

Abstract
Metal-O2 adducts, such as metal-superoxo and -peroxo species, are key intermediates often detected in the catalytic cycles of dioxygen activation by metalloenzymes and biomimetic compounds. The synthesis and spectroscopic characterization of an end-on nickel(II)-superoxo complex with a 14-membered macrocyclic ligand was reported previously. Here we report the isolation, spectroscopic characterization, and high-resolution crystal structure of a mononuclear side-on nickel(III)-peroxo complex with a 12-membered macrocyclic ligand, [Ni(12-TMC)(O2)]+ (1) (12-TMC = 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclododecane). Different from the end-on Ni(II)-superoxo complex, the Ni(III)-peroxo complex is not reactive in electrophilic reactions, but is capable of conducting nucleophilic reactions. The Ni(III)-peroxo complex transfers the bound dioxygen to manganese(II) complexes, thus affording the corresponding nickel(II) and manganese(III)-peroxo complexes. The present results demonstrate the significance of supporting ligands in tuning the geometric and electronic structures and reactivities of metal-O2 intermediates that have been shown to have biological as well as synthetic usefulness in biomimetic reactions.
Metalloenzymes activate dioxygen to carry out a variety of biological reactions including biotransformation of naturally occurring molecules, oxidative metabolism of xenobiotics, and oxidative phosphorylation. One goal in biomimetic research is to understand the mechanistic details of dioxygen activation and oxygenation reactions and the structures of reactive intermediates occurring at the active sites of the metalloenzymes1. In the unified mechanism of dioxygen activation, dioxygen first binds to a reduced metal center that forms metal-superoxo and -peroxo intermediates, followed by O-O bond cleavage leading to the formation of high-valent metal-oxo species that are believed to carry out substrate oxidations1. Among the metal-oxygen intermediates, mononuclear metal-O2 adducts, such as metal-superoxo and -peroxo species, have attracted much attention as key intermediates in the catalytic cycles of dioxygen activation by metalloenzymes, including heme and non-heme iron and copper enzymes2-4. In biomimetic and synthetic chemistry, mononuclear metal-O2 complexes, including titanium, vanadium, chromium, manganese, iron, cobalt, nickel, copper, and the second and third row transition metals, have been synthesized and characterized with various spectroscopic techniques and X-ray crystallography, and their reactivities have been extensively investigated in nucleophilic and electrophilic oxidation reactions5-11. A notable example is the mononuclear copper-O2 species, which shows a diverse and rich chemistry in structures, spectroscopic properties, and reactivities10-16. X-ray crystal structures of side-on (η2) and end-on (η1) Cu(II)-superoxo and side-on (η2) Cu(III)-peroxo complexes were successfully obtained17-19, and the mode of O2 coordination (e.g., side-on vs end-on O2-binding) and the electronic nature of the Cu-O2 core (e.g., Cu(II)-superoxo vs Cu(III)-peroxo) were found to vary depending on the supporting ligands of copper complexes20-23.
In the case of mononuclear Ni-O2 intermediates, side-on and end-on nickel(II)-superoxo and side-on nickel(II)-peroxo complexes have been characterized by spectroscopic, X-ray crystallographic, and computational methods24-27, and the Ni(II)-superoxo complexes showed electrophilic reactivity, such as the oxidation of PPh3 to OPPh324-26. However, to the best of our knowledge, the crystal structure and reactivity of a mononuclear Ni(III)-peroxo complex have not yet been reported. In this study, we have examined the effects of supporting ligands on the mode of O2 coordination and the electronic structure of the Ni-O2 moiety in mononuclear Ni-O2 complexes, by varying the ring–size of a macrocyclic ligand coordinated to [Ni(II)(14-TMC)(O2)]+ (2) (14-TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) that was characterized as an end-on Ni(II)-superoxo complex by spectroscopic and computational methods25. We now report for the first time the synthesis, spectroscopic and electronic properties, and crystal structure of a mononuclear side-on (η2) nickel(III)-peroxo complex stabilized by a 12-membered macrocyclic ligand, [Ni(III)(12-TMC)(O2)]+ (1) (12-TMC = 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclododecane). The reactivities of the Ni(III)-peroxo complex in electrophilic and nucleophilic reactions and peroxo group transfer to other metal complexes have been discussed as well.
