensley et al. 1995; Smith et al. 1997), advanced glycation end products (Vitek et al. 1994), and oxidation of nucleic acids (Gabbita et al. 1998; Lovell and Markesbery 2001; Mecocci et al. 1994; Nunomura et al. 1999, 2001). Further, the markers of oxidative stress have been localized to the regions of the AD brain that have pathological hallmarks of this dementing disorder (Hensley et al. 1995), consistent with a role of Aβ in the initiation of the free-radical mediated process and consequently to the build up oxidative stress and AD pathogenesis.
In addition to the observation of increased levels of oxidative stress in AD brain, studies from our laboratory and other have shown increase levels of oxidative stress in brain of subjects with mild cognitive impairment brain (MCI), a transition stage between AD and normal aging, that shows neuropathological hallmarks similar to AD such as temporal lobe atrophy, low CSF Aβ levels (Chertkow et al. 2001), but different cognitive effects, e.g., mild current memory loss without dementia or significant impairment of other cognitive functions (Morris and Cummings 2005; Petersen et al. 1999). Like AD, brain from MCI subjects also showed increased levels of oxidative stress markers for protein oxidation [indexed by protein carbonyls and 3-ntirotyrosine (3-NT)], lipid peroxidation [indexed by protein bound-4-hydroxy-2-nonenal (HNE), isoprostanes, and neuroprostanes], and nucleic acid oxidation [indexed by 2,6-diamino-4-hydroxy-5-formamidopyrimidine (fapyguanine), 8-hydroxyadenine, 4,6-diamino-5-formamidopyrimidine (fapyadenine) and 5-hydroxycytosine] (Bader Lange et al. 2008; Butterfield et al. 2006a, b; Keller et al. 2005; Markesbery et al. 2005; Migliore et al. 2005; Wang et al. 2006; Williams et al. 2006). Our laboratory is the first to show that protein-bound HNE levels, and 3-NT are elevated in the MCI brain compared to control IPL and hippocampus (Butterfield et al. 2006a, 2007a; Keller et al. 2005). These results suggest the accumulation of oxidative stress markers (Butterfield et al. 2006b; Ding et al. 2006; Keller et al. 2005) in MCI brain, and are consistent with the notion that oxidative stress could be an early event in the progression of MCI to AD.
A number of studies provided evidence of increased oxidative stress in AD pathogenesis, which could initiate from mitochondria, cytoplasm, and also outside the cells (Butterfield and Lauderback 2002; Cross et al. 1987; Markesbery 1997; Smith et al. 1995). Mitochondria use a major part of oxygen in the electron transport system, during which superoxide radicals are produced, one-five percent of which leak out from mitochondria. Superoxide radicals, in turn, can lead to increased production of highly reactive hydroxyl radicals, peroxynitrite, etc., that can contribute to oxidation of proteins, lipids, carbohydrates and nucleic acids (Butterfield and Stadtman 1997).
Recent studies showed the presence of Aβ(1-42) monomers and oligomers in the mitochondrial membranes isolated from the AD brain and brain of animal models of AD (Caspersen et al. 2005; Crouch et al. 2007; Manczak et al. 2006), which suggest possible alterations in mitochondrial structure and function as one of the mechanism(s) of AD pathogenesis.
Previous studies from our laboratory and others have shown that Aβ has a critical methionine residue at position 35, which is believed to be associated with the toxicity of Aβ peptide (Boyd-Kimball et al. 2004, 2005; Butterfield and Boyd-Kimball 2005; Clementi et al. 2006; Crouch et al. 2006; Kanski et al. 2002; Murray et al. 2005). The substitution of Met by norleucine diminishes the toxic effect of the Aβ that clearly documented the importance of Aβ peptide in AD pathogenesis (Butterfield and Kanski 2002; Kanski et al. 2002).
