

Abstract
The recently discovered enzyme lysine-specific demethylase 1 (LSD1) plays an important role in the epigenetic control of gene expression, and aberrant gene silencing secondary to LSD1 over expression is thought to contribute to the development of cancer. We recently reported a series of (bis)guanidines and (bis)biguanides that are potent inhibitors of LSD1, and induce the re-expression of aberrantly silenced tumor suppressor genes in tumor cells in vitro. We now report a series of isosteric ureas and thioureas that are also potent inhibitors of LSD1. These compounds induce increases in methylation at the histone 3 lysine 4 (H3K4) chromatin mark, a specific target of LSD1, in Calu-6 lung carcinoma cells. In addition, these analogues increase cellular levels of secreted frizzle-related proteins (SFRP) 2 and 5, and transcription factor GATA4. These compounds represent an important new series of epigenetic modulators with the potential for use as antitumor agents.
Keywords: Epigenetics; lysine-specific demethylase 1; histones; histone demethylase; urea, thiourea; gene expression; enzyme inhibitor; secreted frizzle-related protein; GATA4; Calu-6 human anaplastic non-small cell lung carcinoma
Introduction
Chromatin architecture is a key determinant in the regulation of gene expression, and this architecture is strongly influenced by post–translational modifications of histones.1, 2 Histone protein tails contain lysine residues that interact with the negative charges on the DNA backbone. These lysine-containing tails, consisting of up to 40 amino acid residues, protrude through the DNA strand, and act as a site for post-translational modification of chromatin, allowing alteration of higher order nucleosome structure.3 Multiple post-translational modifications of histones can mediate epigenetic remodeling of chromatin, with acetylation being the best characterized process.4 Transcriptional repression is associated with specific CpG island DNA methylation and recruitment of histone deacetylases (HDACs) to gene promoters that cooperate in the epigenetic silencing of specific genes.5, 6 Normal mammalian cells exhibit an exquisite level of control of chromatin architecture by maintaining a balance between histone acetyltransferase (HAT) and HDAC activity.7
In cancer, CpG island DNA promoter hypermethylation in combination with other chromatin modifications, including decreased activating marks and increased repressive marks on histone proteins 3 and 4, have been associated with the silencing of tumor suppressor genes.8 The important role of promoter CpG island methylation and its relationship to covalent histone modifications has recently been reviewed.9 As was mentioned above, the N-terminal lysine tails of histones can undergo numerous posttranslational modifications, including phosphorylation, ubiquitination, acetylation and methylation.4, 10, 11 To date, 17 lysine residues and 7 arginine residues on histone proteins have been shown to undergo methylation,12 and lysine methylation on histones can signal transcriptional activation or repression, depending on the specific lysine residue involved.13-15 All known histone lysine methyltransferases contain a conserved SET methyltransferase domain, and it has been shown that aberrant methylation of histones due to SET domain deregulation is linked to carcinogenesis.16 Histone methylation, once thought to be an irreversible process, has recently been shown to be a dynamic process regulated by the addition of methyl groups by histone methyltransferases and removal of methyl groups from mono- and dimethyllysines by lysine specific demethylase 1 (LSD1), and from mono-, di, and trimethyllysines by specific Jumonji C (JmjC) domain-containing demethylases.10, 11, 17, 18 Additional demethylases in the JmjC demethylase class are continuing to be identified.19, 20 Recent evidence suggests that LSD1 is required for maintenance of global DNA methylation,21 indicating that the LSD1-mediated demethylation is a general mechanism for transcriptional control.
A key positive chromatin mark found associated with promoters of active genes is histone 3 dimethyllysine 4 (H3K4me2).22, 23 LSD1, also known as BHC110 and KDM1,10, 24 catalyzes the oxidative demethylation of histone 3 methyllysine 4 (H3K4me1) and H3K4me2, and is associated with transcriptional repression.10 H3K4me2 is a transcription-activating chromatin mark at gene promoters, and demethylation of this mark by LSD1 may prevent expression of tumor suppressor genes important in human cancer.25 Thus, LSD1 is emerging as an important new target for the development of specific inhibitors as a new class of antitumor drugs.26
To date, only a few existing compounds have been shown to act as inhibitors of LSD1. The active site structure of LSD1 has considerable sequence homology to monoamine oxidases A and B (MAO A and B), and to N1-acetylpolyamine oxidase (APAO) and spermine oxidase (SMO).10, 24, 27 It has been shown that classical MAO inhibitors phenelzine and tranylcypromine inactivate nucleosomal demethylation by the recombinant LSD1/CoRest complex, and increase global levels of H3K4me2 in the P19 cell line.24, 27 The synthetic substrate analogue aziridinyl-K4H31-21 reversibly inhibited LSD1 with an IC50 of 15.6 μM, while propargyl-K4H31-21 produced time-dependent inactivation with a Ki of 16.6 μM.28 Propargyl-K4H31-21 was later shown to inactivate LSD1 through formation of a covalent adduct with the enzyme-bound flavin cofactor.27, 29 McCafferty et al. recently described the synthesis of a series of trans-2-arylcyclopropylamine analogues that inhibit LSD1 with Ki values between 188 and 566 μM.30 However, in all but one instance, these analogues were 1-2 orders of magnitude more potent against MAO A and MOA B. Most recently, Ueda and coworkers identified small molecule tranylcypromine derivatives that are selective for LSD1 over MAO-A and MAO-B,31 and Binda et al. described similar tranylcypromine analogues that exhibited partial selectivity between LSD1 and the newly identified histone demethylase LSD2.32
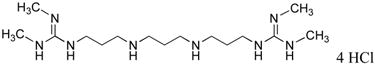
LSD1 was identified in part because its C-terminal domain shares significant sequence homology with the amine oxidases acetylpolyamine oxidase (APAO) and spermine oxidase (SMO).10, 33 Several groups have identified amines, guanidines or similar analogues that act as selective modulators of these 2 amine oxidases.33-39 We previously reported the synthesis of a novel series of (bis)guanidines and (bis)biguanides40 that are potent antitrypanosomal agents in vitro, with IC50 values against Trypanosoma brucei as low as 90 nM. Because of their structural similarity to guanidine-based inhibitors of APAO and SMO, we sought to determine whether (bis)guanidines 1a-g and (bis)biguanides 2a-f (Figure 1) were inhibitors of LSD1, and whether this inhibition had any influence on selected chromatin marks in tumor cells. Nine of the 13 compounds tested were found to inhibit LSD1 activity by >50% at 1 μM.25 The two most potent LSD1 inhibitors exhibited non-competitive kinetics at concentrations up to 2.5 μM. A 48 hr exposure of HCT116 human colon carcinoma cells to increasing concentrations of analogues 1c and 2d (Figure 1) produced significant global increases in both H3K4me1 and H3K4me2, while not affecting global H3K9me2 levels. These analogues also induced the re-expression of multiple, aberrantly silenced genes important in the development of colon cancer, including members of the secreted frizzle-related proteins (SFRPs) and the GATA family of transcription factors.
Figure 1.

(Bis)guanidine and (bis)biguanides with potent antitrypanosomal activity in vitro.
Because of the promising cellular effects of 1c and 2d, the synthesis and evaluation of additional analogues was proposed. To access a library of more diverse analogues related to 1c and 2d, we adapted our previously published syntheses40 to produce a series of 30 isosteric (bis)alkylureas or (bis)alkylthioureas (compounds 3-33, Table 1), and these analogues were evaluated for the ability to inhibit LSD1 and induce increases in global H3K4me2 in vitro.
Table 1.
Structures of compounds 1c, 2d and 3-33, and inhibition of LSD1 in vitro following treatment with each analogue at 10 μM.
| Compound | Compound | %LSD1 activity remaining |
|---|---|---|
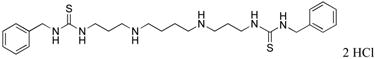
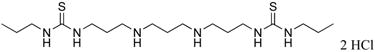
 |
1c | 14.1 (from ref. 25) |
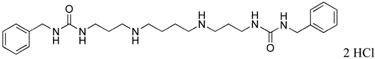
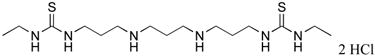
 |
2d | 17.2 |
 |
3 | 74.8 |
 |
4 | 49.5 |
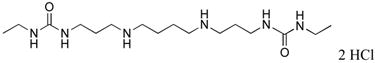 |
5 | 49.2 |
 |
6 | 40 |
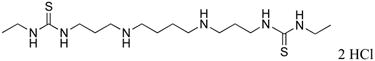
 |
7 | 100 |
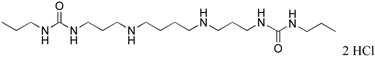 |
8 | 79 |
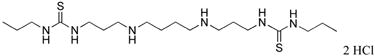 |
9 | 89.6 |
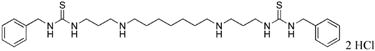 |
10 | 35.9 |
| 11 | 95.9 | |
| 12 | 91.5 | |
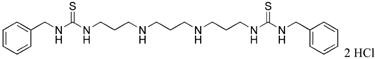 |
13 | 52.1 |
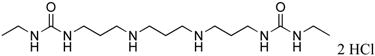 |
14 | 65.5 |
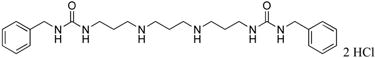 |
15 | 60.5 |
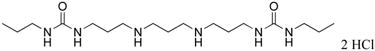 |
16 | 51.3 |
 |
17 | 100 |
 |
18 | 36.2 |
 |
19 | 92.9 |
 |
20 | 92.2 |
 |
21 | 74.6 |
 |
22 | 88.6 |
 |
23 | 51.5 |
 |
24 | 77.3 |
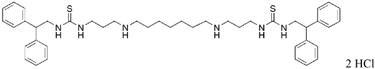 |
25 | 19.5 |
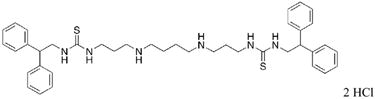 |
26 | 17.1 |
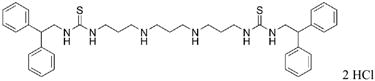 |
27 | 24.8 |
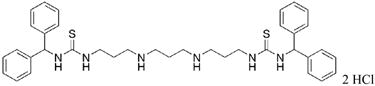 |
28 | 51.1 |
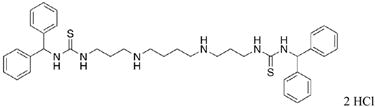 |
29 | 34.4 |
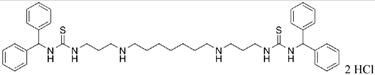 |
30 | 28.9 |
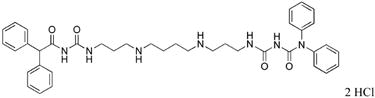 |
31 | 26.1 |
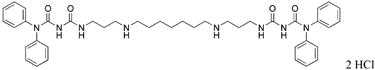 |
32 | 70 |
 |
33 | 91.5 |
Chemistry
Preparation of compounds 3-33 depended on the availability of the appropriate isocyanates and isothiocyanates. All of these intermediates were commercially available, with the exception of isocyanate 35c and isothiocyanates 37a-c, which were synthesized as shown in Scheme 1. Isocyanate 35c could be made in a single step by reacting the requisite diphenylalkylamine 34c (m = 2) with trichloroacetic anhydride (toluene, N2, reflux) for 5 hours.41, 42 To produce the corresponding isothiocyanates 37a-c (m = 0, 1 or 2, respectively), amines 34a-c were allowed to react with carbon disulfide in the presence of triethylamine in THF at 5°C.43 The reaction was allowed to warm to room temperature, and after 3 hours the intermediate dithiocarbamates 36a-c could be isolated. Reaction of the intermediate dithiocarbamates 36a-c with tosyl chloride in THF then afforded the desired isothiocyanates 37a-c.43
Scheme 1.

To access a library of isosteric urea and thiourea analogues related to 1c and 2d, we employed our previously published synthesis40 of precursor molecules 41a-c, as shown in Scheme 2. The appropriate diamine 38a, 38b or 38c was (bis)cyanoethylated (acrylonitrile, EtOH, reflux) to afford the corresponding (bis)cyano intermediates 39a-c. The central nitrogens in 39a-c were then N-Boc protected ((Boc)2O, CH2Cl2/Aq. NaHCO3)44 to form 40a-c, and the cyano groups were reduced (Raney Ni) to yield the desired diamines 41a-c.40, 45 Compounds 41a-c were then reacted with the appropriate isocyanates or isothiocyanates 42d-z, 42aa-ee, 35c and 37a-c42 to produce the corresponding protected (bis)ureas or (bis)thioureas 43d-z and 43aa-ee, followed by acid removal of the N-Boc protection groups (HCl in EtOAc)44 to afford the desired urea or thiourea products 3-30.
Scheme 2.

In order to access isosteric derivatives of the (bis)biguanide lead compound 2d, the synthetic route outlined in Scheme 3 was devised. Initially, primary amines of general structure 44 were reacted with N-chlorocarbonyl isocyanate 45,41, 46 with the intention of forming the alkyl N-chlorocarbonylurea 46. However, this reaction could not be controlled, even at low temperature, to produce the monoalkylated derivative 46, but immediately formed the bis-alkylated (bis)biguanide 47. In order to form the desired monoalkylated product, it was necessary to use a less reactive secondary amine in the initial step. Thus diphenylamine 48 was added to N-chlorocarbonyl isocyanate 45,41, 46 and the mixture was stirred for 20 minutes to form a mixture of 49 and the bis-alkylated product 50. Addition of compounds 41a, b or c40 to this reaction mixture in the presence of triethylamine produced the bis-N-Boc-protected precursors 51-53, which were separated from 50 by silica gel chromatography. Acid-catalyzed removal of the N-Boc protecting groups44 in 51-53 then afforded the desired target compounds 31-33. Importantly, the syntheses described in Schemes 1-3 can be adapted to produce a wide variety of analogues with chemical diversity in the length of the alkyl chains, and in the terminal alkyl- or aralkyl substituents.
Scheme 3.

