

Abstract
Purpose
Hemodialysis grafts fail because of venous neointimal hyperplasia formation caused by adventitial fibroblasts which have become myofibroblasts (α-smooth muscle actin positive cells) and migrate to the neointima. There is increased expression of hypoxia inducible factor-1 alpha (HIF-1α in venous neointimal hyperplasia formation in experimental animal model and clinical samples. We hypothesized that under hypoxic stimulus (HIF-1α fibroblasts will convert to myofibroblasts through a matrix metalloproteinase-2 (MMP-2) mediated pathway.
Materials and methods
Murine AKR-2B fibroblasts were made hypoxic or normoxic for 24, 48, and 72 hours. Protein expression for HIF-1α, α-smooth muscle actin, MMP-2, MMP-9, TIMP-1, and TIMP-2 was performed to determine the kinetic changes of these proteins. Immunostaining for α-smooth muscle actin, collagen, and fibronectin was performed.
Results
At all time points, there was significantly increased expression of HIF-1α in the hypoxic fibroblasts when compared to normoxic fibroblasts (P<0.05). There was significantly increased expression α-smooth muscle actin at all time points which peaked by 48 hours in hypoxic fibroblasts when compared to normoxic fibroblasts (P<0.05). There was a significant increase in the expression of active MMP-2 by 48-72 hours and a significant increase in tissue inhibitor of metalloproteinase-1 (TIMP-1) by 48-72 hours by hypoxic fibroblasts (P<0.05). By 72 hours, there was significant increase in TIMP-2 expression (P<0.05). Immunohistochemical analysis demonstrated increased expression for α-smooth muscle actin, collagen, and fibronectin as the length of hypoxia increased.
Conclusions
Under hypoxia, fibroblasts will convert to myofibroblasts through a MMP-2 mediated pathway which may provide insight into the mechanism of venous neointimal hyperplasia.
Introduction
In the United States, over half the patients undergoing chronic hemodialysis require polytetrafluoroethylene (PTFE) grafts for vascular access [1]. Although arteriovenous fistulas are the preferred permanent hemodialysis vascular access, hemodialysis PTFE grafts are still being used. The primary patency of PTFE hemodialysis grafts has been reported to be 23% at 1 year and 4% at 2 years [2]. Furthermore, primary patency of angioplasty of stenosis involving the vein-to-PTFE graft anastomosis is poor at 6 months [3]. Data suggests that failure of hemodialysis vascular access grafts results primarily from venous neointimal hyperplasia, with subsequent development of venous stenosis and thrombosis at the vein-to- graft anastomosis [4]. At a cellular level, venous neointimal hyperplasia formation is caused by adventitial fibroblasts which have migrated to the neointima and become myofibroblasts (α-smooth muscle actin positive cells) [5, 6].
The mechanisms which initiate the signaling pathways that promote venous neointimal hyperplasia are multifactorial but recently several lines of evidence suggest that hypoxia may be a key component of venous neointimal hyperplasia formation in hemodialysis graft failure [7, 8]. Hypoxia inducible factor-1 alpha (HIF-1α) is a transcription factor which increases under hypoxic stimulus. A recent study suggests that reducing hypoxia may reduce venous neointimal hyperplasia formation possibly by reducing the phenotypic switch of fibroblast to myofibroblasts [9]. These observations suggest that hypoxia and therefore HIF-1α may play an important role in venous neointimal hyperplasia formation in hemodialysis vascular access grafts and in the subsequent graft failure.