Identification of oxidatively modified brain proteins in subjects with AD and MCI
Our laboratory is the first to use redox proteomics approaches to identify oxidatively modified proteins in brain of subjects with AD and MCI. Employing redox proteomics we identified a large number of oxidatively modified proteins in AD and MCI brain that play key roles in various cellular functions. Many of the proteomics-identified brain proteins belong to energy metabolism pathways. Oxidative modification of the proteins alters conformation and function of proteins (Lauderback et al. 2001a; Subramaniam et al. 1997). Previous studies in AD brain showed that HNE modification of the glutamate transporter (Glt-1 or EAAT), glutathione-S-transferase (GST), and multi-drug resistant protein (MRP-1) (Lauderback et al. 2001a; Sultana and Butterfield 2004) leads to decrease function of these proteins and consequently lead to excitotoxic cell death owing to the decrease clearance of glutamate or increased accumulation of HNE, a highly reactive product of lipid peroxidation.
Alterations of mitochondrial function in AD and MCI
AD brain shows abnormal mitochondrial morphology, and impaired mitochondrial energy metabolism has been well-documented in AD (Atamna and Frey 2007; Caspersen et al. 2005; Crouch et al. 2007, 2008). A large number of studies suggest that the rate of cerebral metabolism is reduced in AD. This decreased cerebral metabolism has been also found in pre-AD stages, i.e., MCI and early AD (EAD), as revealed by PET studies (Dimou et al. 2009). The decrease in the cerebral energy correlated with the altered expression and decreased activity of mitochondrial energy related proteins such as pyruvate dehydrogenase complex (PDHC), alpha-ketoglutarate dehydrogenase complex (KGDHC), isocitrate dehydrogenase (Bubber et al. 2005). In contrast, succinate dehydrogenase (SD) and malate dehydrogenase (MDH) activities were found to be significantly increased in AD brain (Bubber et al. 2005). Further, in vitro studies showed that incubation of isolated mitochondria with Aβ peptides decreased the activity of KGDHC and PDHC (Casley et al. 2002). Most oxidatively modified brain enzymes have decreased activity (Butterfield et al. 2006a), and it is not clear why certain enzymes show an increased activity upon oxidation. The alteration in the enzyme activity can be explained based on protein expression differences or the oxidation of the proteins, which expose more hydrophobic sites to the aqueous environment with subsequent changes in conformation and aggregation (Butterfield and Stadtman 1997). Depending on the site where an oxidative modification occurs the activity of a particular enzyme may be down regulated or upregulated, and we speculate that the increased MDH and SD activity in AD result from oxidative modification-mediated changes in the active site. This speculation is based on our finding of increased MDH activity in MCI brain (Reed et al. 2008). Further, decreased glucose utilization has been reported in skin fibroblasts of AD patients (Sims 1990).
Oxidatively modified brain mitochondrial proteins in AD
As noted above, a large number of brain proteins were identified as oxidatively modified in AD. Among these brain proteins, GAPDH, aconitase, VDAC, ATP synthase-alpha chain, lactate dehydrogenase (LDH), beta-actin, and alpha-tubulin, are either mitochondrial proteins or are known to interact with mitochondria (Aksenov et al. 2000; Castegna et al. 2002, 2003; Reed et al. 2008; Sultana et al. 2006a, b).
VDAC is a part of the outer mitochondrial membrane and a component of the mitochondrial permeability transition pore (MPTP). VDAC has been shown to conduct movement of metabolites like ATP in and out of mitochondria by passive diffusion, participate in synaptic communication, and be involved in the early stages of apoptosis (Shoshan-Barmatz et al. 2006). ATP production and mitochondrial calcium buffering are essential for normal synaptic transmission (Csordas and Hajnoczky 2003; Mattson and Liu 2002). There are three different forms of VDAC proteins, i.e., VDAC-1, VDAC-2 and VDAC-3. We found VDAC-1 protein as an oxidatively modified protein, and further the levels of VDAC-1 were reported to be significantly decreased in AD brain (Sultana et al. 2006a, b). A previous in vitro study demonstrated that VDAC1-deficient mice show deficits in learning behavior and synaptic plasticity; hence, oxidation and dysfunction of this protein correlated well with the impairment of learning and memory as reported in AD (Weeber et al. 2002). Moreover, VDAC regulates release of several apoptotic factors such as cytochrome C, apoptosis inducing factor, smac, and caspases from mitochondria; therefore, VDAC is essential for regulation of the apoptotic signals in the cell, and oxidation of VDAC-1 may trigger apoptosis (Lorenzo et al. 1999; Susin et al. 1999). Taken together, we propose that oxidation of VDAC-1 protein in AD brain leads to mitochondrial depolarization and altered signal transduction pathways, which could be crucial in synaptic transmission and plasticity. In addition, this alteration may also induce apoptotic events leading to cell death observed in AD.