Biological Evaluation
The ability of the target (bis)ureas and (bis)thioureas to inhibit LSD1 was determined in an assay procedure utilizing the recombinant human enzyme. Expression and purification of LSD1 were conducted as previously reported.10, 25 Enzymatic activity of LSD1 in the presence of target compounds was determined using luminol-dependent chemiluminescence to measure the production of H2O2, as previously described.25, 33 The results of these experiments are summarized in Table 1, and graphically in Figure 2. As previously observed, compound 2d at 10 μM reduced LSD1 activity by 82.9%. Among the 31 urea and thiourea isosteres 3-33, six compounds were essentially inactive (i.e. produced <20% inhibition), while 11 analogues (ureas 4 and 5, thioureas 6, 10, 18, 25, 26, 27, 29 and 30 and the disubstituted carbamoylurea 31) reduced LSD1 activity by 50% or greater at 10 μM concentration (Figure 2). The three most effective LSD1 inhibitors, compounds 25-27, were chosen for additional studies as outlined below. Subsequent experiments were conducted in the Calu-6 human anaplastic non-small cell lung carcinoma line because it has a highly reproducible response to epigenetic modulation, and because it is known that various tumor suppressor genes are silenced in this line. In order for synthetic analogues to be effective at the cellular level, any observed decreases in cellular LSD1 activity should be accompanied by an increase in global H3K4me1 and H3K4me2 content. Thus, the ability of compounds 25, 26 and 27 to produce increases in global H3K4me1 and H3K4me2 levels was measured as previously described.10 The results of these studies are shown in Figure 3. At 24 hours, analogues 25 and 27 produced significant increases in both H3K4me1 (Figure 3, Panel A) and H3K4me2 (Figure 3, Panel B), while analogue 26 induced a significant increase in H3K4me1, but decreased the relative amount of H3K4me2. A similar pattern was observed at 48 hours (Figure 3, Panels C and D). Compound 25 produced the most dramatic increases in H3K4me1 and H3K4me2 at both 24 and 48 hours. The reduction in H3K4me2 and corresponding increase in H3K4me1 by 26 at both 24 and 48 hours cannot be readily explained, and is the subject of continuing investigation. However, this anomolous finding seem to correlate with the observed cytotoxicity of 26 (see below). These data strongly suggest that intracellular inhibition of LSD1 by 25-27 leads to significant increases in methylation at the H3K4 chromatin mark. It is noteworthy that in HCT116 human colon tumor cells, compounds 25-27 all produced at least a 2-fold increase in global H3K4me2 (data not shown), with the most effective analogue being compound 25 (17.4-fold increase).
Figure 2.
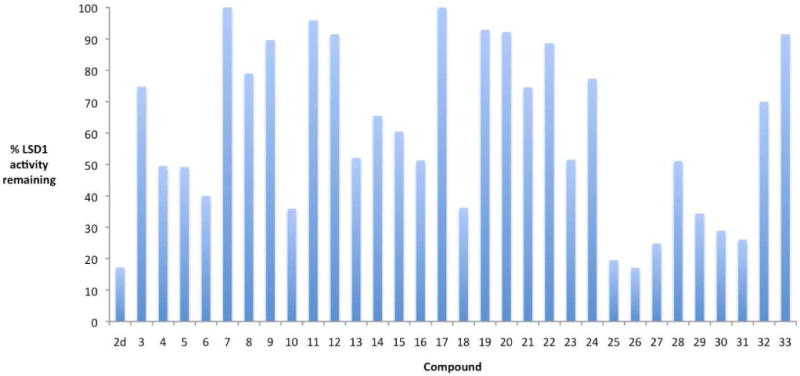
Effect of compounds 2d and 3-33 on LSD1 activity in vitro. Percent of LSD1 activity remaining was determined following 24-hour treatment with 10 μM of each test compound as determined by the luminol-dependent chemiluminescence method.
Figure 3.
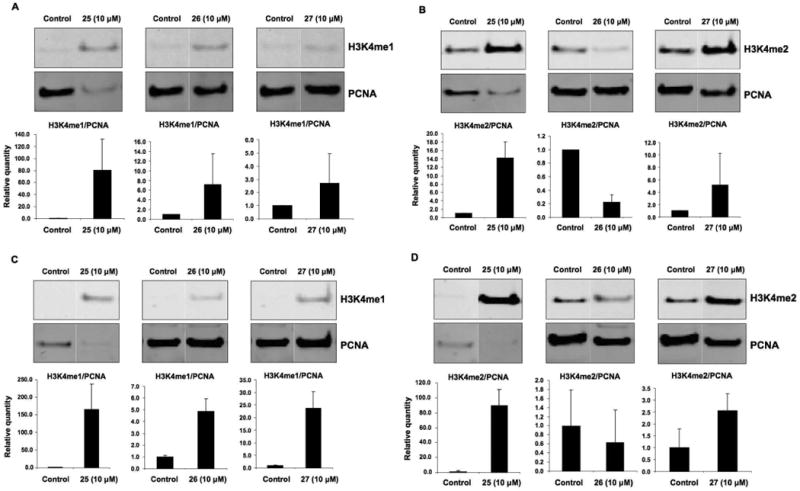
Effect of compounds 25-27 on the expression of global H3K4me1 and H3K4me2. Calu-6 human anaplastic non-small cell lung carcinoma cells were treated with a 10 μM concentration of 25, 26 or 27 for 24 h (panel A and B) or 48 h (panel C and D) as described in the methods section. Panel A and C shows global H3K4me1 expression and panel B and D shows global H3K4me2 expression. Proliferating cell nuclear antigen (PCNA) was used as a loading control. Shown are Western blot images from a single representative experiment performed in triplicate. Relative protein expression levels were determined by quantitative Western analysis using the Odyssey infrared detection system shown as bar graphs. The results represent the mean of three treatments ± SD. The protein expression level for control samples was set to a value of 1.
The ability of compounds 25-27 to induce the re-expression of aberrantly silenced tumor suppressor genes in vitro was next measured using the Calu-6 human lung carcinoma cell line. The tumor suppressor genes SFRP2 and GATA4 were chosen because they are known to be under expressed in human lung cancer, and because they are thought to play a role in tumorigenesis when silenced. Thus, the genes coding for these proteins are well-documented LSD1 targets. Cells were treated for 24 hours with either a 5 or 10 μM concentration of 25, 26 or 27, after which the levels of secreted frizzle-related protein (SFRP) 2, a soluble modulator of Wnt signaling, and the zinc-finger transcription factor GATA4, were determined by quantitative PCR (qPCR). The results of these studies are shown in Figure 4. All three compounds produced increases in SFRP2 expression that appeared to be dose dependent for 25 and 27 (Figure 4A). Compound 27 produced the largest increase in SFRP2 expression at 10 μM (4.8-fold increase). Compounds 25 and 26 did not produce significant increases in GATA4 levels at 5 and 10 μM (Figure 4B), and compound 27 induced a 1.3-fold increase in GATA4 mRNA at 10 μM, and had no significant effect at 5 μM (Figure 3B). The increase in GATA4 mRNA caused by 10 μM 27 is reproducible, but is not statistically significant (P > 0.05).
Figure 4.
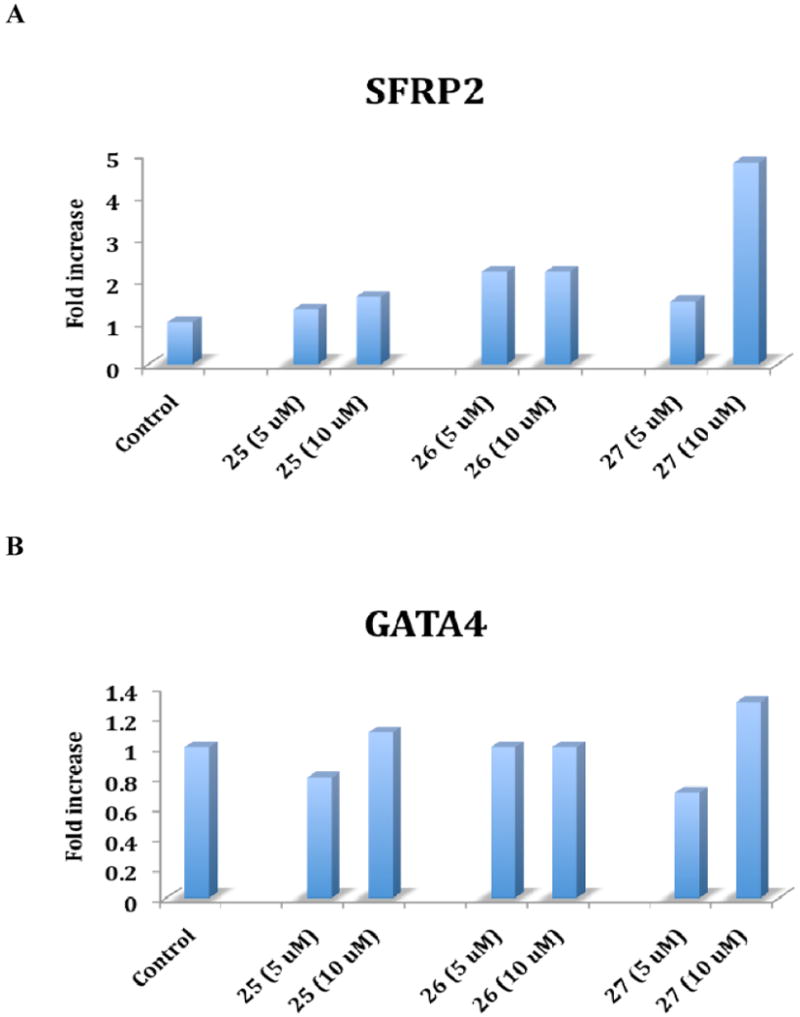
Effect of compounds 25-27 on the re-expression of secreted frizzle-related protein 2 (SFRP2, Panel A) and the transcription factor GATA4 (Panel B) mRNA. Calu-6 human anaplastic non-small cell lung carcinoma cells were treated with either a 5 or 10 μM concentration of 25, 26 or 27 for 24 hours as described in the methods section. cDNA was then synthesized from mRNA, amplified and measured by qPCR. Each data point is the average of 3 determinations that differed in all cases by 5% or less.
The (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) reduction assay was used to determine the effects of compounds 25-27 on cell viability in the Calu-6 cell line. Cells were treated with increasing concentrations of each test compound for 96 hours prior to measurement of cell viability, and growth inhibition (GI50) values were then determined from the resulting dose-response curve. As seen in Figure 5, compounds 25, 26 and 27 produced moderate reduction in cell viability, with GI50 values of 10.3, 38.3 and 9.4 μM, respectively.
Figure 5.
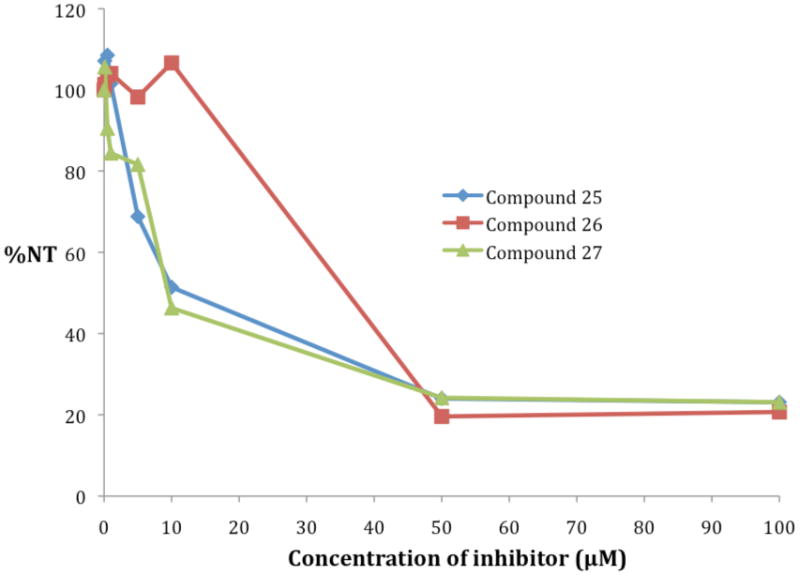
Effect of compounds 25-27 on Calu-6 human anaplastic non-small cell lung carcinoma cell viability as measured by standard MTS assay. Cells were treated with increasing concentrations of each test compound for 96 hours prior to measurement of cell viability. %NT refers to the percent of viable cells remaining at time T (96 hours) as compared to the number of cells seeded, N0. Each data point is the average of 3 determinations that differed in all cases by 5% or less.
Discussion
The potential LSD1 inhibitors 3-33 were synthesized using pathways that are facile and relatively inexpensive, and that can be used to introduce chemical diversity into the resulting urea and thiourea analogues, thus making them suitable for generation of a library of related ureas and thioureas. Our initial series of guanidine and biguanide derivatives25 represented the first novel small molecule inhibitors of LSD1 with potential for development as therapeutic agents. The current studies suggest that replacement of the imine NH functionality of the terminal guanidine in 1c with oxygen or sulfur is an allowable isosteric change, and active analogues in both the urea and thiourea series were identified (Figure 2). However, the sulfur isosteric replacement is likely more acceptable, since the 6 best LSD1 inhibitors (6, 10, 18, and 25-27) were all thioureas. A more bulky aromatic substituent on the terminal nitrogen, as in 25-27, appears to impart greater activity than the smaller alkyl or benzyl substituents found in 6, 10 and 18. There did not appear to be predictable differences in activity between analogues with 3, 4 or 7 carbon central chains, suggesting that this parameter may not have a great influence on inhibition of the enzyme. This is especially apparent among 25-27 (terminal N-substituent = 2,2-diphenylethyl), which have 7, 4 and 3 carbon central regions, respectively, but vary in activity by less than 5%. However, by contrast, among compounds 28-30 (terminal N-substituent = 1,1-diphenylmethyl), inhibitory potency did appear to be proportional to the length of the internal carbon chain. In addition to the urea and thiourea derivatives, the carbamoylurea 31, designed as an analogue of 2d, also produces potent inhibition of LSD1 (73.9% at 10 μM). Additional analogues will need to be synthesized and evaluated to generate a more accurate set of structure/activity relationships for this series of compounds.
The inhibitory effects of 25-27 on LSD1 (Figure 2), combined with the observed methylation levels at the H3K4 chromatin mark (Figure 3A-D) strongly suggest that LSD1 is inhibited in the Calu-6 tumor cell line, resulting in increases in the substrates H3K4me1 and H3K4me2. The anomolous reduction in H3K4me2 at 24 and 48 hours caused by 26 are unexpected, and have yet to be explained. In future studies, we will attempt to elucidate the mechanism of this effect. In addition, the effects of 25-27 on other histone demethylases, including LSD2,47 the Jumonji C (JmjC) domain-containing demethylases10, 11, 17, 18 and the recently discovered JmjC demethylasee PHF848, 49 need to be determined. Additional experiments are being undertaken to determine the inhibitor selectivity of these analogues.