At a cellular level, hypoxia inducible factor-1 alpha (HIF-1α) is known to regulate the expression of many genes which have been shown to be increased in venous neointimal hyperplasia formation including macrophage migration inhibition factor (MIF), matrix metalloproteinases (MMPs), tissue inhibitors of metalloproteinases (TIMPs), and others [5, 10-12]. Recent studies have demonstrated in a rat and porcine model of hemodialysis vascular access failure that there is increased MMP-2 expression with cellular migration from the adventitia and media leading to venous stenosis formation [5, 7, 13, 14]. There is increased expression of pro MMP-2 and pro MMP-9 in venous neointimal hyperplasia removed from patients with failed hemodialysis vascular access [5, 7, 13, 14]. Moreover, inhibiting MMPs has been shown to decrease venous neointimal hyperplasia in a porcine model of hemodialysis graft failure [15]. Taken collectively, these observations led us to hypothesize that venous neointimal hyperplasia formation in hemodialysis grafts occurs due to localized areas of hypoxia in the adventitia which results in inducing a phenotypic switch of fibroblast to myofibroblasts through a MMP-2 mediated pathway (Fig. 1). In the present paper, we investigated the in vitro role of hypoxia on the phenotypic switch of fibroblast to myofibroblasts. We determined the kinetic changes in MMP-2, MMP-9, TIMP-1, and TIMP-2 production and α-smooth muscle actin (α-SMA) production which are a marker for myofibroblasts. Understanding the mechanism of this phenotypic switch could help develop insight into the mechanism responsible for hemodialysis graft failure and help develop therapies aimed at inhibiting venous neointimal hyperplasia formation.
Figure 1.
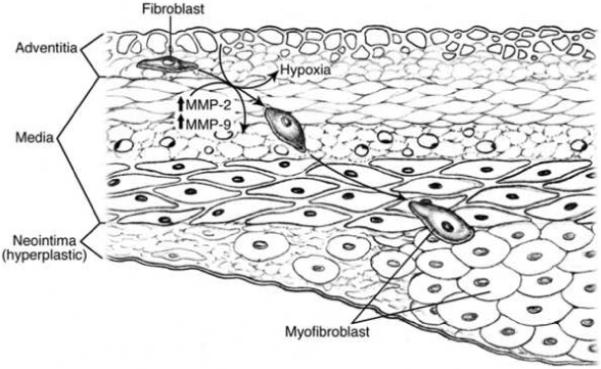
Central hypothesis of phenotypic switch of fibroblasts to myofibroblasts under hypoxic stimulus
Materials and methods
Study design
Murine embryonic AKR-2B fibroblasts were subjected to hypoxia or normoxia for 24, 48, and 72 hours and the protein expression of α-SMA, HIF-1α, MMP-2, MMP-9, TIMP-1, and TIMP-2 was determined. Immunostaining for α-SMA, collagen, and fibronectin was performed.
AKR-2B fibroblast protein expression in vitro under hypoxic conditions
Murine AKR-2B fibroblasts were cultured under hypoxic and normoxic conditions for 24, 48, and 72 hours. Fibroblasts were grown to 80% confluency in complete media Dulbecco’s Modified Eagle Medium containing 10%, vol/vol, fetal bovine serum, 4 mmol/L L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. For inducing hypoxia, the cells were placed in a water jacketed CO2 incubator that maintains a sub-ambient O2 level (3%) by the regulated injection of N2 (Hera Cell 150) [5]. Control fibroblast cells were placed in a similar incubator which was maintained at 5% CO2 and 20% O2 level. After the appropriate amount of time, cells were placed on ice and stimulation was halted by the addition of ice-cold phosphate buffer stock. Cells were washed 3 times with ice-cold phosphate buffer stock and lysed with cold radioimmunoprecipitation buffer (20mM Tris-HCl, pH 7.5, 0.15 MnCl, 1% Triton Z-100, 1 mM phenylmethylsulfonyl fluoride, 1 mM Na3VO4, 1 mM ethylenediaminetetraacetic acid, 1μg/ml leupeptin, 0.5% aprotinin, and 2 μg/ml pepstatin A). Cell lysates and conditioned media were collected after centrifugation for 15 min at 4°C and stored at −80°C for protein analysis. Experiments were repeated six times at each time point.