GAPDH is a glycolytic enzyme that catalyzes the reversible phosphorylation of glyceraldehyde-3-phosphate to 1,3-bisphosphoglycerate. This enzyme is critical for generation of energy. In addition, this protein interacts with other proteins including VDAC-1 (Tarze et al. 2007). Moreover, decreased activity of GAPDH would lead to elevation of trioses, such as methylglyoxal, which is more reactive than HNE and capable of modifying protein. Studies from our laboratory showed that GAPDH is oxidatively modified in AD that correlated with the reported decreased activity of GAPDH (Sultana et al. 2006b). The decreased level of GAPDH correlates well with the decreased cerebral glucose utilization reported in AD brain (Reed et al. 2008; Sultana et al. 2006b). Since GAPDH interacts with VDAC, alterations in the structure and function of GAPDH may lead to functional impairment of VDAC that may eventually lead to leakage of calcium and cytochrome C from the mitochondria and trigger processes of cell death. Consequently, oxidative modification of VDAC and GAPDH may mediate apoptotic processes and alter mitochondrial and synaptic functions, all of which are known to be impaired in AD.
Beta-actin and alpha-tubulin, in addition to their role as structural proteins, also regulate the function of other proteins via their interaction with them. Mitochondrial tubulin has been shown to exist as an alpha-beta dimer and represents 2.2+−0.5% of total cellular tubulin (Carre et al. 2002). Further, this group also reported that mitochondrial tubulin interacts with VDAC. We found alpha tubulin as oxidatively modified in AD brain, which suggests that alteration in the structure and function of this protein could lead to alteration in the function of membrane permeability transition pore and hence may play a significant role in apoptosis. Beta-actin has been reported to localize in the mitochondrial matrix space and on the internal surface of the mitochondrial inner membrane and to be important for mitochondrial organization and morphogenesis (Etoh et al. 1990). Consequently, oxidation of beta-actin may lead to mitochondrial structural abnormalities that may eventually lead to malfunction of mitochondria, contributing to the AD pathogenesis.
ATP synthase α-chain is a part of complex V of the electron transport chain of mitochondria, involved with oxidative phosphorylation, localized in the inner membrane, and is important for ATP synthesis and release. Recent studies showed that both APP and Aβ bind to ATP synthase and regulates its activity (Schmidt et al. 2008). Thus, such interactions of ATP synthase with AD-relevant APP and Aβ may play important roles in regulating ATP levels in the cell. Previous studies from our laboratory and others have reported a decreased level of complex V in AD and also oxidative modification of ATP synthase (Bosetti et al. 2002; Schagger and Ohm 1995; Sultana et al. 2006b), which suggest that the function of ATP synthase α-chain is altered in AD degenerating neurons. Since complex V is a key entity in the conversion of the mitochondrial proton gradient into chemical energy, such oxidative modification could contribute to the observed decrease in brain glucose metabolism in AD and MCI.
Lactate dehydrogenase is an important enzyme that carries out the reversible conversion of pyruvate to lactate under anaerobic conditions. A recent study using a human astrocyte cell line showed that astrocytes can use lactate to generate ATP via oxidative phosphorylation (Lemire et al. 2008). Since the energy demand of brain is high, this process of utilization of lactate to generate energy is critical to brain function. LDH is also found in the mitochondria and is important in the utilization of lactate to generate ATP. Also, LDH is important for the astrocyte-neuron shuttle and for the effective functioning of neurons (Mangia et al. 2009). Oxidative modification of LDH may impair its ability to utilize lactate for energy production, which could contribute to decreased brain energetics reported in AD.