Compounds 25-27 were next evaluated for the ability to induce the re-expression of SFRP2 and GATA4 mRNA, as determined by qPCR from treated Calu-6 human lung carcinoma cells. In the case of SFRP2, all three analogues induced increases of the protein between 1.3- and 4.8-fold (Figure 4A). These increases appeared to be dose-dependent, except in the case of 26, which induced same level of SFRP2 expression at both 5 and 10 μM. The order of potency in this regard was 27 > 26 > 25. Compound 27 produced 1.3-fold increase in GATA4 expression at 10 μM that was not statistically significant, and 25-27 at all other concentrations produced no effect on GATA4 mRNA. The observed increases in SFRP2 re-expression following treatment with 25-27, and the increase in GATA4 re-expression induced by 10 μM 27, are consistent with the previously reported effects of the parent compounds 1c and 2d.25 The disparity in the ability of 25-27 to induce SFRP2 expression, but not GATA4 expression, suggests that LSD1 inhibition may have variable effects at different gene promoters. A more detailed account of the effects of 3-33 on these and other transcription factors will be reported in a subsequent manuscript.
As discussed above, compounds 25-27 proved to be only moderately cytotoxic in the Calu-6 non-small cell lung carcinoma line in vitro. Compounds 25 and 27 produced the most prominent reduction in cell viability, exhibiting GI50 values of 10.3 and 9.4 μM, respectively. These values are comparable to the GI50 value for other epigenetic modulators, such as the polyaminohydroxamic acid and polyaminobenzamide HDAC inhibitors developed in our laboratory,50, 51 and the parent compound 2d. In addition, these GI50 values are in the range of the histone deacetylase (HDAC) inhibitor MS-275, as measured in three colon tumor cell lines.52 Compound 26 was significantlty less cytotoxic, exhibiting a GI50 value of 38.3 μM. Our data suggests that decreases in H3K4me2 at 24 and 48 hours and/or minimal effects on the re-expression of SFRP2 and GATA4 by 26 could account for this reduced cytotoxicity. It is important to note that epigenetic modulators such as those mentioned above are generally used in combination with traditional cytotoxic agents, and serve to restore the ability of transformed cells to undergo apoptosis.53 As such, cytotoxicity is less of an issue, as long as the compound produces epigenetic effects in tumor cells that can be exploited by traditional cytotoxic agents. We have recently shown that the LSD1 inhibitor 2d alone has little effect in vivo on tumor cell growth in an HCT116 human colon carcinoma mouse xenograft model, but acts synergistically to limit tumor growth in combination with the DNA methyltransferase inhibitor 5-azacytidine.54 Additional studies are now being conducted to determine whether isosteres of 2d such as 25-27 also produce a synergistic effect on tumor cell growth in vivo. Additional biological studies, as well as the synthesis and evaluation of additional LSD1 inhibitors in this and other compound libraries, is an ongoing effort in our laboratory.
Experimental Section
All reagents and dry solvents were purchased from Aldrich Chemical Co. (Milwaukee, WI), Sigma Chemical Co. (St. Louis, MO) or Acros Chemical (Chicago, IL) and were used without further purification except as noted below. Pyridine was dried by passing it through an aluminum oxide column and then stored over KOH. Triethylamine was distilled from potassium hydroxide and stored in a nitrogen atmosphere. Methanol was distilled from magnesium and iodine under a nitrogen atmosphere and stored over molecular sieves. Methylene chloride was distilled from phosphorus pentoxide and chloroform was distilled from calcium sulfate. Tetrahydrofuran was purified by distillation from sodium and benzophenone. Dimethyl formamide was dried by distillation from anhydrous calcium sulfate and was stored under nitrogen. Preparative scale chromatographic procedures were carried out using E. Merck silica gel 60, 230-440 mesh. Thin layer chromatography was conducted on Merck precoated silica gel 60 F-254. Ion exchange chromatography was conducted on Dowex 1×8-200 anion exchange resin. Compounds 41a-c 40, 55 were synthesized as previously described.
All 1H- and 13C-NMR spectra were recorded on a Varian Mercury 400 mHz spectrometer, and all chemical shifts are reported as δ values referenced to TMS or DSS. Infrared spectra were recorded on a Jasco FT-IR spectrophotometer and are referenced to polystyrene. In all cases, 1H-NMR, 13C-NMR and IR spectra were consistent with assigned structures. Mass spectra were recorded on a Kratos MS 80 RFA (EI and CI) or Kratos MS 50 TC (FAB) mass spectrometer. Prior to biological testing, target molecules 3-33 were determined to be 95% pure or greater by HPLC chromatography using an Agilent Series 1100 high-performance liquid chromatograph fitted with a C18 reversed-phase column.
Synthetic H3K4me2 peptides were purchased from Millipore (Billerica, MA). Calu-6 cells were maintained in RPMI medium, both supplemented with 10% fetal bovine serum (Gemini Bio-Products, Woodland, CA) and grown at 37°C in 5% CO2 atmosphere.
3,3-Diphenylpropylisocyanate (35c)
A 4.24 g (0.020 mol) portion of 3,3-diphenylpropylamine was dissolved in 90 mL of dry toluene in a 250 mL round-bottomed flask under a nitrogen atmosphere, and triphosgene (2.98 g, 0.010 mol) was added to the reaction mixture. The reaction mixture was heated under reflux for 5 h and then cooled to room temperature, at which time an additional 0.5 g of triphosgene was added. The reaction was then stirred for an additional 18 h at room temperature. During this time, the formation of product was monitored by TLC using hexane: ethyl acetate (3:1). When the reaction was complete, activated charcoal (0.50 g) was carefully added into reaction mixture to decolorize the solution, which was stirred for 30 min and filtered. The filtrate was concentrated under reduced pressure to give a light pale yellow semi-solid. A 100 mL portion of n-hexane/ethyl ether(1:1 ratio) was then added, and the mixture was stirred for 15 minutes. The solution was filtered and concentrated to afford 4.23 g of viscous material. The crude product was purified by flash chromatography on silica gel eluted with dichloromethane to furnish 3,3-diphenylpropylisocyanate 35c as a white solid (1.31 g, 28% yield). 1H NMR (CDCl3): δ 7.38-7.10 (m, 10H, Ar-H), 4.09 (t, 1H, J = 7.2 Hz, CHPh2), 3.27 (t, 2H, J = 6.4 Hz, CH2NCS), 2.36 (m, 2H, CH2CH2); 13C NMR (CDCl3): δ 143.69, 128.94, 128.01, 126.85 (Ar-C), 48.14, 41.51, 36.87 (CH and CH2).
General procedure for preparation of isothiocyanates 37a-c
3,3-Diphenylpropylisothiocyanate (37c)
In a 250 mL round-bottomed flask under a nitrogen atmosphere, 3,3-diphenylpropylamine 34c (2.10 g, 0.010 mol) was dissolved in 40 mL of freshly distilled THF, 3.64 g (5.0 mL, 0.036 mol) of triethylamine was added, and the mixture was cooled to 5°C in an ice bath. Carbon disulfide (0.76 g, 0.96 mL, 0.10 mol) was then added to the reaction mixture via syringe over 20 min. Following addition of carbon disulfide, the mixture was stirred an additional 30 min, warmed to room temperature and allowed to stir a further 2h. A 1H NMR of an aliquot (after removing the solvent in vacuo) indicated that conversion to the dithiocarbamate salt 36c was complete. 1H NMR (DMSO-d6): δ 8.46 (t, 1H, NH), 7.34-7.12 (m, 8H, Ar-H), 7.06 (t, 2H, Ar-H), 3.94 (t, 1H, CHPh2), 3.34 (m, 2H, CH2NCS), 3.04 (q, 6H, NCH2CH3), 2.24 (m, 2H, CH2CH2), 1.20 (t, 6H, NCH2CH3).
The reaction mixture from above was re-cooled in an ice bath, 2.38 g of tosyl chloride (0.012 mol) was added, and the reaction mixture was allowed to stir for 30 min at 5°C. It was then warmed to room temperature and stirred for an additional 3h. The solvent was removed in vacuo, the reaction was partitioned between 40 mL of 1.0 N HCl and 150 mL of Et2O, and the two-phased mixture was stirred for 10 min. The organic layer was separated and the aqueous layer was extracted with a 100 mL portion of Et2O. The combined organic layers was dried over Na2SO4, and concentrated to produce a viscous oil that solidified during vacuum drying. The product was purified by flash chromatography on silica gel (eluted with CH2Cl2) to give 37c as a white solid (1.48 g, 53% based on 34c, TLC Rf: 0.45 (n-hexane/EtOAc, 9:1). 1H NMR (CDCl3): δ 7.32-7.19 (m, 10H, Ar-H), 4.08 (t, 1H, J = 8.0 Hz, CHPh2), 3.44 (t, 2H, J = 6.8 Hz, CH2NCS), 2.41 (m, 2H, CH2CH2); 13C NMR (CDCl3): δ 143.17, 129.08, 127.97, 126.99 (Ar-C), 48.12, 43.66, 35.69 (CH and CH2).
1,1-Diphenylmethylisothiocyanate (37a)
Isothiocyanate 37a was prepared from 1,1-diphenylethylamine 34a and carbon disulfide using the procedure described above for the synthesis of 37c. The product was isolated as a white solid in 70% yield. TLC Rf: 0.90 (n-hexane/MeCO2Et, 4:1). 1H NMR (CDCl3): δ 7.40-7.31 (m, 10H, Ar-H), 5.99 (s, 1H, CHPh2); 13C NMR (CDCl3): δ 139.43, 129.18, 128.57, 126.85 (Ar-C), 64.82 (CH).
2,2-Diphenylethylisothiocyanate (37b)
Isothiocyanate 37b was prepared from 1,1-diphenylethylamine 34a and carbon disulfide using the procedure described above for the synthesis of 37c. The product was isolated as a white solid in 87% yield. 1H NMR (DMSO-d6): δ 7.36-7.29 (m, 8H, Ar-H), 7.24-7.20 (t, 2H, J = 7.2Hz, Ar-H), 4.45 (t, 1H, J = 8.0 Hz, CHPh2), 4.34 (d, 2H, J = 7.6 Hz, CH2NCS); 13C NMR (DMSO-d6): δ 1 41.64, 129.31, 128.53, 127.67 (Ar-C), 51.18, 48.95 (CH and CH2).
General procedure for preparation of N-Boc protected (bis)thioureas
1,12-bis-{3-[1-(benzyl)thioureado]}-4,9-[N-(tertbutyl)oxycarbonyl)]-4,9-diazadodecane (43d)
In a 100 mL round-bottom flask, a 0.3 g portion of 4,9-[N-(tertbutyl)oxycarbonyl)]-4,9-diaza-1,12-diaminododecane 41b (0.0008 mol) was dissolved in 20 mL of HPLC grade CH2Cl2 under a nitrogen atmosphere and the mixture was cooled to 0°C. A solution of benzylisothiocyanate (240 mg, 0.0016 mol) in 5 mL of CH2Cl2 was then added dropwise with stirring, and the reaction mixture was allowed to stir at room temperature for 5 h. During this time, the formation of product was monitored by TLC (CH2Cl2/MeOH/NH4OH 89:10:1). After completion of the reaction, the CH2Cl2 was removed under reduced pressure to produce a viscous colorless oil. The crude product was purified by flash chromatography on silica gel eluted with CH2Cl2/MeOH/NH4OH (94.5:5:0.5 followed by 89:10:1) to furnish pure 43d (0.46 g, 88% yield) as viscous oil. Rf: 0.46 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CDCl3): δ 7.31 (m, 10H, Ar-H), 6.31 (b, 2H, NH), 4.55 (bs, 4H, NCH2), 3.54 (bs, 4H, NCH2), 3.20 (bs, 4H, NCH2), 3.10 (bs, 4H, NCH2), 1.65 (bs, 4H, CH2CH2), 1.46 (bs, 4H, CH2CH2), 1.38 (s, 18H, C[CH3]3).
1,12-bis-{3-[1-(ethyl)thioureado]}-4,9-[N-(tertbutyl)oxycarbonyl)]-4,9-diazadodecane (43g)
Compound 43g was prepared from 375 mg of 41b (375 mg, 0.0009 mol) and ethylisothiocyanate, according to procedure described above for the synthesis of 43d to afford 43g (512 mg, 95%) as viscous oil. Rf: 0.52 (CH2Cl2/MeOH/NH4OH, 89:10:1); 1H NMR (CDCl3): δ 7.40 (b, 2H, NH), 6.00 (b, 2H, NH), 3.56 (m, 4H, NCH2), 3.34 (b, 4H, NCH2), 3.26 (b, 4H, NCH2), 3.12 (b, 4H, NCH2), 1.71 (b, 4H, CH2CH2), 1.50 (bs, 4H, CH2CH2), 1.40 (s, 18H, C(CH3)3), 1.20 (t, 6H, J = 7.2 Hz, CH3). 13C NMR (CDCl3): δ 80.36 ([CH3]3C), 46.95, 43.34, 41.19, 38.12, 28.63, 27.31, 26.16 (CH2), 14.28 (CH3).
1,12-bis-{3-[1-(propyl)thioureado]}-4,9-[N-(tertbutyl)oxycarbonyl)]-4,9-diazadodecane (43j)
Compound 43j was prepared from 260 mg of 41b (0.0007 mol) and n-propylisothiocyanate according to procedure described above for the synthesis of 43d to afford 43j (380 mg, 96%) as viscous oil. Rf: 0.51 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CDCl3): δ3.50-3.36 (b, 8H, NCH2), 3.28-3.20 (m, 8H, NCH2), 3.26 (b, 4H, NCH2) 1.78 (b, 4H, CH2CH2), 1.52 (bs, 4H, CH2CH2), 1.46 (s, 18H, C[CH3]3), 0.73 (t, 6H, J = 7.2 Hz, CH3). 13C NMR (CDCl3): δ80.36 ([CH3]3C), 46.95, 43.34, 41.19, 38.12, 28.63, 27.31, 26.16 (CH2), 14.28 (CH3).
1,15-bis-{3-[1-(benzyl)thioureado]}-4,12-[N-(tertbutyl)oxycarbonyl)]-4,12-diazapentadecane (43k)
Compound 43k was prepared from 220 mg of 41c (0.0005 mol) and benzylisothiocyanate according to procedure described above for the synthesis of 43d to afford 43k (360 mg, 96%) as viscous oil; 1H NMR (CDCl3): δ 7.39-7.30 (m, 10H, Ar-H), 4.76 (b, 4H, CH2Ph), 3.46 (b, 4H, NCH2), 3.18 (m, 8H, NCH2), 1.52 (b, 4H, CH2CH2), 1.54 (b, 4H, CH2CH2), 1.44 (s, 18H, C(CH3)3), 1.28 (b, 6H, CH2CH2).