Results and discussion
The starting nickel complex, [Ni(12-TMC)(CH3CN)]2+ (3), was synthesized and characterized with UV-vis absorption spectroscopy, electrospray ionization mass spectrometry (ESI MS), and X-ray crystallography (see Supplementary text, Figs. S1 and S2, Tables S1 and S2). The reaction of 3 with 5 equiv H2O2 in the presence of 2 equiv triethylamine (Et3N) in CH3CN at 0 °C produces a green intermediate, 1, that exhibits distinct absorption features that are different from those of 2 (Fig. 1)25. The intermediate persisted for several days at 25 °C, and the greater thermal stability of 1 allowed us to isolate crystals that were used in spectroscopic and structural analyses and reactivity studies. The ESI MS of 1 exhibits a prominent ion peak at a mass-to-charge (m/z) ratio of 318.0 (see Supplementary Fig. S3A), whose mass and isotope distribution pattern correspond to [Ni(12-TMC)(O2)]+ (calculated m/z of 318.2) (see Supplementary Fig. S3A, inset). When the reaction was carried out with isotopically labeled H218O2, a mass peak corresponding to [Ni(12-TMC)(18O2)]+ appeared at m/z of 322.0 (calculated m/z of 322.2) (see Supplementary Fig. S3A, inset). The 4-mass unit upshift upon the substitution of 16O with 18O indicates that 1 contains an O2 unit. The EPR spectrum of a frozen acetonitrile solution of 1 measured at 4.3 K exhibits a rhombic signal with g values of 2.22, 2.17 and 2.06 (see Supplementary Fig. S3B), which is indicative of a (dz2)1 electron configuration typically observed for Ni(III) species28, Ni(II)-superoxo complexes24-26, and a six-coordinated bis(μ-superoxo)Ni2(II) complex29. The room temperature magnetic moment of 2.13 μB, determined using the 1H NMR Evans method30, is consistent with an S = 1/2 ground state of 1.
Figure 1. Characterization of 1.
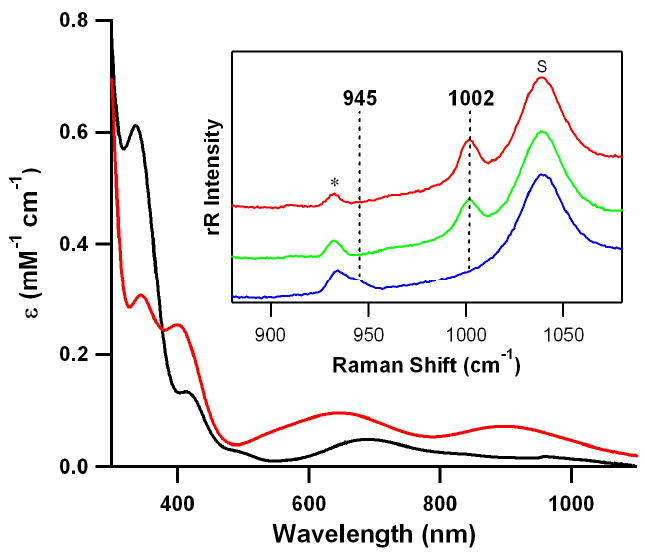
Electronic absorption spectra of 1 (red solid line) and 2 (black solid line) in CH3CN at 0 °C. Inset shows resonance Raman spectra of 1 (32 mM) obtained upon excitation at 442 nm in CD3CN at −20 °C; isolated 1 (red line) and in situ-generated 1 prepared with H216O2 (green line) and H218O2 (blue line). The peak marked with “s” is ascribed to d3-acetonitrile solvent and an asterisk denotes a band derived from a nickel complex bearing 12-TMC ligand.
The resonance Raman spectrum of 1 was collected using 442-nm excitation at −20 °C. 1 prepared with H216O2 exhibits an isotopically sensitive band at 1002 cm−1 that shifts to 945 cm−1 when H218O2 is used, consistent with its assignment as an O-O stretching vibration on the basis of its 16Δ−18Δ value of 57 cm−1 (16Δ−18Δ (calcd) = 57 cm−1) (Fig. 1, inset). Interestingly, the observed O-O stretching frequency (1002 cm−1) of 1 is significantly lower than that (1131 cm−1) of 225, but is between the superoxo (i.e., νOO of ∼1050–1200 cm-1) and peroxo (i.e., νOO of ∼800–930 cm-1) categories31. It is worth noting that the O-O stretching frequency (1002 cm−1) of 1 is quite close to those observed in Cu(III)-peroxo species (∼970 cm-1)18,22. Thus, the resonance Raman spectroscopic data suggests that the O2 in 1 has significant peroxo character, O22−.