Oxidatively modified brain mitochondrial proteins in MCI
A number of overlapping mitochondria-relevant proteins have been found to be common targets of oxidation between AD and MCI brain, including, ATP synthase alpha, lactate dehydrogenase and beta-actin (Fig. 1) (Aksenov et al. 2000; Castegna et al. 2002, 2003; Reed et al. 2008; Sultana et al. 2006a, b). The significance of oxidative dysfunction of these brain proteins for diminished brain function and neuronal loss was discussed above. In addition to these proteins one other mitochondrial protein was identified as oxidatively modified in brain of subjects with MCI, i.e., malate dehydrogenase (MDH) (Reed et al. 2008). MDH catalyzes the reversible oxidation of malate to oxaloacetate by NAD+ in the TCA cycle. The significance of increased activity of MDH in MCI brain is unclear, but conceivably this observation may reflect a compensatory response of the brain to oxidative damage to other key energy-related proteins in this prodromal stage of AD.
Fig. 1.
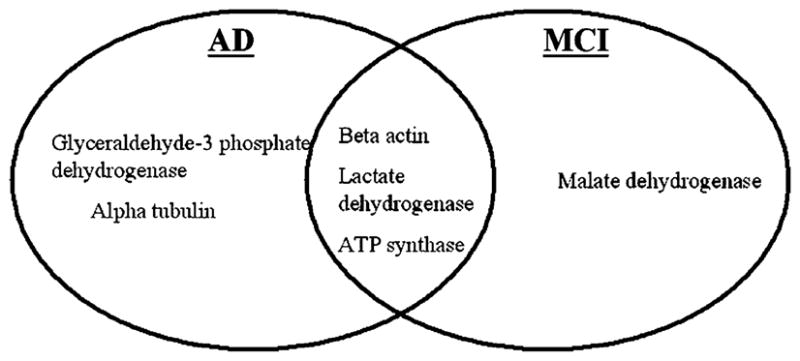
Oxidatively modified mitochondria-relevant proteins in AD and MCI brain
Conclusions
Based on the proteins that were found to oxidatively modified proteins in AD and MCI, it is possible that the oxidation of key mitochondrial proteins contributes to the observed decrease in energetics in the AD and MCI brain compared to age-matched controls and may also be participate in the triggering of apoptotic processes that eventually lead to modification of important cellular functions vital to neuronal survival, leading to the AD signs and symptoms.
The precise mechanistic link between mitochondrial oxidative damage and role of oligomeric Aβ has not been established. However, based on the above observations, it is tempting to speculate that Aβ(1-42) is transported to the mitochondrial membrane as monomeric or oligomeric forms. Once Aβ(1-42) is inserted into the membrane, it may initiate the process of lipid peroxidation (Butterfield and Boyd-Kimball 2005; Butterfield et al. 2007a; Kanski et al. 2002; Lauderback et al. 2001b; Varadarajan et al. 2001) that may eventually lead to damage of both mitochondrial membrane proteins and lipids (Fig. 2). The perturbation in the mitochondrial membrane due to damage occurring at protein and lipid levels might lead to the reported alterations in the electron transport system and alteration in the mitochondrial membrane fluidity (Aleardi et al. 2005). Moreover, oxidative modification of these proteins may result in increase leakage of superoxide radicals from mitochondria, which may eventually lead to increase production of reactive oxygen and nitrogen species and consequently to increased oxidative stress and AD pathogenesis (Fig. 2). Additional studies are necessary to test these ideas, and our laboratory together with several other laboratories is working in this direction. Based on the proteomic studies conducted so far, the notion that mitochondrial alterations appear to be involved in the AD pathogenesis and progression of AD seem highly likely, and, consequently, targeting mitochondria to treat or slow onset of AD may be a promising therapeutic approach for this devastating dementing disorder.
Fig. 2.