1,11-bis-{3-[1-(benzyl)thioureado]}-4,8-[N-(tertbutyl)oxycarbonyl)]-4,8-diazaundecane (43n)
Compound 43n was prepared from 291 mg of 41a (0.0008 mol) and benzylisothiocyanate according to the procedure described above for 43d to afford 43n (373 mg, 73%) as viscous oil. Rf: 0.87 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CDCl3): δ 7.35 (m, 10H, Ar-H), 4.58 (bs, 4H, N-CH2), 3.58 (bs, 4H, N-CH2), 3.21 (b, 4H, N-CH2), 3.10 (b, 4H, N-CH2), 1.72 (b, 6H, CH2CH2), 1.40 (s, 18H, C[CH3]3).
1,11-bis-{3-[1-(propyl)thioureado]}-4,8-[N-(tertbutyl)oxycarbonyl)]-4,8-diazaundecane (43r)
Compound 43r was prepared from 291 mg of 41a (0.0008 mol) and n-propylisothiocyanate according to the procedure described above for 43d to afford 43r (379 mg, 86%) as viscous oil. Rf: 0.57 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CDCl3): δ 7.29 (bs, 1H, NH), 6.44 (s, 2H, NH), 3.43 (bs, 4H, N-CH2), 3.01-3.15 (b, 12H, N-CH2), 1.61 (bs, 6H, CH2CH2), 1.47 (m, J = 7.2 Hz, 4H, CH2CH3), 1.31 (s, 18H, C[CH3]3), 0.81 (t, J = 7.2 Hz, 6H, CH2CH3).
1,11-bis-{3-[1-(n-ethyl)thioureado]}-4,8-[N-(tertbutyl)oxycarbonyl)]-4,8-diazaundecane (43s)
Compound 43s was prepared from 291 mg of 41a (0.0008 mol) and ethylisothiocyanate according to the procedure described above for 43d to afford 43s (347 mg, 83%) as viscous oil. Rf: 0.72 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CDCl3): δ 7.24 (bs, 2H, NH), 6.22 (bs, 2H, NH), 3.49 (bs, 4H, CH2N), 3.29 (bs, 4H, N-CH2), 3.20 (b, 4H, N-CH2), 3.07 (b, 4H, N-CH2), 1.62-1.74 (b, 6H, CH2CH2), 1.37 (s, 18H, C[CH3]3), 1.14 (t, 6H, CH2CH3).
1,11-bis-{3-[1-(3,3-diphenylpropyl)thioureado]}- 4,8-[N-(tertbutyl)oxycarbonyl)]-4,8-diazaundecane (43u)
Compound 43u was prepared from 155 mg of 41a (0.0004 mol) and 37c according to procedure described above for the synthesis of 43d to afford 43u (290 mg, 81%) as a white solid. Rf: 0.44 (CH2Cl2/MeOH/NH4OH, 89:10:1); 1H NMR (CDCl3): δ 7.29-7.15 (m, 22H, Ar-H, and NH), 5.88 (b, 2H, NH), 4.04 (t, 2H, J = 7.6 Hz, CHPh2), 3.53 (b, 4H, NCH2), 3.28 (b, 4H, NCH2), 3.23 (b, 4H, NCH2), 3.12 (b, 8H, NCH2), 2.36 (q, 4H, J = 8.0 Hz, NCH2), 1.70 (m, 2H, CH2CH2), 1.47 (b, 4H, CH2CH2), 1.40 (s, 20H, C[CH3]3).
1,12-bis-{3-[1-(3,3-diphenylpropyl)thioureado]}- 4,9-[N-(tertbutyl)oxycarbonyl)]-4,9-diazadodecane (43w)
Compound 43w was prepared from 161 mg of 41b (0.0004 mol) and 37c according to procedure described above for the synthesis of 43d to afford 43w (322 mg, 89%) as a white solid. Rf: 0.52 (CH2Cl2/MeOH/NH4OH, 89:10:1); 1H NMR (CDCl3): δ 7.25-7.16 (m, 22H, Ar-H, and NH), 5.88 (b, 2H, NH), 4.02 (t, 2H, J = 8.0 Hz, CHPh2), 3.17 (b, 8H, NCH2), 3.09 (b, 4H, NCH2), 2.37 (q, 4H, J = 7.6 Hz, CH2CH), 1.76-1.65 (m, 8H, CH2CH2), 1.41 (s, 18H, C[CH3]3).
1,15-bis-{3-[1-(3,3-diphenylpropyl)thioureado]}- 4,12-[N-(tertbutyl)oxycarbonyl)]-4,12-diazapentadecane (43y)
Compound 43y was prepared from 178 mg of 41c (0.0004 mol) and 37c according to procedure described above for the synthesis of 43d to afford 43y (305 mg, 80%) as a white solid. Rf: 0.57 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CDCl3): δ 7.28-7.15 (m, 20H, Ar-H), 5.88 (b, 2H, NH), 4.02 (t, 2H, J = 8.0 Hz, CHPh2), 3.54 (b, 4H, NCH2), 3.28 (b, 4H, NCH2), 3.23 (b, 4H, NCH2), 3.08 (t, 4H, J = 7.2 Hz, NCH2), 2.36 (q, 4H, J = 7.6 Hz, CH2CH), 1.69 (bs, 4H, CH2CH2), 1.50 (b, 4H, CH2CH2), 1.40 (s, 18H, C[CH3]3), 1.28 (m, 6H, CH2CH2).
1,15-bis-{3-[1-(2,2-diphenylethyl)thioureado]}- 4,12-[N-(tertbutyl)oxycarbonyl)]-4,12-diazapentadecane (43z)
Compound 43z was prepared from 223 mg of 41c (0.0005 mol) and 37b according to procedure described above for the synthesis of 43d to afford 43z (288 mg, 79%) as a white solid. Rf: 0.68 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CDCl3): δ 7.31-7.19 (m, 20H, Ar-H), 5.75 (b, 2H, NH), 4.28 (b, 2H, CHPh2), 4.02 (b, 4H, NCH2), 3.54 (b, 4H, NCH2), 3.25 (b, 4H, NCH2), 3.09 (t, 4H, J = 7.2 Hz, NCH2), 1.69 (bs, 4H, CH2CH2), 1.49 (b, 4H, CH2CH2), 1.40 (bs, 18H, C[CH3]3), 1.24 (m, 6H, CH2CH2).
1,12-bis-{3-[1-(2,2-diphenylethyl)thioureado]}- 4,9-[N-(tertbutyl)oxycarbonyl)]-4,9-diazadodecane (43aa)
Compound 43aa was prepared from 161 mg of 41b (0.0004 mol) and 37b according to procedure described above for the synthesis of 43d to afford 43aa (295 mg, 84%) as a white solid. Rf: 0.60 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CDCl3): δ 7.32-7.20 (m, 20H, Ar-H), 5.77 (b, 2H, NH), 4.29 (b, 2H, CHPh2), 4.02 (b, 4H, NCH2), 3.56 (bs, 4H, NCH2), 3.26 (bs, 4H, NCH2), 3.12 (bs, 4H, NCH2), 1.70 (b, 4H, CH2CH2), 1.48 (b, 4H, CH2CH2), 1.41 (s, 18H, C[CH3]3).
1,11-bis-{3-[1-(2,2-diphenylethyl)thioureado]}- 4,9-[N-(tertbutyl)oxycarbonyl)]-4,9-diazadodecane (43bb)
Compound 43bb was prepared from 193 mg (0.0005 mol) of 41b and 37b according to procedure described above for the synthesis of 43d to afford 43bb (350 mg, 80%) as a white solid. Rf: 0.63 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CDCl3): δ 7.32-7.20 (m, 20H, Ar-H), 5.77 (bs, 2H, NH), 4.29 (bs, 2H, CHPh2), 4.02 (bs, 4H, NCH2), 3.56 (bs, 4H, NCH2), 3.26 (bs, 4H, NCH2), 3.12 (t, 4H, J = 7.2 Hz, NCH2), 1.71 (b, 4H, CH2CH2), 1.41 (b, 20H, CH2 and C[CH3]3).
1,11-bis-{3-[1-(1,1-diphenylmethyl)thioureado]}- 4,8-[N-(tertbutyl)oxycarbonyl)]-4,8-diazaundecane (43cc)
Compound 43cc was prepared from 192 mg of 41a (0.0005 mol) and 37a according to procedure described above for the synthesis of 43d to afford 43cc as a white solid (350 mg, 83%), Rf: 0.63 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CDCl3): δ 7.34-7.27 (m, 20H, Ar-H), 6.43 (d, 2H, J = 5.2 Hz, NCH), 6.02 (b, 2H, NH), 3.52 (d, 4H, J = 5.2 Hz, NCH2), 3.06 (m, 8H, NCH2), 1.66 (bs, 6H, CH2CH2), 1.36 (bs, 18H, C[CH3]3).
1,12-bis-{3-[1-(1,1-diphenylmethyl)thioureado]}- 4,9-[N-(tertbutyl)oxycarbonyl)]-4,9-diazadodecane (43dd)
Compound 43dd was prepared from 201 mg of 41b (0.0005 mol) and 37a according to procedure described above for the synthesis of 43d to afford 43dd (380 mg, 89%) as white solid. Rf: 0.60 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CDCl3): δ 7.40 (b, 2H, NH), 7.34-7.27 (m, 20H, Ar-H), 6.43 (d, 2H, J = 5.2 Hz, NCH), 6.02 (b, 2H, NH), 3.52 (d, 4H, J = 5.2 Hz, NCH2), 3.06 (bs, 8H, NCH2), 1.63 (m, 4H, CH2CH2), 1.42 (bs, 4H, CH2CH2), 1.36 (s, 18H, C[CH3]3).
1,15-bis-{3-[1-(1,1-diphenylmethyl)thioureado]}- 4,12-[N-(tertbutyl)oxycarbonyl)]-4,12-diazapentadecane (43ee)
Compound 43ee was prepared from 223 mg of 41c and 37a according to procedure described above for the synthesis of 43d to afford 43ee (408 mg, 91%) as a white solid. Rf: 0.77 (CH2Cl2/MeOH/NH4OH, 89:10:1); 1H NMR (CDCl3): δ 7.45 (b, 2H, NH), 7.33-7.26 (m, 20H, Ar-H), 6.41 (d, 2H, J = 2.8 Hz, NCH), 6.03 (b, 2H, NH), 3.51 (m, 4H, NCH2), 3.04 (m, 8H, NCH2), 1.54 (bs, 4H, CH2CH2), 1.45 (b, 4H, CH2CH2), 1.35 (bs, 18H, C[CH3]3), 1.23 (m, 6H, CH2CH2).
General procedure for preparation of N-Boc protected (bis)ureas
1,12-bis-{3-[1-(benzyl)ureado]}-4,9-[N-(tertbutyl)oxycarbonyl)]-4,9-diazadodecane (43e)
In a 100 mL round-bottom flask, a 0.35 g portion of 4,9-[N-(tertbutyl)oxycarbonyl)]-4,9-1,12-diamino-diazadodecane 41b (0.0009 mol) was dissolved in 20 mL of HPLC grade CH2Cl2 under a nitrogen atmosphere and the mixture was cooled to 0°C. A solution of benzylisocyanate (0.235 g, 0.0018 mol) in 5 mL of CH2Cl2 was then added dropwise with stirring, and the reaction mixture was allowed to stir at room temperature for 24 h. During this time, the formation of product was monitored by TLC (CH2Cl2:MeOH:NH4OH 89:10:1). When the starting material had been consumed, the CH2Cl2 was removed under reduced pressure to afford a viscous colorless material. The crude product was purified by flash chromatography on silica gel eluted with CH2Cl2:MeOH:NH4OH (97:2.5:0.5 followed by 94.5:5.0:0.5) to furnish pure 43e (0.50 g, 86% yield) as viscous oil. Rf: 0.54 (CH2Cl2/MeOH/NH4OH, 89:10:1); 1H NMR (CDCl3): δ 3.20-3.02 (m, 16H, NCH2), 1.64 (b, 4H, CH2CH2), 1.48 (b, 4H, CH2CH2), 1.43 (s, 18H, C[CH3]3), 1.11 (t, 6H, J = 6.4 Hz, CH3).
1,12-bis-{3-[1-(ethyl)ureado]}-4,9-[N-(tertbutyl)oxycarbonyl)]-4,9-diazadodecane (43f)
Compound 43f was prepared from 368 mg of 41b (0.0009 mol) and ethylisocyanate according to the procedure described above for 43e to afford 43f (480 mg, 96%) as viscous oil. Rf: 0.54 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CDCl3): δ 3.20-3.02 (m, 16H, NCH2), 1.64 (b, 4H, CH2CH2), 1.48 (b, 4H, CH2CH2), 1.43 (s, 18H, C[CH3]3), 1.11 (t, 6H, J = 6.4 Hz, CH3).
1,15-bis-{3-[1-(benzyl)ureado]}-4,12-[N-(tertbutyl)oxycarbonyl)]-4,12-diazapentadecane (43h)
Compound 43h was prepared from 230 mg of 41b (0.0005 mol) and benzylisocyanate according to the procedure described above for 43e to afford 43h (350 mg, 96%) as viscous oil. Rf: 0.50 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CD3OD): δ 7.30 (m, 10H, Ar-H), 4.29 (s, 4H, CH2Ph), 3.21-3.15 (m, 8H, NCH2), 3.12 (t, 4H, J = 7.2 Hz, NCH2), 1.70 (bs, 4H, CH2CH2), 1.50 (bs, 4H, CH2CH2), 1.44 (s, 18H, C[CH3]3), 1.32 (bs, 6H, CH2CH2).
1,12-bis-{3-[1-(propyl)ureado]}-4,9-[N-(tertbutyl)oxycarbonyl)]-4,9-diazadodecane (43i)
Compound 43i was prepared from 260 mg of 41c (0.0005 mol) and n-propylisocyanate according to the procedure described above for 43e to afford 43i (356 mg, 94%) as viscous oil. Rf: 0.54 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CD3OD): δ 3.22 (m, 8H, NCH2), 3.09 (t, 4H, J = 6.4 Hz, NCH2), 3.05 (t, 4H, J = 7.6 Hz, NCH2), 1.70 (b, 4H, CH2CH2), 1.50 (m, 8H, CH2CH2), 1.45 (s, 18H, C[CH3]3), 0.90 (t, 6H, J = 7.6 Hz, CH3).