The X-ray crystal structure of 1-(ClO4)·CH3CN revealed the mononuclear side-on 1:1 nickel complex of O2 in a distorted octahedral geometry arising from the triangular NiO2 moiety with a small bite angle of 43.04(11)° (Fig. 2). Notably, the O-O bond length (1.386(4) Å) of 1 is longer than those of Ni(II)-superoxo complexes, such as 2 (1.301 Å, from DFT calculations)25 and Ni(II)(O2) with a β-diketiminato ligand (1.347 Å)26; the O-O bond length of 1 is between those of metal-superoxo compounds (∼1.2–1.3 Å) and metal-peroxo compounds (∼1.4–1.5 Å)31, but closer to the metal-peroxo category. For comparison, the O-O bond length of 1.392 Å was reported in a Cu(III)-peroxo complex22. In addition, the Ni-O average bond of 1 (1.889 Å) is shorter than that of 2 (1.984 Å, from DFT calculations)25, supporting a Ni(III) formulation. In conjunction with the low O-O stretching frequency measured by Raman spectroscopy, the structural data, such as O-O and Ni-O bond distances, suggest that 1 can be formulated as a Ni(III)-peroxo species, Ni(III)-(O22−).
Figure 2. X-ray crystal structure of 1.
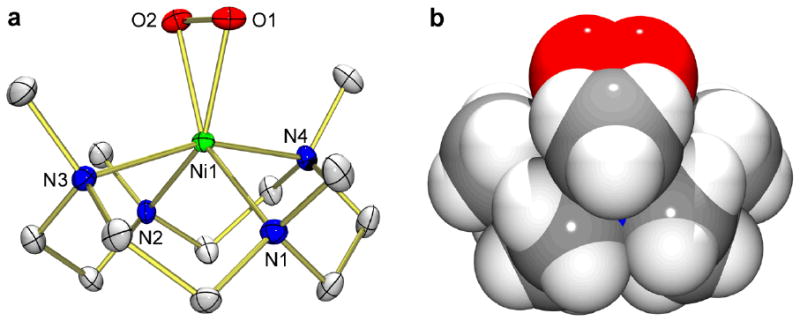
a, ORTEP plot of [Ni(12-TMC)(O2)]+ (1) with thermal ellipsoids drawn at the 30 % probability level. Hydrogen atoms are omitted for clarity. b, Side view (space-filling representation) of 1, derived from the crystal structure determination. Selected bond lengths (Å) and angles (°): Ni-O1 1.884(3), Ni-O2 1.894(3), Ni-N1 2.027(3), Ni-N2 2.038(3), Ni-N3 2.160(3), Ni-N4 2.158(3), O1-O2 1.386(4); O1-Ni-O2 43.04(11), Ni-O1-O2 68.87(16), Ni-O2-O1 68.09(15). The crystallographic data have been deposited with the Cambridge Crystallographic Data Center under CCDC 719999.
Ni K-edge X-ray absorption spectroscopy was then performed to directly probe the oxidation state of nickel and the ligand field (LF) of the nickel center. The normalized Ni K-edge X-ray absorption spectra of 1 and 2 are presented in Fig. 3. The inset shows the expanded pre-edge region. The pre-edge features are due to an electric dipole-forbidden quadrupole-allowed 1s→3d transition32. The energy position of the pre-edge transition is dominantly affected by changes in LF at the absorbing Ni center and increases with increase in LF23. The pre-edge transitions in 1 and 2 occur at 8332.3 eV and 8331.6 eV, respectively, indicating an increase in LF on going from 2 to 1 (Fig. 3). The Ni K-edge energy position increases with increase in QNi, the charge on the absorbing Ni center in the complex. Typically, for Ni complexes, the K-edge first-maxima do not show a large change with change in QNi33; however, as seen in Fig. 3 the first-maximum is shifted ∼1.8 eV in going from 2 to 1, clearly indicating an increase in QNi in 1 compared to 2. Ni K-edge EXAFS data and their analyses show that the first shell coordination has increased from 5 in 2 to 6 in 1 (see Supplementary Figs. S4–S6, Tables S3 and S4), yet the Ni-O bond distance is ∼0.06 Å shorter in 1 than in 2. Together, the Ni K-edge and EXAFS data support a unique Ni(III)-(O22−) description for 1. These data combined with the crystal structure indicate that the O2 binds side-on to the Ni center in 1, whereas the O2 is end-on bound in 2 as previously reported25. This binding mode difference is accompanied by an electronic structure change from Ni(II)-(O2−) in 2 to Ni(III)-(O22−) in 1.
Figure 3. Ni K-edge x-ray absorption spectra of 1 (–) and 2 (–).