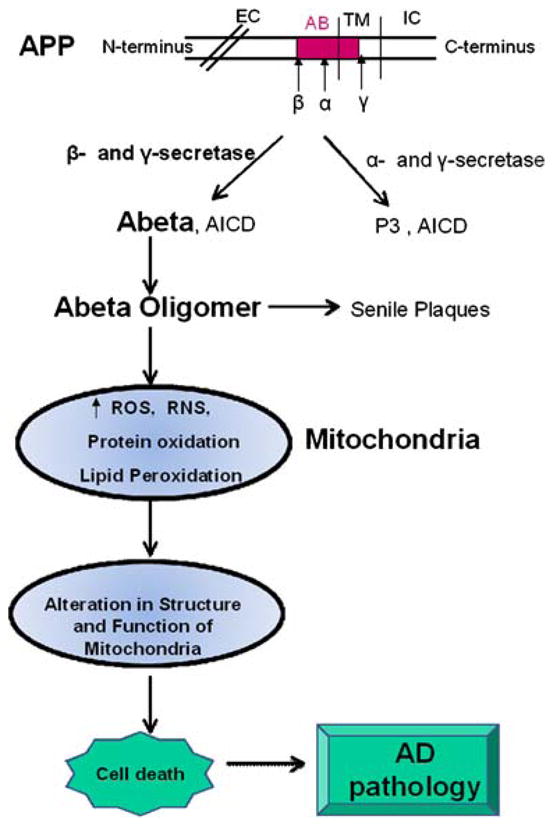
Consequence of Aβ(1-42) on mitochondria and AD pathogenesis. See text. EC extra cellular, IC intracellualr, TM transmembrane, ROS reactive oxygen species, RNS reactive nitrogen species
Acknowledgments
This work was supported in part by NIH grants to D.A.B. [AG-10836; AG-05119].
Contributor Information
Rukhsana Sultana, Department of Chemistry, University of Kentucky, Lexington, KY 40506-0055, USA, Center of Membrane Sciences, University of Kentucky, Lexington, KY 40506-0059, USA, Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY 40536, USA.
D. Allan Butterfield, Email: dabcns@uky.edu, Department of Chemistry, University of Kentucky, Lexington, KY 40506-0055, USA, Center of Membrane Sciences, University of Kentucky, Lexington, KY 40506-0059, USA, Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY 40536, USA.
References
- Aksenov M, Aksenova M, Butterfield DA, Markesbery WR. J Neurochem. 2000;74:2520–2527. doi: 10.1046/j.1471-4159.2000.0742520.x. [DOI] [PubMed] [Google Scholar]
- Aleardi AM, Benard G, Augereau O, Malgat M, Talbot JC, Mazat JP, Letellier T, Dachary-Prigent J, Solaini GC, Rossignol R. J Bioenerg Biomembr. 2005;37:207–225. doi: 10.1007/s10863-005-6631-3. [DOI] [PubMed] [Google Scholar]
- Atamna H, Frey WH., 2nd Mitochondrion. 2007;7:297–310. doi: 10.1016/j.mito.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Bader Lange ML, Cenini G, Piroddi M, Abdul HM, Sultana R, Galli F, Memo M, Butterfield DA. Neurobiol Dis. 2008;29:456–464. doi: 10.1016/j.nbd.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosetti F, Brizzi F, Barogi S, Mancuso M, Siciliano G, Tendi EA, Murri L, Rapoport SI, Solaini G. Neurobiol Aging. 2002;23:371–376. doi: 10.1016/s0197-4580(01)00314-1. [DOI] [PubMed] [Google Scholar]
- Boyd-Kimball D, Mohmmad Abdul H, Reed T, Sultana R, Butterfield DA. Chem Res Toxicol. 2004;17:1743–1749. doi: 10.1021/tx049796w. [DOI] [PubMed] [Google Scholar]
- Boyd-Kimball D, Sultana R, Mohmmad-Abdul H, Butterfield DA. Peptides. 2005;26:665–673. doi: 10.1016/j.peptides.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE. Ann Neurol. 2005;57:695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Boyd-Kimball D. Biochim Biophys Acta. 2005;1703:149–156. doi: 10.1016/j.bbapap.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Kanski J. Peptides. 2002;23:1299–1309. doi: 10.1016/s0196-9781(02)00066-9. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Lauderback CM. Free Radic Biol Med. 2002;32:1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Stadtman ER. Adv Cell Aging Gerontol. 1997;2:161–191. [Google Scholar]
- Butterfield DA, Drake J, Pocernich C, Castegna A. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR. Neurobiol Dis. 2006a;22:223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Reed T, Perluigi M, De Marco C, Coccia R, Cini C, Sultana R. Neurosci Lett. 2006b;397:170–173. doi: 10.1016/j.neulet.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Reed T, Newman SF, Sultana R. Free Radic Biol Med. 2007a;43:658–677. doi: 10.1016/j.freeradbiomed.2007.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Reed TT, Perluigi M, De Marco C, Coccia R, Keller JN, Markesbery WR, Sultana R. Brain Res. 2007b;1148:243–248. doi: 10.1016/j.brainres.2007.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carre M, Andre N, Carles G, Borghi H, Brichese L, Briand C, Braguer D. J Biol Chem. 2002;277:33664–33669. doi: 10.1074/jbc.M203834200. [DOI] [PubMed] [Google Scholar]