1,15-bis-{3-[1-(benzyl)ureado]}-4,12-[N-(tertbutyl)oxycarbonyl)]-4,12-diazapentadecane (43l)
Compound 43l was prepared from 225 mg of 41c (0.0005 mol) and ethylisocyanate according to the procedure described above for 43e to afford 43l (280 mg, 94%) as viscous oil. Rf: 0.37 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CD3OD): δ 3.28-3.12 (m, 8H, NCH2), 3.10-3.06 (m, 8H, NCH2), 1.68 (b, 4H, CH2CH2), 1.54 (b, 4H, CH2CH2), 1.44 (s, 18H, C[CH3]3), 1.30 (b, 6H, CH2CH2), 1.08 (t, 6H, J = 7.2 Hz, CH2CH2).
1,15-bis-{3-[1-(propyl)ureado]}-4,12-[N-(tertbutyl)oxycarbonyl)]-4,12-diazapentadecane (43m)
Compound 43m was prepared from 225 mg of 41c (0.0005 mol) and propylisocyanate according to the procedure described above for 43e to afford 43m (280 mg, 92%) as viscous oil. Rf: 0.35 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CD3OD): δ 3.25-3.16 (m, 8H, NCH2), 3.09-3.02 (m, 8H, NCH2), 1.68 (b, 4H, CH2CH2), 1.52 (b, 8H, CH2CH2), 1.44 (s, 18H, C[CH3]3), 1.30 (b, 6H, CH2CH2), 0.90 (t, 6H, J = 7.2 Hz, CH2CH2).
1,11-bis-{3-[1-(ethyl)ureado]}-4,8-[N-(tertbutyl)oxycarbonyl)]-4,8-diazaundecane (43o)
Compound 43o was prepared from 287 mg of 41a (0.0007 mol) and ethylisothiocyanate according to the procedure described above for 43e to afford 43o (245 mg, 62%) as viscous oil, Rf: 0.63 (CH2Cl2/MeOH/NH4OH, 89:10:1); 1H NMR (CDCl3): δ 5.59 (bs, 1H, NH), 4.60 (bs, 1H, NH), 3.08-3.31 (m, 16H, N-CH2), 1.58-1.78 (m, 6H, CH2CH2), 1.43 (s, 18H, C[CH3]3), 1.10 (t, J = 7.2 Hz, 6H, CH2CH3).
1,11-bis-{3-[1-(benzyl)ureado]}-4,8-[N-(tertbutyl)oxycarbonyl)]-4,8-diazaundecane (43p)
Compound 43p was prepared from 302 mg of 41a (0.0008 mol) and benzylisothiocyanate according to the procedure described above for 43e to afford 43p (485 mg, 95%) as viscous oil, Rf: 0.63 (CH2Cl2/MeOH/NH4OH, 89:10:1); 1H NMR (CDCl3): δ 7.10-7.30 (m, 10H, Ar-H), 4.20 (bs, 4H, PhCH2), 2.98-3.20 (m, 12H, N-CH2), 1.65 (p, 2H, CH2CH2), 1.55 (p, J = 6.4 Hz, 4H, CH2CH2), 1.39 (s, 18H, C[CH3]3).
1,11-bis-{3-[1-(n-propyl)ureado]}-4,8-[N-(tertbutyl)oxycarbonyl)]-4,8-diazaundecane (43q)
Compound 43q was prepared from 291 mg of 41a (0.0008 mol) and n-propylisothiocyanate according to the procedure described above for 43e to afford 43q (359 mg, 91%) as viscous oil. Rf: 0.63 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CDCl3): δ 5.60 (bs, 1H, NH), 4.70 (bs, 1H, NH), 3.05-3.28 (m, 16H, N-CH2), 1.60-1.78 (m, 6H, CH2CH2), 1.47 (m, J = 7.2, 4H, CH2CH3), 1.42 (s, 18H, C[CH3]3), 0.88 (t, J = 7.2 Hz, 6H, CH2CH3).
1,11-bis-{3-[1-(3,3-diphenylpropyl)ureado]}-4,8-[N-(tertbutyl)oxycarbonyl)]-4,8-diazaundecane (43t)
Compound 43t was prepared from 194 mg (0.0005 mol) of 41a and 35c according to the procedure described above for 43e to afford 43t (420 mg, 98%) as a viscous oil. Rf: 0.58 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CDCl3): δ 7.29-7.15 (m, 20H, Ar-H), 5.50 (b, 2H, NH), 3.96 (t, 2H, J = 8.0 Hz, CHPh2), 3.25 (t, 4H, J = 6.4 Hz, NCH2), 3.10 (b, 12H, NCH2), 2.23 (q, 4H, J = 7.2 Hz, NCH2), 1.72 (b, 2H, CH2CH2), 1.61 (b, 4H, CH2CH2), 1.42 (s, 18H, C[CH3]3).
1,12-bis-{3-[1-(3,3-diphenylpropyl)ureado]}-4,9-[N-(tertbutyl)oxycarbonyl)]-4,9-diazadodecane (43v)
Compound 43v was prepared from 193 mg of 41b (0.0005 mol) and 35c according to the procedure described above for 43e to afford 43v (386 mg, 92%) as viscous oil. 1H NMR (CDCl3): δ 7.29-7.13 (m, 22H, Ar-H, and NH), 5.50 (b, 2H, NH), 3.96 (t, 2H, J = 8.0 Hz, CHPh2), 3.25 (t, 4H, J = 6.4 Hz, NCH2), 3.10 (m, 12H, NCH2), 2.24 (b, 4H, CH2CH2), 1.60 (b, 4H, CH2CH2), 1.42 (s, 22H, CH2CH2 and C[CH3]3).
1,15-bis-{3-[1-(3,3-diphenylpropyl)ureado]}-4,12-[N-(tertbutyl)oxycarbonyl)]-4,12-diazapentadecane (43x)
Compound 43x was prepared from 158 mg of 41c (0.0004 mol) and 35c according to the procedure described above for 43e to afford 43x (310 mg, 95%) as viscous oil; Rf: 0.50 (CH2Cl2/MeOH/NH4OH, 89:10:1). 1H NMR (CDCl3): δ 7.30-7.12 (m, 20H, Ar-H), 5.50 (b, 2H, NH), 4.40 (b, 2H, NH), 3.97 (t, 2H, J = 7.2 Hz, CHPh2), 3.25 (t, 4H, J = 6.4Hz, NCH2), 3.10 (bs, 12H, NCH2), 2.26 (q, 4H, J = 8.0 Hz, CH2CH2), 1.60 (bs, 4H, CH2CH2), 1.42 ((s, 18H, C[CH3]3), 1.24 (bs, 6H, CH2CH2).
General procedure for preparation of N-Boc protected (bis)carbamylureas 51-53
1,12-bis-{5-[1-(N,N-diphenyl)carbamyl]ureado}-4,9-[N-(tertbutyl)oxycarbonyl)]-4,9-diazadodecane (51)
A 0.34 g portion of N,N-diphenylamine 48 (0.34 g, 0.0002 mol) in 5 mL of dry CH2Cl2 was added dropwise into a cold solution of N-chlorocarbonylisocyanate 45 (0.22 g, 0.0002 mol) in 5.0 mL of CH2Cl2, and the reaction was stirred for 30 min under N2 atmosphere. A solution of 41b (0.3 g, 0.0008 mol) and NEt3 (0.3 g, 0.0003 mol) in 10 mL of CH2Cl2 was then added via syringe, and the reaction mixture was allowed to stir at room temperature for 18 h. During this time, the progress for formation of product was monitored by TLC (CH2Cl2/MeOH/NH4OH, 89:10:1). The dichloromethane was removed under reduced pressure to produce a viscous material, which was purified by flash chromatography on silica gel eluted with CH2Cl2/MeOH/NH4OH (89:10:1) to furnish pure 51 as a white solid (140 mg, 21%). 1H NMR (CDCl3): δ 8.44 (s, 2H, NH), 7.41-7.39 (m, 8H, Ar-H), 7.29-7.23 (m, 12H, Ar-H), 6.75 (s, 2H, NH), 3.25-3.08 (m, 12H, NCH2), 1.78-1.69 (m, 6H, CH2CH2), 1.43 (s, 18H, C[CH3]3).
1,15-bis-{5-[1-(N,N-diphenyl)carbamyl]ureado}-4,12-[N-(tertbutyl)oxycarbonyl)]-4,12-diazapentadecane (52)
Compound 52 was made from 48, 45 and 41c according to the procedure described above for the synthesis of 51 to afford pure 52 (140 mg, 21%) as a viscous material. Rf: 0.88 (CH2Cl2/MeOH/NH4OH 89:10:1). 1H NMR (CDCl3): δ 8.40 (s, 2H, NH), 7.37-7.28 (m, 8H, Ar-H), 7.20-7.12 (m, 12H, Ar-H), 6.76 (s, 2H, NH), 3.23-3.06 (m, 12H, NCH2), 1.72 (m, 4H, CH2CH2), 1.39 (bs, 22H, CH2CH2 and C[CH3]3). HRMS (CSI-MS m/z) calcd for C48H62N8O8 [M+] = 878.47; found 879.40 [M+H].
1,11-bis-{5-[1-(N,N-diphenyl)carbamyl]ureado}-4,8-[N-(tertbutyl)oxycarbonyl)]-4,8-diazaundecane (53)
Compound 55 was made from 48, 45 and 41a according to the procedure described above for the synthesis of 51 to afford pure 53 (115 mg, 18%) as a white solid. Rf: 0.90 (CH2Cl2/MeOH/NH4OH, 89:10:1); 1H NMR (CDCl3): δ 8.44 (s, 2H, NH), 7.41-7.37 (m, 8H, Ar-H), 7.31-7.27 (m, 12H, Ar-H), 6.77 (s, 2H, NH), 3.30-3.12 (m, 12H, NCH2), 1.75 (t, 4H, J = 6.8 Hz, CH2CH2), 1.47 (b, 4H, CH2CH2), 1.43 (s, 18H, C[CH3]3), 1.27 (b, 6H, CH2CH2).
General procedure for cleavage of N-Boc protecting group
1,12-bis-{3-[1-(benzyl)thioureado]}-4,9-diazadodecane (3)
In a 100 mL round-bottom flask, a 0.4 g portion of 43d (402 mg, 0.0006 mol) was dissolved in 30 mL of HPLC grade EtOAc under a nitrogen atmosphere, and 4.0 mL of a 1.0 M solution of HCl in EtOAc was added. The reaction mixture was allowed to stir at room temperature for 48 h, during which time the formation of product was monitored by TLC (CH2Cl2/MeOH/NH4OH 89:10:1 or 78:20:2). The product precipitated as a white crystalline solid during the course of the reaction. When completion of the reaction was confirmed by TLC, the solvent was removed under reduced pressure to produce a white powder. The solid product was stirred with 30 mL of fresh EtOAc, and the solvent was decanted. The solid so obtained was vacuum dried to give pure 3 as a white solid (315 mg, 95% yield). An analytical sample was obtained by purification on silica gel (CH2Cl2:MeOH:NH4OH 89:10:1). 1H NMR (CD3OD): δ 7.32-7.10 (m, 10H, Ar-H), 4.67 (s, 4H, CH2Ph), 3.71 (t, 4H, J = 5.6 Hz, NCH2), 3.01 (bs, 8H, NCH2), 1.95 (m, 4H, CH2CH2), 1.78 (bs, 4H, CH2CH2). MS (CI m/z) calcd for C26H40N6S2 [M+.] = 500.28; found 501.4 [M+H].
1,12-bis-{3-[1-(benzyl)ureado]}-4,9-diazadodecane (4)
Compound 4 was prepared from 480 mg (0.0007 mol) of 43e according to procedure described above for the synthesis of 3 to afford 370 mg (94%) of 4 as a white solid; 1H NMR (D2O): δ 7.32 (m, 4H, Ar-H), 7.26 (m, 6H, Ar-H), 4.22 (s, 4H, CH2Ph), 3.16 (t, 4H, J = 6.4 Hz, NCH2), 2.88 (t, 4H, J = 7.2 Hz, NCH2), 2.81 (bs, 4H, NCH2), 1.25 (p, 4H, J = 6.4 and 7.2 Hz, CH2CH2), 1.57 (m, 4H, CH2CH2). 13C NMR (D2O): δ 160.89 (C=O), 139.83, 129.00, 127.45, 126.97 (Ar-C), 46.96, 45.12, 43.66, 36.44, 26.71, 22.92 (CH2).
1,12-bis-{3-[1-(ethyl)ureado]}-4,9-diazadodecane (5)
Compound 5 was prepared from 448 mg (0.0008 mol) of 43f according to procedure described above for the synthesis of 3 to afford 330 mg (96%) of 5 as a white solid. 1H NMR (D2O): δ 3.15 (t, 4H, J = 5.6 Hz, N-CH2), 3.05-2.98 (m, 12H, NCH2), 1.79 (p, 4H, J = 7.2 Hz, CH2CH2), 1.71 (bs, 4H, CH2CH2), 1.01 (t, 6H, J = 7.2 Hz, CH3). 13C NMR (D2O): δ 160.88 (C=O), 47.06, 45.31, 36.67, 35.27, 26.70, 23.06 (CH2), 14.63 (CH3).
1,12-bis-{3-[1-(ethyl)thioureado]}-4,9-diazadodecane (6)
Compound 6 was prepared from 470 mg (0.0008 mol) of 43g according to procedure described above for the synthesis of 3 to afford 314 mg (87%) of 6 as a white solid. 1H NMR (D2O): δ 3.51 (bs, 4H, NCH2), 3.31 (bs, 4H, NCH2), 3.06 (bs, 4H, NCH2), 1.93 (p, 4H, J = 6.4 Hz, CH2CH2), 1.75 (bs, 4H, CH2CH2), 1.12 (t, 6H, J = 6.0 Hz, CH3). 13C NMR (DMSO-d6): δ 154.38, 153.98 (C=O), 47.13, 45.00, 40.92, 26.13, 23.15 (CH2), 13.49 (CH3).
1,15-bis-{3-[1-(benzyl)ureado]}-4,12-diazapentadecane (7)
Compound 7 was prepared from 320 mg (0.0005 mol) of 43h according to procedure described above for the synthesis of 3 to afford 250 mg (95%) of 7 as a white solid. 1H NMR (D2O): δ 7.35 (m, 4H, Ar-H), 7.28 (m, 6H, Ar-H), 4.25 (s, 4H, CH2Ph), 3.18 (t, 4H, J = 5.6 Hz, NCH2), 2.88 (t, 4H, J = 7.2 Hz, NCH2), 2.81 (t, 4H, J = 8.0 Hz, NCH2), 1.76 (p, 4H, J = 7.2 Hz, CH2CH2), 1.53 (m, 4H, CH2CH2), 1.26 (bs, 6H, CH2CH2). 13C NMR (D2O): δ 160.91 (C=O), 139.83, 129.01, 127.48, 126.99 (Ar-C), 47.78, 44.97, 43.68, 36.47, 27.87, 26.71, 25.64, 25.59 (CH2).