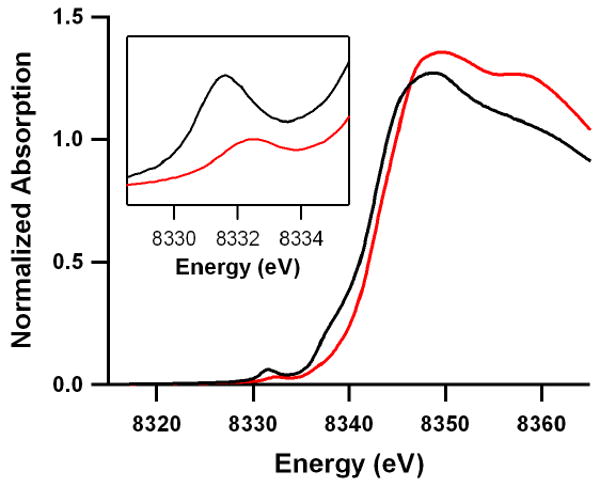
The inset shows the expanded pre-edge region. The Ni K-edge XAS spectrum of 2 was reported previously in ref. 18. Due to large differences in the beamline optics, cryostat temperatures, and detection methods, the data were re-measured in the present study for accurate comparison of 1 and 2 under identical conditions.
Density functional theory (DFT) calculations were performed to understand the role of the macrocyclic ring in determining the geometric and electronic structures of 1 and 2 (see Supplementary Fig. S7 and Table S5 for structural details). The geometry optimized structures of 1 and 2 show that the smaller 12-membered ring in 1 leads to contraction of the trans Ni-N bond angles. This displaces the Ni out of the N1N2N3N4 plane and allows for its facile side-on overlap with the O2. The side-on coordination of the O2 in 1 allows for a stronger overlap of its π* orbital with the Ni 3dx2-y2 orbital. This stronger σ overlap leads to the relative destabilization of the Ni 3dx2-y2 orbital, transfers charge from the Ni 3dx2-y2 orbital to the O2, and results in a stable Ni(III)-(O22−) species. We therefore conclude, based on the spectroscopic results, X-ray crystallography, and DFT calculations, that 1 is best described as a mononuclear Ni(III)-peroxo complex. The role of the supporting ligand, 12-TMC, is to sterically allow O2 to bind in a side-on fashion, resulting in more sigma anti bonding with 3dx2-y2 orbital as indicated above. It also stabilizes the high oxidation state of Ni3+, by the strong interaction of Ni-Nequatorial bond (2.059 Å) in 1 compared to Ni-Nequatorial bond (2.10 Å from DFT calculations) in 2 (see Supplementary Table S5). Thus, as discussed in the mononuclear Cu-O2 chemistry10-17,20-23, the geometric and electronic structures of Ni-O2 species are modulated by the nature of supporting ligands (Fig. 4).
Figure 4. Formation of Ni(III)-peroxo vs Ni(II)-superoxo intermediates.

The geometric and electronic structures of Ni-O2 intermediates are determined by the ring-size of macrocyclic ligands, such as a Ni(III)-peroxo complex with a 12-membered macrocyclic ligand and a Ni(II)-superoxo complex with a 14-membered macrocyclic ligand.
The activity of 2 was investigated in electrophilic and nucleophilic reactions. First, the electrophilic character of 1 was tested in the oxidation of PPh3, thioanisole, and xanthene. Upon addition of the substrates to the reaction solution of 2 in CH3CN at 25 °C, the intermediate remained intact without showing any absorption spectral changes, and product analysis of the reaction solutions revealed that no oxygenated products were formed in the reactions. These results demonstrate that 2 is not capable of conducting electrophilic oxidation under the reaction conditions. In contrast, Ni(II)-superoxo complexes have shown reactivities in electrophilic reactions, such as the oxidation of PPh325,26 and xanthene34. The nucleophilic character of 1 was investigated in aldehyde deformylation, with precedents that metal(III)-peroxo complexes with heme and non-heme ligands react with aldehydes to give the corresponding deformylated products6,35,36. Upon addition of 2-phenylpropionaldehyde (2-PPA) to 1 in CH3CN at 25 °C, the characteristic UV-vis absorption bands of 1 disappeared with a first-order decay profile (see Supplementary Fig. S8A), and pseudo-first-order rate constants increased proportionally with the aldehyde concentration (k2 = 4.0 × 10−2 M−1 s−1 at 25 °C) (see Supplementary Fig. S8B). Similar results were obtained in the reactions of cyclohexanecarboxaldehyde (CCA) but with a faster rate (k2 = 2.0 × 10-1 M−1 s−1 at −10 °C) (see Supplementary Fig. S9). Product analysis of the resulting solutions revealed the formation of acetophenone (92 % based on 1) and cyclohexene (85 % based on 1) in the reactions of 2-PPA and CCA, respectively. The reactivity of 1 was further investigated using substituted benzaldehydes with a series of electron-donating and -withdrawing substituents at the para-position of the phenyl group (para-Y-Ph-CHO; Y = Me, F, H, Br, Cl) (see Supplementary Fig. S10). A positive ρ+ value of 6.1 in the Hammett plot was obtained that is consistent with the process involving nucleophilic character of the Ni(III)-O2 unit in the oxidation of aldehydes.