- Casley CS, Canevari L, Land JM, Clark JB, Sharpe MA. J Neurochem. 2002;80:91–100. doi: 10.1046/j.0022-3042.2001.00681.x. [DOI] [PubMed] [Google Scholar]
- Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Faseb J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Free Radic Biol Med. 2002;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- Castegna A, Thongboonkerd V, Klein JB, Lynn B, Markesbery WR, Butterfield DA. J Neurochem. 2003;85:1394–1401. doi: 10.1046/j.1471-4159.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- Chertkow H, Bergman H, Schipper HM, Gauthier S, Bouchard R, Fontaine S, Clarfield AM. Can J Neurol Sci. 2001;28(Suppl 1):S28–S41. doi: 10.1017/s0317167100001189. [DOI] [PubMed] [Google Scholar]
- Clementi ME, Pezzotti M, Orsini F, Sampaolese B, Mezzogori D, Grassi C, Giardina B, Misiti F. Biochem Biophys Res Commun. 2006;342:206–213. doi: 10.1016/j.bbrc.2006.01.137. [DOI] [PubMed] [Google Scholar]
- Cross CE, Halliwell B, Borish ET, Pryor WA, Ames BN, Saul RL, McCord JM, Harman D. Ann Intern Med. 1987;107:526–545. doi: 10.7326/0003-4819-107-4-526. [DOI] [PubMed] [Google Scholar]
- Crouch PJ, Barnham KJ, Duce JA, Blake RE, Masters CL, Trounce IA. J Neurochem. 2006;99:226–236. doi: 10.1111/j.1471-4159.2006.04050.x. [DOI] [PubMed] [Google Scholar]
- Crouch PJ, Cimdins K, Duce JA, Bush AI, Trounce IA. Rejuvenation Res. 2007;10:349–357. doi: 10.1089/rej.2007.0592. [DOI] [PubMed] [Google Scholar]
- Crouch PJ, Harding SM, White AR, Camakaris J, Bush AI, Masters CL. Int J Biochem Cell Biol. 2008;40:181–198. doi: 10.1016/j.biocel.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Cruts M, van Duijn CM, Backhovens H, Van den Broeck M, Wehnert A, Serneels S, Sherrington R, Hutton M, Hardy J, St George-Hyslop PH, Hofman A, Van Broeckhoven C. Hum Mol Genet. 1998;7:43–51. doi: 10.1093/hmg/7.1.43. [DOI] [PubMed] [Google Scholar]
- Csordas G, Hajnoczky G. J Biol Chem. 2003;278:42273–42282. doi: 10.1074/jbc.M305248200. [DOI] [PubMed] [Google Scholar]
- Dimou E, Booij J, Rodrigues M, Prosch H, Attems J, Knoll P, Zajicek B, Dudczak R, Mostbeck G, Kuntner C, Langer O, Bruecke T, Mirzaei S. Curr Alzheimer Res. 2009;6:312–319. doi: 10.2174/156720509788486563. [DOI] [PubMed] [Google Scholar]
- Ding Q, Markesbery WR, Cecarini V, Keller JN. Neurochem Res. 2006;31:705–710. doi: 10.1007/s11064-006-9071-5. [DOI] [PubMed] [Google Scholar]
- Drake J, Link CD, Butterfield DA. Neurobiol Aging. 2003;24:415–420. doi: 10.1016/s0197-4580(02)00225-7. [DOI] [PubMed] [Google Scholar]
- Etoh S, Matsui H, Tokuda M, Itano T, Nakamura M, Hatase O. Biochem Int. 1990;20:599–606. [PubMed] [Google Scholar]