1,12-bis-{3-[1-(n-propyl)ureado]}-4,9-diazadodecane (8)
Compound 8 was prepared from 330 mg (0.0006 mol) of 43i according to procedure described above for the synthesis of 3 to afford 228 mg (90%) of 8 as a white solid. 1H NMR (D2O): δ 3.14 (t, 4H, J = 6.4 Hz, NCH2), 3.00-2.95 (m, 12H, NCH2), 1.79 (p, 4H, J = 6.4 Hz, CH2CH2), 1.70 (bs, 4H, CH2CH2), 1.40 (q, 4H, J = 6.4 Hz, CH2CH2), 0.79 (t, 6H, J = 7.2 Hz, CH3). 13C NMR (D2O): δ 160.98 (C=O), 47.05, 45.31, 42.03, 36.67, 26.70, 23.05, 22.82 (CH2), 10.77 (CH3).
1,12-bis-{3-[1-(n-propyl)thioureado]}-4,9-diazadodecane (9)
Compound 9 was prepared from 350 mg (0.0006 mol) of 43j according to procedure described above for the synthesis of 3 to afford 240 mg (86%) of 9 as a white solid. 1H NMR (D2O): δ 3.59 (b, 4H, NCH2), 3.23 (b, 4H, NCH2), 3.07-3.00 (m, 8H, NCH2), 1.92 (p, 4H, J = 7.2 and 6.4 Hz, CH2CH2), 1.75 (b, 4H, CH2CH2), 1.57-1.48 (m, 4H, CH2CH2), 0.85 (t, 6H, J = 7.2 Hz, CH3).
1,15-bis-{3-[1-(benzyl)thioureado]}-4,12-diazapentadecane (10)
Compound 10 was prepared from 340 mg (0.0005 mol) of 43k according to procedure described above for the synthesis of 3 to afford 214 mg (77%) of 10 as a white solid. 1H NMR (D2O): δ 7.37-7.30 (m, 10H, Ar-H), 4.58 (b, 4H, CH2Ph), 3.58 (b, 4H, NCH2), 3.10-2.80 (m, 8H, NCH2), 1.85 (b, 4H, CH2CH2), 1.59 (b, 4H, CH2CH2), 1.32 (b, 6H, CH2CH2).
1,15-bis-{3-[1-(ethyl)ureado]}-4,12-diazapentadecane (11)
Compound 11 was prepared from 255 mg (0.0004 mol) of 43l according to procedure described above for the synthesis of 3 to afford 178 mg (89%) of 11 as a white solid. 1H NMR (D2O): δ 3.16 (t, 4H, J = 7.2Hz, NCH2), 3.08 (q, 4H, J = 7.6 Hz, NCH2), 2.99 (m, 8H, NCH2), 1.79 (p, 4H, J = 7.2 Hz, CH2CH2), 1.62 (bs, 4H, CH2CH2), 1.32 (s, 6H, CH2CH2), 1.02 (t, 6H, J = 7.2 Hz, CH3). 13C NMR (D2O): δ 160.92 (C=O), 47.079, 45.15, 36.67, 35.25, 27.89, 26.69, 25.66 (CH2), 14.65 (CH3).
1,15-bis-{3-[1-(n-propyl)ureado]}-4,12-diazapentadecane (12)
Compound 12 was prepared from 255 mg (0.0004 mol) of 43m according to procedure described above for the synthesis of 3 to afford 180 mg (89%) of 12 as a white solid. 1H NMR (D2O): δ 3.16 (t, 4H, J = 5.6 Hz, NCH2), 2.99 (m, 12H, NCH2), 1.79 (p, 4H, J = 7.2 Hz, CH2CH2), 1.62 (m, 4H, CH2CH2), 1.42 (q, 4H, J = 6.4 Hz, CH2CH2), 1.32 (bs, 6H, CH2CH2), 0.81 (t, 6H, J = 7.2 Hz, CH3);). 13C NMR (D2O): δ 161.03 (C=O), 47.79, 45.14, 42.00, 36.67, 27.89, 26.71, 25.65, 22.86 (CH2), 10.77 (CH3).
1,11-bis-{3-[1-(benzyl)thioureado]}-4,8-diazaundecane (13)
Compound 13 was prepared from 373 mg (0.0005 mol) of 43n according to procedure described above for the synthesis of 3 to afford 302 mg (99%) of 13 as white solid. 1H NMR (DMSO-d6): δ 9.09 (bs, 2H, NH), 8.21 (t, 2H, NH), 8.00 (bs, 2H, NH), 7.20-7.32 (m, 10H, Ar-H), 4.64 (bs, 4H, N-CH2), 3.48 (bs, 4H, N-CH2), 2.97 (bs, 4H, N-CH2), 2.87 (bs, 4H, N-CH2), 2.02 (p, 2H, CH2CH2), 1.86 (p, 4H, CH2CH2). 13C NMR (DMSO-d6): δ 128.92, 127.91, 127.45 (Ar-C), 47.43, 45.30, 44.60, 41.33, 26.37, 23.01 (CH2).
1,11-bis-{3-[1-(ethyl)ureado]}-4,8-diazaundecane (14)
Compound 14 was prepared from 245 mg (0.0005 mol) of 43o according to procedure described above for the synthesis of 3 to afford 178 mg (96%) of 14 as white solid. 1H NMR (DMSO-d6): δ 9.14 (bs, 2H, NH), 6.00 (bs, 4H, NH), 2.88-3.08 (m, 12H, CH2N), 2.82 (bs, 4H, CH2N), 2.02 (bs, 2H, CH2CH2), 1.71 (bs, 4H, CH2CH2), 0.95 (t, J = 7.2 Hz, 6H, CH2CH3). 13C NMR (DMSO-d6): δ 159.16 (C=O), 45.25, 44.51, 36.84, 34.80, 27.51, 22.98 (CH2), 16.34 (CH3).
1,11-bis-{3-[1-(benzyl)ureado]}-4,8-diazaundecane (15)
Compound 15 was prepared from 485 mg (0.0007 mol) of 43p according to procedure described above for the synthesis of 3 to afford 364 mg (99%) of 15 as white solid. 1H NMR (DMSO-d6): δ 9.21 (bs, 6H, NH), 7.17-7.30 (m, 10H, Ar-H), 4.19 (s, 4H, N-CH2), 3.08 (bs, 4H, N-CH2), 2.94 (bs, 2H, N-CH2), 2.82 (bs, 4H, N-CH2), 2.02 (b, 2H, CH2CH2), 1.74 (b, 4H, CH2CH2). 13C NMR (DMSO-d6): δ 159.24 (C=O), 141.49, 128.89, 127.63, 127.20 (Ar-C), 45.27, 44.53, 43.57, 37.02, 27.45, 22.93 (CH2).
1,11-bis-{3-[1-(n-propyl)ureado]}-4,8-diazaundecane (16)
Compound 16 was prepared from 359 mg (0.0006 mol) of 43q according to procedure described above for the synthesis of 3 to afford 303 mg (99%) of 16 as a white solid. 1H NMR (DMSO-d6): δ 9.24 (bs, 6H, NH), 2.82-3.06 (m, 16H, N-CH2), 2.04 (b, 2H, CH2CH2), 1.73 (b, 4H, CH2CH2), 1.33 (m, J = 7.2 Hz, 4H, CH2CH3), 0.80 (t, J = 7.2 Hz, 4H, CH2CH3). 13C NMR (DMSO-d6): δ 159.29 (C=O), 45.22, 44.52, 41.88, 36.97, 27.39, 23.78, 22.92 (CH2), 12.04 (CH3).
1,11-bis-{3-[1-(n-propyl)ureado]}-4,8-diazaundecane (17)
Compound 17 was prepared from 379 mg (0.0006 mol) of 43r according to procedure described above for the synthesis of 3 to afford 317 mg (99%) of 17 as white solid. 1H NMR (DMSO-d6): δ 9.46 (b, 2H, NH), 9.16 (b, 2H, NH), 7.82 (b, 2H, NH), 2.85-3.90 (b, 16H, N-CH2), 1.84 (b, 2H, CH2CH2), 1.59 (b, 4H, CH2CH2), 1.44 (m, 4H, CH2CH3), 0.85 (t, 6H, CH2CH3). 13C NMR (DMSO-d6): δ 45.30, 44.58, 26.37, 22.92, 22.71, 22.00 (CH2), 12.10 (CH3).
1,11-bis-{3-[1-(ethyl)thioureado]}-4,8-diazaundecane (18)
Compound 18 was prepared from 347 mg (0.0006 mol) of 43s according to procedure described above for the synthesis of 3 to afford 282 mg (99%) of 18 as white solid; 1H NMR (DMSO-d6): δ 9.10 (bs, 2H, NH), 7.78 (bs, 2H, NH), 7.70 (bs, 2H, NH), 3.43 (bs, 4H, N-CH2), 3.32 (bs, 4H, N-CH2), 2.97 (bs, 4H, N-CH2), 2.86 (bs, 4H, N-CH2), 2.02 (b, 2H, CH2CH2), 1.83 (b, 4H, CH2CH2), 1.02 (t, J = 7.2 Hz, 6H, CH2CH3); 13C NMR (DMSO-d6): δ 45.27, 44.58, 38.83, 31.99, 26.38, 22.98 (CH2), 15.12 (CH3).
1,11-bis-{3-[1-(3,3-diphenylpropyl)ureado]}-4,8-diazaundecane (19)
Compound 19 was prepared from 400 mg (0.0005 mol) of 43t according to procedure described above for the synthesis of 3 to afford 290 mg (86%) of 19 as a white solid. 1H NMR (DMSO-d6): δ 9.10 (bs, 4H, NH), 7.27-7.21 (m, 16H, Ar-H), 7.18-7.10 (m, 4H, Ar-H), 3.96 (t, 2H, J = 7.2 Hz, CHPh2), 3.02 (t, 4H, J = 6.4 Hz, NCH2), 2.92 (b, 4H, NCH2), 2.84 (t, 4H, J = 7.2 Hz, NCH2), 2.79 (bs, 4H, NCH2), 2.09 (q, 4H, J = 8.0 Hz, CH2CH2), 1.99 (m, 2H, CH2CH2), 1.69 (m, 4H, CH2CH2). 13C NMR (DMSO-d6): δ 159.22 (CO), 145.50, 129.08, 128.28, 126.72 (Ar-C), 48.51, 45.25, 44.49, 38.77, 36.88, 36.07, 27.45, 22.95 (CH and CH2).
1,11-bis-{3-[1-(3,3-diphenylpropyl)thioureado]}-4,8-diazaundecane (20)
Compound 20 was prepared from 260 mg (0.0003 mol) of 43u according to procedure described above for the synthesis of 3 to afford 205 mg (92%) of 20 as a white solid. 1H NMR (DMSO-d6): δ 9.10 (b, 4H, NH), 7.91 (b, 2H, NH), 7.32-7.14 (m, 20H, Ar-H), 6.10 (b, 2H, NH), 4.04 (t, 2H, J = 7.6 Hz, CHPh2), 3.45 (b, 4H, NCH2), 3.24 (b, 4H, NCH2), 2.98 (b, 4H, NCH2), 2.88 (b, 4H, NCH2), 2.61 (m, 4H, CH2CH2), 2.04 (m, 2H, CH2CH2), 1.85 (m, 4H, CH2CH2); 13C NMR (DMSO-d6): δ 145.36, 129.11, 128.31, 126.78 (Ar-C), 48.62, 45.29, 44.60, 42.80, 41.02, 34.97, 26.34, 22.96 (CH and CH2).
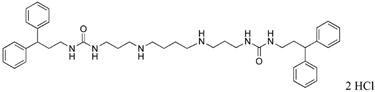
1,12-bis-{3-[1-(3,3-diphenylpropyl)ureado]}-4,9-diazadodecane (21)
Compound 21 was prepared from 370 mg (0.42 mmol) of 43v according to procedure described above for the synthesis of 3 to afford 285 mg (90%) of 21 as a white solid; 1H NMR (DMSO-d6): δ 9.00 (bs, 4H, NH), 7.21-7.12 (m, 20H, Ar-H, and NH), 3.96 (t, 2H, J = 7.2 Hz, CHPh2), 3.02 (t, 4H, J = 6.4 Hz, NCH2), 2.84 (t, 4H, J = 6.4 Hz, NCH2), 2.79 (b, 12H, NCH2), 2.09 (q, 4H, J = 7.2 Hz, CH2CH2), 1.69 (t, 4H, J = 6.4 Hz, CH2CH2), 1.63 (b, 4H, CH2CH2); 13C NMR (DMSO-d6): δ 159.27 (C=O), 145.49, 129.08, 128.28, 126.73 (Ar-C), 48.49, 46.50, 45.10, 38.76, 36.88, 36.07, 27.46, 23.23 (CH and CH2). MS (EI m/z) calculated for C42H56N6O2 [M+.] = 676.45; found 677.40 [M+H].
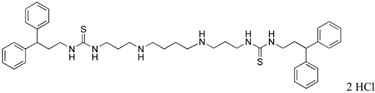
1,12-bis-{3-[1-(3,3-diphenylpropyl)thioureado]}-4,9-diazadodecane (22)
Compound 22 was prepared from 260 mg (0.0003 mol) of 43w according to procedure described above for the synthesis of 3 to afford 205 mg (92%) of 22 as a white solid. 1H NMR (DMSO-d6): δ 9.02 (bs, 4H, NH), 8.02 (b, 2H, NH), 7.30-7.12 (m, 22H, Ar-H, and NH), 4.03 (t, 2H, J = 7.6 Hz, CHPh2), 3.43 (bs, 4H, NCH2), 3.23 (bs, 4H, NCH2), 2.85 (b, 8H, NCH2), 2.24 (m, 4H, CH2CH2), 1.84 (b, 4H, CH2CH2), 1.67 (b, 4H, CH2CH2). 13C NMR (DMSO-d6): δ 145.34, 129.14, 128.31, 126.79 (Ar-C), 48.58, 46.59, 45.13, 41.18, 34.93, 26.29, 23.26 (CH and CH2).