More interestingly, we have observed a complete intermolecular O2-transfer from 1 to different transition metal complexes, such as [Mn(II)(14-TMC)]2+ (4) (Fig. 5). Addition of 4 to a solution of 1 afforded changes in the absorption spectrum which are consistent with transfer of the O2 from 1 to 4, thereby producing 3 and a manganese(III)-peroxo complex, [Mn(III)(14-TMC)(O2)]+ (5) (Fig. 6)36. Well-defined isosbestic points were observed at 416 and 687 nm in the titration reaction (Fig. 6). The intermolecular O2-transfer from 1 to 4 was further confirmed by ESI MS of the reaction solution, in which the mass peak corresponding to 1 disappeared with a concomitant appearance of mass peaks corresponding to 3 and 5 (see Supplementary Fig. S11). When the O2-transfer reaction was carried out under an 18O2 atmosphere, the product 5 did not contain the isotopically labeled 18O2 group, demonstrating that molecular oxygen was not involved in the peroxo-transfer reaction. Since the peroxo-transfer reaction was fast at 25 °C in CH3CN, kinetic studies were performed in acetone at −50 °C. Upon addition of 10 equiv of 4 to the solution of 1, 1 disappeared with a first-order decay profile (see Supplementary Fig. S12A). Pseudo-first-order fitting of the kinetic data allowed us to determine the kobs value to be 1.7(2) × 10−3 s−1 at −50 °C (see Supplementary Fig. S12A, inset). The first-order rate constants increased proportionally with the concentration of 4, giving a second-order rate constant of 0.2 M−1 s−1 at −50 °C (see Supplementary Fig. S12B). The rates were dependent on reaction temperatures, from which a linear Eyring plot was obtained between −60 and −30 °C to give the activation parameters of ΔH‡ = 49 kJ mol−1 and ΔS‡ = −76 J mol−1 K−1 (see Supplementary Fig. S12C). The observed second order kinetics and significant negative entropy value support that a bimolecular mechanism is operating in the O2-transfer reaction, where the formation of the [(12-TMC)Ni–O2–Mn(14-TMC)]3+ intermediate is presumed to be the rate-determining step (Fig. 5). Finally, we found that the reverse reaction, which is the peroxo-transfer from 5 to 3, does not occur. In this section, we have shown the first example of the complete O2-transfer between metal complexes. The observation of the complete O2-transfer from a Ni(III)-peroxo complex to a Mn(II) complex is different from the behavior of other systems where the formation of homo- or hetero-dinuclear complexes comprising [M2(μ-O)2]n+, [M2(O2)]n+, or [MM′(μ-O)2]n+ cores in the reactions of mononuclear M-O2 adducts (M = Cu and Ni) and a second metal complex, M or M′, is observed18,24,37,38. The formation of (Porp)FeIII-(O22−)-CuII(L) complexes has also been observed in the reactions of Fe(III)-peroxo porphyrins with Cu(II)(L) complexes as chemical models of cytochrome c oxidase39. Detailed mechanistic investigations are underway in this laboratory to elucidate the difference between the complete O2-transfer and the O2-bridged dinuclear formation occurring in the reactions of mononuclear M-O2 and second metal complexes.
Figure 5. Reaction scheme showing an intermolecular O2-transfer between metal complexes.

An O2 group is transferred from [Ni(III)(12-TMC)(O2)]+ (1) to [Mn(II)(14-TMC)]2+ (4) via a [(12-TMC)Ni–O2–Mn(14-TMC)]3+ transition state, and the final products are [Ni(II)(12-TMC)]2+ (3) and [Mn(III)(14-TMC)(O2)]+ (5).
Figure 6. Spectral evidence for an intermolecular O2-transfer from 1 to 4.