- Gabbita SP, Lovell MA, Markesbery WR. J Neurochem. 1998;71:2034–2040. doi: 10.1046/j.1471-4159.1998.71052034.x. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Good PF, Werner P, Hsu A, Olanow CW, Perl DP. Am J Pathol. 1996;149:21–28. [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M, Aksenova M, Gabbita SP, Wu JF, Carney JM, et al. J Neurochem. 1995;65:2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- Kanski J, Aksenova M, Schoneich C, Butterfield DA. Free Radic Biol Med. 2002;32:1205–1211. doi: 10.1016/s0891-5849(02)00821-3. [DOI] [PubMed] [Google Scholar]
- Katzman R, Saitoh T. Faseb J. 1991;5:278–286. [PubMed] [Google Scholar]
- Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR. Neurology. 2005;64:1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Mann DM, Harris JM, Chartier-Harlin MC, Cumming A, Coates J, Lemmon H, StClair D, Iwatsubo T, Lendon C. J Med Genet. 2001;38:353–355. doi: 10.1136/jmg.38.6.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. J Neurochem. 2001a;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- Lauderback CM, Hackett JM, Keller JN, Varadarajan S, Szweda L, Kindy M, Markesbery WR, Butterfield DA. Biochemistry. 2001b;40:2548–2554. doi: 10.1021/bi002312k. [DOI] [PubMed] [Google Scholar]
- Lemire J, Mailloux RJ, Appanna VD. PLoS One. 2008;3:e1550. doi: 10.1371/journal.pone.0001550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo HK, Susin SA, Penninger J, Kroemer G. Cell Death Differ. 1999;6:516–524. doi: 10.1038/sj.cdd.4400527. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Markesbery WR. Arch Neurol. 2001;58:392–396. doi: 10.1001/archneur.58.3.392. [DOI] [PubMed] [Google Scholar]
- Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- Mangia S, Simpson IA, Vannucci SJ, Carruthers A. J Neurochem. 2009;109(Suppl 1):55–62. doi: 10.1111/j.1471-4159.2009.06003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markesbery WR. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- Markesbery WR, Kryscio RJ, Lovell MA, Morrow JD. Ann Neurol. 2005;58:730–735. doi: 10.1002/ana.20629. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Liu D. Neuromolecular Med. 2002;2:215–231. doi: 10.1385/NMM:2:2:215. [DOI] [PubMed] [Google Scholar]
- Mecocci P, MacGarvey U, Beal MF. Ann Neurol. 1994;36:747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- Migliore L, Fontana I, Trippi F, Colognato R, Coppede F, Tognoni G, Nucciarone B, Siciliano G. Neurobiol Aging. 2005;26:567–573. doi: 10.1016/j.neurobiolaging.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Neely MD, Quinn JF, Beal MF, Markesbery WR, Roberts LJ, Morrow JD. Free Radic Biol Med. 2002;33:620–626. doi: 10.1016/s0891-5849(02)00807-9. [DOI] [PubMed] [Google Scholar]
- Morris JC, Cummings J. J Alzheimers Dis. 2005;7:235–239. doi: 10.3233/jad-2005-7306. [DOI] [PubMed] [Google Scholar]
- Murray IV, Sindoni ME, Axelsen PH. Biochemistry. 2005;44:12606–12613. doi: 10.1021/bi050926p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, Smith MA. J Neurosci. 1999;19:1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- Oda T, Wals P, Osterburg HH, Johnson SA, Pasinetti GM, Morgan TE, Rozovsky I, Stine WB, Snyder SW, Holzman TF, et al. Exp Neurol. 1995;136:22–31. doi: 10.1006/exnr.1995.1080. [DOI] [PubMed] [Google Scholar]