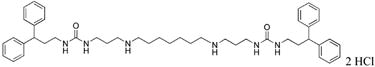
1,15-bis-{3-[1-(3,3-diphenylpropyl)ureado]}-4,12-diazapentadecane (23)
Compound 23 was prepared from 290 mg (0.0003 mol) of 43x according to procedure described above for the synthesis of 3 to afford 225 mg (88%) of 23 as a white solid. 1H NMR (DMSO-d6): δ 8.94 (bs, 4H, NH), 7.27-7.21 (m, 16H, Ar-H), 7.13-7.10 (m, 4H, Ar-H), 3.96 (t, 2H, J = 7.2Hz, CHPh2), 3.02 (t, 4H, J = 6.9 Hz, NCH2), 2.84 (t, 4H, J = 7.2Hz, NCH2), 2.77 (bs, 8H, NCH2), 2.09 (d, 4H, J = 7.2 Hz, CH2CH2), 1.69 (t, 4H, J = 6.4Hz, CH2CH2), 1.56 (bs, 4H, CH2CH2), 1.21 (bs, 6H, CH2CH2). 13C NMR (DMSO-d6): δ 159.35 (C=O), 145.49, 129.07, 128.28, 126.72 (Ar-C), 48.49, 47.24, 45.09, 38.75, 36.82, 36.07, 28.55, 27.48, 26.37, 25.91 (CH and CH2).
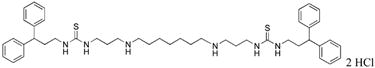
1,15-bis-{3-[1-(3,3-diphenylpropyl)thioureado]}-4,12-diazapentadecane (24)
Compound 24 was prepared from 287 mg (0.0003 mol) of 43y according to procedure described above for the synthesis of 3 to afford 230 mg (92%) of 24 as a white solid. 1H NMR (DMSO-d6): δ 8.87 (bs, 4H, NH), 7.89 (bs, 4H, NH), 7.32-7.25 (m, 16H, Ar-H), 7.18-7.14 (m, 4H, Ar-H), 4.10 (b, 2H, CHPh2), 3.44 (b, 4H, NCH2), 3.23 (b, 4H, NCH2), 2.87 (m, 8H, NCH2), 2.25 (d, 4H, J = 7.6 Hz, CH2CH2), 1.83 (t, 4H, J = 7.2Hz, CH2CH2), 1.68 (m, 4H, CH2CH2), 1.28 (b, 6H, CH2CH2). 13C NMR (DMSO-d6): δ 145.37, 129.11, 128.30, 126.70 (Ar-C), 48.61, 47.30, 45.15, 41.42, 34.93, 28.58, 26.41, 25.94 (CH and CH2).
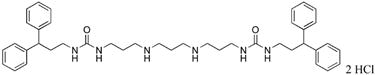
1,15-bis-{3-[1-(2,2-diphenylethyl)thioureado]}-4,12-diazapentadecane (25)
Compound 25 was prepared from 260 mg (0.0003 mol) of 43z according to procedure described above for the synthesis of 3 to afford 201 mg (90%) of 25 as a white solid. 1H NMR (DMSO-d6): δ 8.91 (bs, 3H, NH), 7.70 (b, 1H, NH), 7.52 (b, 1H, NH), 7.26 (bs, 16H, Ar-H), 7.16 (bs, 4H, Ar-H), 4.36 (b, 2H, CHPh2), 4.04 (b, 4H, NCH2), 3.45 (b, 4H, NCH2), 2.78 (b, 8H, NCH2), 1.78 (b, 4H, CH2CH2), 1.58 (b, 4H, CH2CH2), 1.25 (b, 6H, CH2CH2). 13C NMR (DMSO-d6): δ 181.50 (C=S), 143.36, 129.16, 128.62, 127.07 (Ar-C), 50.44, 48.78, 47.30, 45.09, 28.59, 26.40, 26.25, 25.91(CH2).
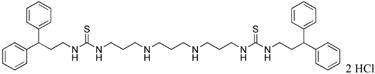
1,12-bis-{3-[1-(2,2-diphenylethyl)thioureado]}-4,9-diazadodecane (26)
Compound 26 was prepared from 280 mg (0.0003 mol) of 43aa according to procedure described above for the synthesis of 3 to afford 214 mg (89%) of 26 as a white solid. 1H NMR (DMSO-d6): δ 9.05 (b, 4H, NH), 7.79 (b, 2H, NH), 7.53 (bs, 2H, NH), 7.28 (bs, 16H, Ar-H), 7.14 (m, 4H, Ar-H), 4.36 (bs, 2H, CHPh2), 4.02 (bs, 4H, NCH2), 3.42 (bs, 4H, NCH2), 2.81 (b, 8H, NCH2), 1.80 (bs, 4H, CH2CH2), 1.66 (bs, 4H, CH2CH2). 13C NMR (DMSO-d6): δ 183.29 (C=S), 143.39, 129.16, 128.63, 127.07 (Ar-C), 50.46, 48.76, 46.57, 45.09, 41.21, 26.25, 23.16 (CH and CH2).
1,11-bis-{3-[1-(2,2-diphenylethyl)thioureado]}-4,8-diazaundecane (27)
Compound 27 was prepared from 330 mg (0.0004 mol) of 43bb according to procedure described above for the synthesis of 3 to afford 220 mg (79%) of 27 as a white solid. 1H NMR (DMSO-d6): δ 9.13 (b, 4H, NH), 7.77 (bs, 2H, NH), 7.50 (bs, 2H, NH), 7.27 (bs, 16H, Ar-H), 7.16 (bs, 4H, Ar-H), 4.35 (bs, 2H, CHPh2), 4.04 (b, 4H, NCH2), 3.66 (bs, 4H, NCH2), 3.42 (bs, 4H, NCH2), 2.94 (bs, 4H, NCH2), 2.80 (bs, 4H, NCH2), 2.01 (b, 2H, CH2CH2), 1.79 (bs, 4H, CH2CH2). 13C NMR (DMSO-d6): δ 183.20 (C=S), 143.38, 129.17, 128.63, 127.07 (Ar-C), 50.45, 48.68, 46.24, 44.57, 41.05, 26.28, 22.98 (CH and CH2).
1,11-bis-{3-[1-(1,1-diphenylmethyl)thioureado]}-4,8-diazaundecane (28)
Compound 28 was prepared from 335 mg (0.0004 mol) of 43cc according to procedure described above for the synthesis of 3 to afford 227 mg (80%) of 28 as a white solid. 1H NMR (DMSO-d6): δ 8.90 (b, 4H, NH), 8.29 (b, 2H, NH), 7.40-7.22 (m, 20H, Ar-H), 6.72 (b, 2H, CH), 4.56 (b, NH), 3.52 (b, 4H, NCH2), 2.97 (m, 8H, NCH2), 2.02 (b, 2H, CH2), 1.87 (b, 4H, CH2CH2). 13C NMR (DMSO-d6): δ 183.22 (C=S), 143.43, 129.08, 127.89, 127.56 (Ar-C), 61.28 (CH), 45.37, 44.59, 41.34, 26.33, 23.00 (CH2).
1,12-bis-{3-[1-(1,1-diphenylmethyl)thioureado]}-4,9-diazadodecane (29)
Compound 29 was prepared from 354 mg (0.0004 mmol) of 43dd according to procedure described above for the synthesis of 3 to afford 262 mg (87%) of 29 as a white solid. 1H NMR (DMSO-d6): δ 8.95 (b, 4H, NH), 8.30 (bs, 2H, NH), 7.30 (m, 20H, Ar-H), 6.72 (b, 2H, CHPh2), 3.51 (b, 4H, NCH2), 2.88 (b, 8H, NCH2), 1.87 (b, 4H, CH2CH2), 1.66 (b, 4H, CH2CH2). 13C NMR (DMSO-d6): δ 183.26 (C=S), 143.42, 129.08, 127.89, 127.57 (Ar-C), 61.30 (CH), 46.60, 45.22, 41.42, 26.38, 23.28 (CH2).
1,15-bis-{3-[1-(1,1-diphenylmethyl)thioureado]}-4,12-diazapentadecane (30)
Compound 30 was prepared from 390 mg (0.0004 mol) of 43ee according to procedure described above for the synthesis of 3 to afford 298 mg (89%) of 30 as a white solid. 1H NMR (DMSO-d6): δ 8.90 (b, 4H, NH), 8.35 (b, 2H, NH), 7.30 (bs, 20H, Ar-H), 6.73 (bs, 2H, CHPh2), 3.51 (bs, 4H, NCH2), 2.89 (bs, 4H, NCH2), 2.81 (bs, 4H, NCH2), 1.87 (bs, 4H, CH2CH2), 1.60 (bs, 4H, CH2CH2), 1.26 (b, 6H, CH2CH2). 13C NMR (DMSO-d6): δ 183.29 (C=S), 143.45, 129.06, 127.89, 127.55 (Ar-C), 61.30 (CH), 47.32, 45.23, 41.42, 28.59, 26.41, 25.93 (CH2).
1,12-bis-{5-[1-(N,N-diphenyl)carbamyl]ureado}-4,9-diazadodecane (31)
Compound 31 was prepared from 51 (130 mg, 0.0002 mol) according to procedure described above for the synthesis of 3 to afford 85 mg of 31 (75%) as a white solid. 1H NMR (DMSO-d6): δ 9.08 (bs, 4H, NH), 8.31 (t, 2H, J = 5.6 Hz, NH), 7.86 (s, 2H, NH), 7.41-7.37 (m, 8H, Ar-H), 7.30-7.26 (m, 12H, Ar-H), 3.21 (m, 4H, NCH2), 2.84 (bs, 8H, NCH2), 1.81 (m, 4H, CH2CH2), 1.67 (bs, 4H, CH2CH2). 13C NMR (DMSO-d6): δ 154.36, 153.97 (C=O), 142.41, 130.19, 128.45, 127.77 (Ar-C), 46.52, 45.13, 37.08, 26.70, 23.26(CH2). MS (EI m/z) calcd for C38H46N8O4 [M+.] = 678.36; found 679.32 [M+H].
1,15-bis-{5-[1-(N,N-diphenyl)carbamyl]ureado}-4,12-diazapentadecane (32)
Compound 32 was prepared from 52 (90 mg, 0.0001 mol) according to procedure described above for the synthesis of 3 to afford 42 mg of 32 (55%) as a white solid. 1H NMR (DMSO-d6): δ 8.92 (b, 4H, NH), 8.32 (bs, 2H, NH), 7.88 (bs, 2H, NH), 7.40-7.31 (m, 20H, Ar-H), 3.21 (bs, 4H, NCH2), 2.83 (bs, 8H, NCH2), 1.81 (bs, 4H, CH2CH2), 1.60 (bs, 4H, CH2CH2), 1.27 (bs, 6H, CH2CH2). 13C NMR (DMSO-d6): δ 154.39, 154.00 (C=O), 142.42, 130.01, 128.44, 127.79 (Ar-C), 45.27, 45.15, 37.04, 28.60, 26.73, 26.42, 25.96(CH2).
1,11-bis-{5-[1-(N,N-diphenyl)carbamyl]ureado}-4,8-diazaundecane (33)
Compound 33 was prepared from 53 (110 mg, 0.0001 mol) according to procedure described above for the synthesis of 3 to afford 66 mg of 33 (75%) as a white solid. 1H NMR (DMSO-d6): δ 9.14 (bs, 4H, NH), 8.32 (t, 2H, J = 5.6 Hz, NH), 7.87 (s, 2H, NH), 7.42-7.38 (m, 8H, Ar-H), 7.31-7.27 (m, 12H, Ar-H), 3.20 (m, 4H, NCH2), 2.97 (bs, 4H, NCH2), 2.84 (bs, 4H, NCH2), 2.02 (m, 2H, CH2CH2), 1.81 (m, 4H, CH2CH2); 13C NMR (DMSO-d6): δ 154.38, 153.98 (C=O), 142.42, 130.21, 128.46, 127.79 (Ar-C), 45.29, 44.56, 37.04, 26.74, 23.03(CH2).
Expression, Purification and Demethylase Assay of Recombinant Proteins
Full-length human LSD1 cDNA was subcloned into the pET15b bacterial expression vector (Novagen, Madison, WI) in frame with an N-terminal 6× HIS-tag and transformed into the BL21(DE3) strain of Escherichia coli. Following selection, expression and purification of recombinant LSD1 protein were performed as previously described.10 Briefly, expression of LSD1-HIS protein was induced by 1 mM IPTG for 6 h at 25 °C. The HIS-tagged protein was purified using Ni-NTA affinity purification resin and column as recommended by the manufacturer (Qiagen, Valencia, California). Bound protein was eluted by imidazole and the eluate was dialyzed in PBS at 4°C. Enzymatic activity of LSD1 was examined using luminol-dependent chemiluminescence to measure the production of H2O2, as previously described.33 In brief, LSD1 activity was assayed in 50 mM Tris, pH 8.5, 50 mM KCl, 5 mM MgCl, 5 nmol luminol, and 20 μg/ml horseradish peroxidase with the indicated concentrations of H3K4me2 (1–21 aa) peptide as substrate. The integral values were calibrated against standards containing known concentrations of H2O2, and the activities expressed as pmols H2O2/mg protein/min. Reaction mixtures were incubated with or without 5 μg purified LSD1 in 50 mM Tris, pH 8.5, 50 mM KCl, 5 mM MgCl, 0.5% BSA, and 5% glycerol for 3 hr at 37°C. This reaction mixture was analyzed by Western blotting using antibodies (Millipore) that specifically recognize the dimethyl group of H3K4.
Western Blotting
Cytoplasmic and nuclear fractions were prepared for Western blot analysis using the NE-PER™ Nuclear and Cytoplasmic Extraction Kit (Pierce, Rockford, IL). Primary antibodies against H3K4me2 were from Millipore. The pCNA monoclonal antibody was purchased from Oncogene Research Products (Cambridge, MA). Dye-conjugated secondary antibodies were used for quantification of Western blot results using the Odyssey Infrared Detection system and software (LI-COR Biosciences, Lincoln, NE).
RNA isolation and qPCR
RNA was extracted using TRIzol reagents (Invitrogen, Carlsbad, CA). First-strand cDNA was synthesized using SuperScript III reverse transcriptase with an oligo(dT) primer (Invitrogen). qPCR was performed using the following primers: SFRP2 sense, 5′AAG CCT GCA AAA ATA AAA ATG ATG; SFRP2 antisense, 5′TGT AAA TGG TCT TGC TCT TGG TCT (annealing at 57.4°C); GATA4 sense, 5′GGC CGC CCG ACA CCC CAA TCT; GATA4 antisense, 5′ ATA GTG ACC CGT CCC ATC TCG (annealing at 64°C). qPCR was performed in a MyiQ single color real-time PCR machine (Bio-Rad, Hercules, CA) with GAPDH as an internal control.