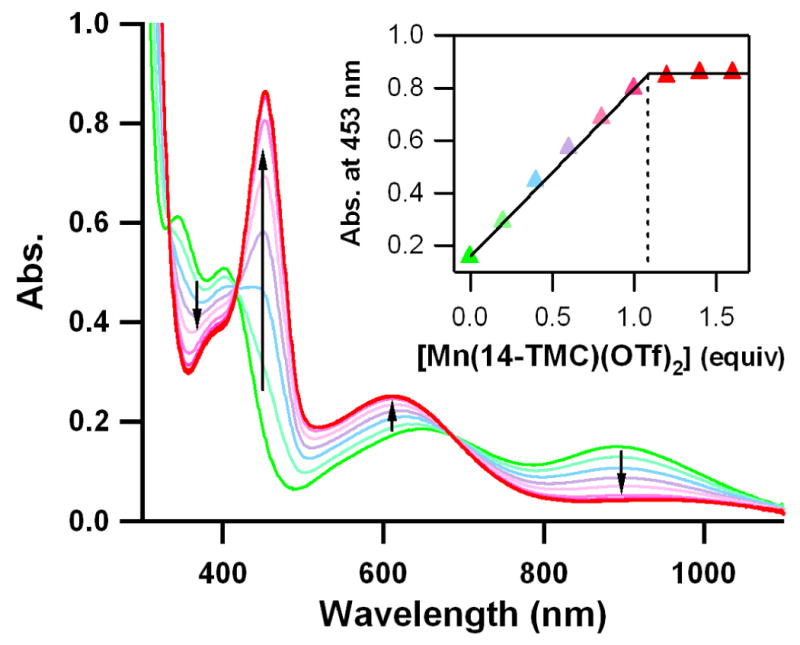
UV-vis spectral changes showing the formation of [Mn(14-TMC)(O2)]+ (5) (red) and the disappearance of [Ni(III)(12-TMC)(O2)]+ (1) (green) by addition of [Mn(II)(14-TMC)]2+ (4) to a solution of 1 in increments of 0.2 equiv in CH3CN at 25 °C. Inset shows the spectroscopic titration at 453 nm for the formation of 5 as a function of the equiv of 4 added to a solution of 1 in increments of 0.2 equiv.
In conclusion, a mononuclear side-on Ni(III)-peroxo complex was successfully synthesized by varying the supporting ligand of the previously reported Ni(II)-superoxo complex (i.e., the ring-size of the macrocyclic ligand). Combined with the precedents of mononuclear Cu-O2 intermediates14,17,18, the present results highlight the importance of supporting ligands in determining geometric and electronic structures of mononuclear Ni-O2 complexes (i.e., Ni(III)-peroxo vs Ni(II)-superoxo species). Whether other factors, such as solvents, influence the geometric and electronic structures of Ni-O2 complexes will be the subject of future studies. The reactivities of Ni(III)-peroxo and Ni(II)-superoxo complexes were compared in electrophilic and nucleophilic reactions. In contrast to Ni(II)-superoxo complexes, which show reactivities in oxidative electrophilic reactions24, the Ni(III)-peroxo complex is not reactive in electrophilic reactions but is capable of deformylating aldehydes through nucleophilic reactions. The observation of complete O2-transfer between metal complexes is unprecedented; whether this is a general feature in other metal-O2 adducts requires further experimental and computational study.
Methods
See experimental section in supplementary information for detailed experimental conditions and procedures, spectroscopic and kinetics analyses, and computational calculations.
Supplementary Material
Acknowledgments
The research was supported by KOSEF/MEST of Korea through the CRI and WCU (R31-2008-000-10010-0) Programs (W.N.), the Ministry of Education, Culture, Sports, Science and Technology of Japan through the Global COE program and Priority Area (No. 20050029) (T.O.), and NIH grant DK-31450 (E.I.S.). SSRL operations are funded by the Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology program is supported by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the Department of Energy, Office of Biological and Environmental Research.
Footnotes
Author contributions: J.C., E.I.S., and W.N. conceived and designed the experiments; J.C., R.S., J.A., S.Y.K., and M.K. performed the experiments; J.C., R.S., J.A., M.K., and T.O. analyzed the data; J.C., R.S., E.I.S., and W.N. co-wrote the paper.