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Arch Neurol. 1999;56:303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- Reed T, Perluigi M, Sultana R, Pierce WM, Klein JB, Turner DM, Coccia R, Markesbery WR, Butterfield DA. Neurobiol Dis. 2008;30:107–120. doi: 10.1016/j.nbd.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Saido TC. Aβ Metabolism and Alzheimer’s Disease. Landes Bioscience; Georgetown, TX: 2003. [Google Scholar]
- Salmon DP, Thomas RG, Pay MM, Booth A, Hofstetter CR, Thal LJ, Katzman R. Neurology. 2002;59:1022–1028. doi: 10.1212/wnl.59.7.1022. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- Schagger H, Ohm TG. Eur J Biochem. 1995;227:916–921. doi: 10.1111/j.1432-1033.1995.tb20219.x. [DOI] [PubMed] [Google Scholar]
- Schmidt C, Lepsverdize E, Chi SL, Das AM, Pizzo SV, Dityatev A, Schachner M. Mol Psychiatry. 2008;13:953–969. doi: 10.1038/sj.mp.4002077. [DOI] [PubMed] [Google Scholar]
- Shoshan-Barmatz V, Israelson A, Brdiczka D, Sheu SS. Curr Pharm Des. 2006;12:2249–2270. doi: 10.2174/138161206777585111. [DOI] [PubMed] [Google Scholar]
- Sims NR. Ann Neurol. 1990;27:691–693. doi: 10.1002/ana.410270621. [DOI] [PubMed] [Google Scholar]
- Smith MA, Sayre LM, Monnier VM, Perry G. Trends Neurosci. 1995;18:172–176. doi: 10.1016/0166-2236(95)93897-7. [DOI] [PubMed] [Google Scholar]
- Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam R, Roediger F, Jordan B, Mattson MP, Keller JN, Waeg G, Butterfield DA. J Neurochem. 1997;69:1161–1169. doi: 10.1046/j.1471-4159.1997.69031161.x. [DOI] [PubMed] [Google Scholar]
- Sultana R, Butterfield DA. Neurochem Res. 2004;29:2215–2220. doi: 10.1007/s11064-004-7028-0. [DOI] [PubMed] [Google Scholar]
- Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR, Butterfield DA. Neurobiol Aging. 2006a;27:1564–1576. doi: 10.1016/j.neurobiolaging.2005.09.021. [DOI] [PubMed] [Google Scholar]
- Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Neurobiol Dis. 2006b;22:76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Brenner C, Larochette N, Prevost MC, Alzari PM, Kroemer G. J Exp Med. 1999;189:381–394. doi: 10.1084/jem.189.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarze A, Deniaud A, Le Bras M, Maillier E, Molle D, Larochette N, Zamzami N, Jan G, Kroemer G, Brenner C. Oncogene. 2007;26:2606–2620. doi: 10.1038/sj.onc.1210074. [DOI] [PubMed] [Google Scholar]
- Varadarajan S, Kanski J, Aksenova M, Lauderback C, Butterfield DA. J Am Chem Soc. 2001;123:5625–5631. doi: 10.1021/ja010452r. [DOI] [PubMed] [Google Scholar]
- Vitek MP, Bhattacharya K, Glendening JM, Stopa E, Vlassara H, Bucala R, Manogue K, Cerami A. Proc Natl Acad Sci U S A. 1994;91:4766–4770. doi: 10.1073/pnas.91.11.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB. J Biol Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- Wang J, Markesbery WR, Lovell MA. J Neurochem. 2006;96:825–832. doi: 10.1111/j.1471-4159.2005.03615.x. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Levy M, Sampson MJ, Anflous K, Armstrong DL, Brown SE, Sweatt JD, Craigen WJ. J Biol Chem. 2002;277:18891–18897. doi: 10.1074/jbc.M201649200. [DOI] [PubMed] [Google Scholar]
- Williams TI, Lynn BC, Markesbery WR, Lovell MA. Neurobiol Aging. 2006;27:1094–1099. doi: 10.1016/j.neurobiolaging.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Zhang YW, Xu H. Curr Mol Med. 2007;7:687–696. doi: 10.2174/156652407782564462. [DOI] [PubMed] [Google Scholar]