Determination of cell viability
Calu-6 human anaplastic non-small cell lung carcinoma cells were maintained in culture using RPMI medium plus 10% fetal bovine serum. For the (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) reduction assay, 4000 cells/well were seeded in 100 μl medium in a 96-well plate and the cells were allowed to attach at 37°C in 5% CO2 for one day. The medium was aspirated and cells were treated with 100 μl of fresh medium containing appropriate concentrations of each test compound. The cells were incubated for 4 days at 37°C in 5% CO2. After 4 days 20 μL of the MTS reagent solution (Promega CellTiter 96 Aqueous One Solution Cell Proliferation Assay) was added to the medium. The cells were incubated for another 2 hours at 37°C under 5% CO2 environment. Absorbance was measured at 490 nm on a microplate reader equipped with SOFTmax PRO 4.0 software to determine the cell viability.
Acknowledgments
The excellent technical assistance of Ms. Amy Hacker-Prietz is gratefully acknowledged. The research described in this manuscript was supported by NIH Grant 1R01CA149095 (PMW) 1RO1CA98454 (RAC) and 1RO1CA51085 (RAC) and by grants from the Samuel Waxman Cancer Research Foundation (RAC, PMW) and the Susan B. Komenn for the Cure Foundation (RAC). In addition, a portion of this work was supported by a generous grant from Progen Pharmaceuticals, Ltd., Brisbane, Queensland, Australia (PMW, RAC). Professor Casero also serves as a consultant to Progen Pharmaceuticals.
Abbreviations
- LSD1
lysine-specific demethylase 1
- H3K4
histone 3 lysine 4
- JmjC
Jumonji C domain-containing demethylase
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- SET
(Su(var)3-9 enhancer of zeste
- APAO
acetylpolyamine oxidase
- SMO
spermine oxidase
- SFRP
secreted frizzle-related protein
- PCNA
proliferating cell nuclear antigen
References
- 1.Marks PA, Richon VM, Breslow R, Rifkind RA. Histone deacetylase inhibitors as new cancer drugs. Curr Opin Oncol. 2001;13(6):477–483. doi: 10.1097/00001622-200111000-00010. [DOI] [PubMed] [Google Scholar]
- 2.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 3.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 4.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1(4):287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 5.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 6.Robertson KD. DNA methylation, methyltransferases, and cancer. Oncogene. 2001;20(24):3139–3155. doi: 10.1038/sj.onc.1204341. [DOI] [PubMed] [Google Scholar]
- 7.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 Acetylation Controls Chromatin Structure and Protein Interactions. Science. 2006;311(5762):844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 8.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction. Nat Rev Cancer. 2006;6(2):107–116. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 9.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 11.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125(3):467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 12.Bannister AJ, Kouzarides T. Reversing histone methylation. Nature. 2005;436(7054):1103–1106. doi: 10.1038/nature04048. [DOI] [PubMed] [Google Scholar]
- 13.Kouzarides T. Histone methylation in transcriptional control. Curr Opin Genet Dev. 2002;12(2):198–209. doi: 10.1016/s0959-437x(02)00287-3. [DOI] [PubMed] [Google Scholar]
- 14.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6(11):838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15(18):2343–2360. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- 16.Schneider R, Bannister AJ, Kouzarides T. Unsafe SETs: histone lysine methyltransferases and cancer. Trends Biochem Sci. 2002;27(8):396–402. doi: 10.1016/s0968-0004(02)02141-2. [DOI] [PubMed] [Google Scholar]
- 17.Tsukada Y, Zhang Y. Purification of histone demethylases from HeLa cells. Methods. 2006;40(4):318–326. doi: 10.1016/j.ymeth.2006.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huarte M, Lan F, Kim T, Vaughn MW, Zaratiegui M, Martienssen RA, Buratowski S, Shi Y. The fission yeast JMJ2 reverses histone H3 lysine 4 tri-methylation. J Biol Chem. 2007 doi: 10.1074/jbc.M703897200. [DOI] [PubMed] [Google Scholar]
- 19.Liang G, Klose RJ, Gardner KE, Zhang Y. Yeast Jhd2p is a histone H3 Lys4 trimethyl demethylase. Nat Struct Mol Biol. 2007;14(3):243–245. doi: 10.1038/nsmb1204. [DOI] [PubMed] [Google Scholar]
- 20.Secombe J, Li L, Carlos L, Eisenman RN. The Trithorax group protein Lid is a trimethyl histone H3K4 demethylase required for dMyc-induced cell growth. Genes Dev. 2007;21(5):537–551. doi: 10.1101/gad.1523007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G, Gaudet F, Li E, Chen T. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009;41(1):125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- 22.Liang G, Lin JC, Wei V, Yoo C, Cheng JC, Nguyen CT, Weisenberger DJ, Egger G, Takai D, Gonzales FA, Jones PA. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc Natl Acad Sci U S A. 2004;101(19):7357–7362. doi: 10.1073/pnas.0401866101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol. 2004;6(1):73–77. doi: 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- 24.Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhattar R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol. 2006;13(6):563–567. doi: 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 25.Huang Y, Greene E, Murray Stewart T, Goodwin AC, Baylin SB, Woster PM, Casero RA., Jr Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc Natl Acad Sci U S A. 2007;104(19):8023–8028. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stavropoulos P, Hoelz A. Lysine-specific demethylase 1 as a potential therapeutic target. Expert Opin Ther Targets. 2007;11(6):809–820. doi: 10.1517/14728222.11.6.809. [DOI] [PubMed] [Google Scholar]
- 27.Schmidt DM, McCafferty DG. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry. 2007;46(14):4408–4416. doi: 10.1021/bi0618621. [DOI] [PubMed] [Google Scholar]
- 28.Culhane JC, Szewczuk LM, Liu X, Da G, Marmorstein R, Cole PA. A mechanism-based inactivator for histone demethylase LSD1. J Am Chem Soc. 2006;128(14):4536–4537. doi: 10.1021/ja0602748. [DOI] [PubMed] [Google Scholar]
- 29.Szewczuk LM, Culhane JC, Yang M, Majumdar A, Yu H, Cole PA. Mechanistic Analysis of a Suicide Inactivator of Histone Demethylase LSD1. Biochemistry. 2007;46:6892–6902. doi: 10.1021/bi700414b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gooden DM, Schmidt DM, Pollock JA, Kabadi AM, McCafferty DG. Facile synthesis of substituted trans-2-arylcyclopropylamine inhibitors of the human histone demethylase LSD1 and monoamine oxidases A and B. Bioorg Med Chem Lett. 2008;18(10):3047–3051. doi: 10.1016/j.bmcl.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ueda R, Suzuki T, Mino K, Tsumoto H, Nakagawa H, Hasegawa M, Sasaki R, Mizukami T, Miyata N. Identification of cell-active lysine specific demethylase 1-selective inhibitors. J Am Chem Soc. 2009;131(48):17536–17537. doi: 10.1021/ja907055q. [DOI] [PubMed] [Google Scholar]
- 32.Binda C, Valente S, Romanenghi M, Pilotto S, Cirilli R, Karytinos A, Ciossani G, Botrugno OA, Forneris F, Tardugno M, Edmondson DE, Minucci S, Mattevi A, Mai A. Biochemical, Structural, and Biological Evaluation of Tranylcypromine Derivatives as Inhibitors of Histone Demethylases LSD1 and LSD2. J Am Chem Soc. 2010;132 doi: 10.1021/ja101557k. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Murray-Stewart T, Devereux W, Hacker A, Frydman B, Woster PM, Casero RA., Jr Properties of purified recombinant human polyamine oxidase, PAOh1/SMO. Biochem Biophys Res Commun. 2003;304(4):605–611. doi: 10.1016/s0006-291x(03)00636-3. [DOI] [PubMed] [Google Scholar]
- 34.Ferioli ME, Berselli D, Caimi S. Effect of mitoguazone on polyamine oxidase activity in rat liver. Toxicol Appl Pharmacol. 2004;201(2):105–111. doi: 10.1016/j.taap.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 35.Casara P, Jund K, Bey P. General synthetic access to alpha-allenyl amines and alpha-allenyl-alpha-amino acids as potential enzyme activated, irreversible inhibitors of PLP-dependent enzymes. Tet Letters. 1984;25:1891–1894. [Google Scholar]
- 36.Bellelli A, Cavallo S, Nicolini L, Cervelli M, Bianchi M, Mariottini P, Zelli M, Federico R. Mouse spermine oxidase: a model of the catalytic cycle and its inhibition by N,N1-bis(2,3-butadienyl)-1,4-butanediamine. Biochem Biophys Res Commun. 2004;322(1):1–8. doi: 10.1016/j.bbrc.2004.07.074. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Hacker A, Murray-Stewart T, Frydman B, Valasinas A, Fraser AV, Woster PM, Casero RA., Jr Properties of recombinant human N1-acetylpolyamine oxidase (hPAO): potential role in determining drug sensitivity. Cancer Chemother Pharmacol. 2005;56(1):83–90. doi: 10.1007/s00280-004-0936-5. [DOI] [PubMed] [Google Scholar]
- 38.Cona A, Manetti F, Leone R, Corelli F, Tavladoraki P, Polticelli F, Botta M. Molecular basis for the binding of competitive inhibitors of maize polyamine oxidase. Biochemistry. 2004;43(12):3426–3435. doi: 10.1021/bi036152z. [DOI] [PubMed] [Google Scholar]
- 39.Stranska J, Sebela M, Tarkowski P, Rehulka P, Chmelik J, Popa I, Pec P. Inhibition of plant amine oxidases by a novel series of diamine derivatives. Biochimie. 2007;89(1):135–144. doi: 10.1016/j.biochi.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 40.Bi X, Lopez C, Bacchi CJ, Rattendi D, Woster PM. Novel alkylpolyaminoguanidines and alkylpolyaminobiguanides with potent antitrypanosomal activity. Bioorg Med Chem Lett. 2006;16(12):3229–3232. doi: 10.1016/j.bmcl.2006.03.048. [DOI] [PubMed] [Google Scholar]
- 41.Gorbatenko VI. Chemistry of chlorocarbonyl isocyanate. Tetrahedron. 1993;49(16):3227–3257. [Google Scholar]
- 42.DePrez P, Lively SE. Derivatives of urea and related diamines, methods for their manufacture, and uses therefor. 20080125424. United States Patent Application. 2008
- 43.Wong R, Dolman SJ. Isothiocyanates from tosyl chloride mediated decomposition of in situ generated dithiocarbamic acid salts. J Org Chem. 2007;72(10):3969–3971. doi: 10.1021/jo070246n. [DOI] [PubMed] [Google Scholar]
- 44.Keller O, Keller WE, van Look G, Wersin G. tert-Butoxycarbonylation of Amino Acids and Their Derivatives: N-tert-Butoxycarbonyl-L-Phenylalanine. Org Syn. 1985;63:160–171. [Google Scholar]
- 45.Bellevue FH, III, Boahbedason M, Wu R, Woster PM, Casero JRA, Rattendi D, Lane S, Bacchi CJ. Structural comparison of alkylpolyamine analogues with potent in vitro antitumor or antiparasitic activity. Bioorganic & Medicinal Chemistry Letters. 1996;6(22):2765. [Google Scholar]
- 46.Hagemann H. Synthesis and reactions of N-Chlorocarbonyl isocyanate. Angewandt Chemie Intl Ed Engl. 1977;16:743–750. [Google Scholar]
- 47.Karytinos A, Forneris F, Profumo A, Ciossani G, Battaglioli E, Binda C, Mattevi A. A novel mammalian flavin-dependent histone demethylase. J Biol Chem. 2009;284(26):17775–17782. doi: 10.1074/jbc.M109.003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loenarz C, Ge W, Coleman ML, Rose NR, Cooper CD, Klose RJ, Ratcliffe PJ, Schofield CJ. PHF8, a gene associated with cleft lip/palate and mental retardation, encodes for an Nepsilon-dimethyl lysine demethylase. Hum Mol Genet. 2010;19(2):217–222. doi: 10.1093/hmg/ddp480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yue WW, Hozjan V, Ge W, Loenarz C, Cooper CD, Schofield CJ, Kavanagh KL, Oppermann U, McDonough MA. Crystal structure of the PHF8 Jumonji domain, an Nepsilon-methyl lysine demethylase. FEBS Lett. 2010;584(4):825–830. doi: 10.1016/j.febslet.2009.12.055. [DOI] [PubMed] [Google Scholar]
- 50.Varghese S, Gupta D, Baran T, Jiemjit A, Gore SD, Casero RA, Jr, Woster PM. Alkyl-substituted polyaminohydroxamic acids: a novel class of targeted histone deacetylase inhibitors. J Med Chem. 2005;48(20):6350–6365. doi: 10.1021/jm0505009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Varghese S, Senanayake T, Murray-Stewart T, Doering K, Fraser A, Casero RA, Woster PM. Polyaminohydroxamic Acids and Polyaminobenzamides as Isoform Selective Histone Deacetylase Inhibitors. Journal of Medicinal Chemistry. 2008;51(8):2447–2456. doi: 10.1021/jm701384x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flis S, Gnyszka A, Flis K, Splawinski J. MS275 enhances cytotoxicity induced by 5-fluorouracil in the colorectal cancer cells. Eur J Pharmacol. 2010;627(1-3):26–32. doi: 10.1016/j.ejphar.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 53.Fouladi M. Histone deacetylase inhibitors in cancer therapy. Cancer Invest. 2006;24(5):521–527. doi: 10.1080/07357900600814979. [DOI] [PubMed] [Google Scholar]
- 54.Huang Y, Stewart TM, Wu Y, Baylin SB, Marton LJ, Perkins B, Jones RJ, Woster PM, Casero RA., Jr Novel Oligoamine Analogues Inhibit Lysine-Specific Demethylase 1 and Induce Reexpression of Epigenetically Silenced Genes. Clin Cancer Res. 2009;15:7217–7228. doi: 10.1158/1078-0432.CCR-09-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bellevue FH, III, Boahbedason M, Wu R, Woster PM, Casero JRA, Rattendi D, Lane S, Bacchi CJ. Structural comparison of alkylpolyamine analogues with potent in vitro antitumor or antiparasitic activity. Bioorganic & Medicinal Chemistry Letters. 1996;6(22):2765–2770. [Google Scholar]