Additional information: Supplementary information and chemical compound information accompany this paper at www.nature.com/naturechemistry. Reprints and permission information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Nam W. Dioxygen activation by metalloenzymes and models. Acc Chem Res. 2007;40:465–465. and review articles in the special issue. [Google Scholar]
- 2.Unno M, Chen H, Kusama S, Shaik S, Ikeda-Saito M. Structural characterization of the fleeting ferric peroxo species in Myoglobin: experiment and theory. J Am Chem Soc. 2007;129:13394–13395. doi: 10.1021/ja076108x. [DOI] [PubMed] [Google Scholar]
- 3.Kovaleva EG, Lipscomb JD. Crystal structures of Fe2+ dioxygenase superoxo, alkylperoxo, and bound product intermediates. Science. 2007;316:453–457. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prigge ST, Eipper BA, Mains RE, Amzel LM. Dioxgyen binds end-on to mononuclear copper in a precatalytic enzyme complex. Science. 2004;304:864–867. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- 5.Klotz IM, Kurtz DM., Jr Metal-dioxygen complexes. Chem Rev. 1994;94:567–568. and review articles in the special issue. [Google Scholar]
- 6.Wertz DL, Valentine JS. Nucleophilicity of iron-peroxo porphyrin complexes. Struct Bonding. 2000;97:37–60. [Google Scholar]
- 7.Girerd JJ, Banse F, Simaan AJ. Characterization and properties of non-heme iron peroxo complexes. Struct Bonding. 2000;97:145–177. [Google Scholar]
- 8.Bakac A. Kinetic and mechanistic studies of the reactions of transition metal-activated oxygen with inorganic substrates. Coord Chem Rev. 2006;250:2046–2058. [Google Scholar]
- 9.Hikichi S, Akita M, Moro-oka Y. New aspects of the cobalt-dioxygen complex chemistry opened by hydrotris(pyrazoly)borate ligands (TpR): unique properties of TpRCo-dioxygen complexes. Coord Chem Rev. 2000;198:61–87. [Google Scholar]
- 10.Mirica LM, Ottenwaelder X, Stack TDP. Structure and spectroscopy of copper–dioxygen complexes. Chem Rev. 2004;104:1013–1045. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- 11.Lewis EA, Tolman WB. Reactivity of dioxygen-copper systems. Chem Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]
- 12.Hatcher LQ, Karlin KD. Oxidant types in copper-dioxygen chemistry: the ligand coordination defines the Cun-O2 structure and subsequent reactivity. J Biol Inorg Chem. 2004;9:669–683. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]
- 13.Itoh S. Mononuclear copper active-oxygen complexes. Curr Opin Chem Biol. 2006;10:115–122. doi: 10.1016/j.cbpa.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 14.Cramer CJ, Tolman WB. Mononuclear Cu–O2 complexes: geometries, spectroscopic properties, electronic structures, and reactivity. Acc Chem Res. 2007;40:601–608. doi: 10.1021/ar700008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rolff M, Tuczek F. How do copper enzymes hydroxylate aliphatic substrates? Recent insights from the chemistry of model systems. Angew Chem Int Ed. 2008;47:2344–2347. doi: 10.1002/anie.200705533. [DOI] [PubMed] [Google Scholar]
- 16.Chen P, Solomon EI. O2 activation by binuclear Cu sites: noncoupled versus exchange coupled reaction mechanisms. Proc Natl Acad Sci USA. 2004;101:13105–13110. doi: 10.1073/pnas.0402114101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujisawa K, Tanaka M, Moro-oka Y, Kitajima N. A monomeric side-on superoxocopper(II) complex: Cu(O2)(HB(3-tBu-5-iPrpz)3) J Am Chem Soc. 1994;116:12079–12080. [Google Scholar]
- 18.Spencer DJE, Aboelella NW, Reynolds AM, Holland PL, Tolman WB. β-Diketiminate ligand backbone structural effects on Cu(I)/O2 reactivity: unique copper–superoxo and bis(μ-oxo) complexes. J Am Chem Soc. 2002;124:2108–2109. doi: 10.1021/ja017820b. [DOI] [PubMed] [Google Scholar]
- 19.Würtele C, et al. Crystallographic characterization of a synthetic 1:1 end-on copper dioxygen adduct complex. Angew Chem Int Ed. 2006;45:3867–3869. doi: 10.1002/anie.200600351. [DOI] [PubMed] [Google Scholar]
- 20.Gherman BF, Cramer CJ. Modeling the peroxide/superoxide continuum in 1:1 side-on adducts of O2 with Cu. Inorg Chem. 2004;43:7281–7283. doi: 10.1021/ic049958b. [DOI] [PubMed] [Google Scholar]
- 21.Aboelella NW, et al. Dioxygen activation at a single copper site: structure, bonding, and mechanism of formation of 1:1 Cu-O2 Adducts. J Am Chem Soc. 2004;126:16896–16911. doi: 10.1021/ja045678j. [DOI] [PubMed] [Google Scholar]
- 22.Reynolds AM, Gherman BF, Cramer CJ, Tolman WB. Characterization of a 1:1 Cu-O2 adduct supported by an anilido imine ligand. Inorg Chem. 2005;44:6989–6997. doi: 10.1021/ic050280p. [DOI] [PubMed] [Google Scholar]
- 23.Sarangi R, et al. X-ray absorption edge spectroscopy and computational studies on LCuO2 species: superoxide–CuII versus peroxide–CuIII bonding. J Am Chem Soc. 2006;128:8286–8296. doi: 10.1021/ja0615223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kieber-Emmons MT, Riordan CG. Dioxygen activation at monovalent nickel. Acc Chem Res. 2007;40:618–625. doi: 10.1021/ar700043n. [DOI] [PubMed] [Google Scholar]
- 25.Kieber-Emmons MT, et al. Identification of an “end-on” nickel–superoxo adduct, [Ni(tmc)(O2)]+ J Am Chem Soc. 2006;128:14230–14231. doi: 10.1021/ja0644879. [DOI] [PubMed] [Google Scholar]
- 26.Yao S, Bill E, Milsmann C, Wieghardt K, Driess M. A “side-on” superoxonickel complex [LNi(O2)] with a square-planar tetracoordinate nickel(II) center and its conversion into [LNi(μ-OH)2NiL] Angew Chem Int Ed. 2008;47:7110–7113. doi: 10.1002/anie.200802234. [DOI] [PubMed] [Google Scholar]
- 27.Matsumoto M, Nakatsu K. Dioxygen-bis-(t-butylisocyanide)nickel. Acta Cryst. 1975;B31:2711–2713. [Google Scholar]
- 28.Haines RI, McAuley A. Synthesis and reactions of nickel(III) complexes. Coord Chem Rev. 1981;39:77–119. [Google Scholar]
- 29.Cho J, et al. Sequential reaction intermediates in aliphatic C–H bond fuctionalization initiated by a bis(μ-oxo)dinickel(III) complex. Inorg Chem. 2006;45:2873–2885. doi: 10.1021/ic0514243. [DOI] [PubMed] [Google Scholar]
- 30.Evans DF, Jakubovic DA. Water-soluble hexadentate Schiff-base lignads as sequestrating agents for iron(III) and gallium(III) J Chem Soc Dalton Trans. 1988:2927–2933. [Google Scholar]
- 31.Cramer CJ, Tolman WB, Theopold KH, Rheingold AL. Varible character of O–O and M–O bonding in side-on (η2) 1:1 metal complexes of O2. Proc Natl Acad Sci USA. 2003;100:3635–3640. doi: 10.1073/pnas.0535926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shulman RG, Yafet Y, Eisenberger P, Blumberg WE. Observation and interpretation of X-ray absorption edges in iron compounds and proteins. Proc Natl Acad Sci USA. 1976;73:1384–1388. doi: 10.1073/pnas.73.5.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colpas GJ, et al. X-ray spectroscopic studies of nickel complexes, with application to the structure of nickel sites in hydrogenases. Inorg Chem. 1991;30:920–928. [Google Scholar]
- 34.Nam W, et al. unpublished results. [Google Scholar]
- 35.Vaz ADN, Roberts ES, Coon MJ. Olefin formation in the oxidative deformylation of aldehydes by cytochrome P-450. Mechanistic implications for catalysis by oxygen-derived peroxide. J Am Chem Soc. 1991;113:5886–5887. [Google Scholar]
- 36.Seo MS, et al. [Mn(tmc)(O2)]+: a side-on peroxido manganese(III) complex bearing a non-heme ligand. Angew Chem Int Ed. 2007;46:377–380. doi: 10.1002/anie.200603414. [DOI] [PubMed] [Google Scholar]
- 37.Kieber-Emmons MT, Schenker R, Yap GPA, Brunold TC, Riordan CG. Spectroscopic elucidation of a peroxo Ni2(μ-O2) intermediate derived from a nickel(I) complex and dioxygen. Angew Chem Int Ed. 2004;43:6716–6718. doi: 10.1002/anie.200460747. [DOI] [PubMed] [Google Scholar]
- 38.Aboelella NW, et al. Mixed metal bis(μ-oxo) complexes with [CuM(μ-O)2]n+ (M = Ni(III) or Pd(II)) cores. Chem Commun. 2004:1716–1717. doi: 10.1039/b404640d. [DOI] [PubMed] [Google Scholar]
- 39.Chufán EE, Puiu SC, Karlin KD. Heme–copper/dioxygen adduct formation, properties, and reactivity. Acc Chem Res. 2007;40:563–572. doi: 10.1021/ar700031t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.