Immunoprecipitation followed by Western Blot
Immunoprecipitation and Western blot analysis was performed to determine the expression of HIF-1α, TIMP-1, and TIMP-2 on cell lysate and conditioned media as previously described [5]. Immunoprecipitation was performed prior to Western blot to concentrate the proteins. Fifty μgs of protein was used for immunoprecipitation using the Protein A Sepharose CL 4B bead (Amersham Biopharm, Piscataway, NJ). Briefly 30 μL of Protein-A Sepharose bead was washed 3X with lysis buffer (10 mM Phosphate buffer, pH 7.5, 150 mM NaCl, 0.1% Triton X 100, 0.1% SDS, 0.5% sodium azide). Next, this was resuspended in lysis buffer which was added to pre-clear the samples. After pre-clearing the samples, 1 μg of appropriate antibody was added to the supernatant and incubated for 4 h. Next, 30 μL of freshly prepared Protein A Sepharose bead was added to the solution and incubated for 2 h. The supernatant was separated by centrifugation at 14,000 RPM for 10 min. After incubation, the supernatant was discarded and 25 μL of 2 X sample loading buffer (125 mM Tris-HCl, 4% sodium dodecyl sulfate, 20% glycerol, 10% 2-mercaptoethanol, 0.004% bromophenol blue) was added to the bead and boiled for 10 min for subsequent sodium dodecyl sulfate polyacrylamide gel electrophoresis separation.
Western blot was performed on polyvinylidene difluoride membrane after sodium dodecyl sulfate - polyacrylamide gel electrophoresis and was blocked for 2 h with 5% milk in TBST (Tris-buffered saline Tween-20) solution (10 mM Tris-HCl, 0.15 M NaCl, 0.05% tween-20, pH 7.5). The same primary antibody was added and incubated overnight at 4° C. The appropriate secondary antibody labeled with HRP (Open Biosystems, Huntsville, AL Cat # SAB 2023. dilution 1:2000) was used for detecting the signal with standard electrochemiluminescent detection procedure. In order to determine that there were no differences in protein loading, IgG Western blot was determined. Antibodies and antisera used included: HIF-1α (Novus Biochemicals, Littleton, CO, Mouse anti-human), α-SMA, TIMP-1 (R & D Systems, Mouse IgG2B), and TIMP-2 (R & D Systems, Mouse IgG1) [5]. For α-SMA (R & D Systems, Mouse IgG1), a regular Western blot was performed and protein loading was confirmed with β-actin.
Sodium dodecyl sulfate - polyacrylamide gel electrophoresis Zymography
To assess matrix metalloproteinase expression, we used zymography on conditioned media. Fifteen micrograms of conditioned media were used. Zymography was performed on the conditioned media as previously described [5].
Bands were semiquantified by reverse image scanning densitometry using Photoshop 5.5 (Adobe, San Jose, CA). An area of the gel image that was devoid of signal was assigned to be the background value. Then, each band of the protein representing HIF-1α, α-SMA, TIMP-1, and TIMP-2 from the normoxic and hypoxic groups at 24, 48, and 72 hours was analyzed for the density above background. To ensure that the quantification was accurate, the area quantified for each band was equal. Next, to ensure that the loading of the protein was equal, the band corresponding to the IgG was determined for each protein (HIF-1α, TIMP-1, and TIMP-2) or β-actin for α-SMA. The density of the protein band was normalized to the IgG band for each protein corresponding to HIF-1α, TIMP-1, and TIMP-2 or β-actin for α-SMA. Next the ratio of the protein representing HIF-1α, TIMP-1, and TIMP-2 was normalized by dividing by the ratio of the IgG protein corresponding to HIF-1α, TIMP-1, and TIMP-2 or β-actin for α-SMA.
Immunohistochemical analysis for α-SMA, collagen, and fibronectin [5]
Immunohistochemical analysis in normoxic and hypoxic cells at different time points was performed. Briefly, cells were plated in Lab-TEK chamber slides until nearly confluent and cultured at normoxic or hypoxic conditions. Media was removed and cells were washed in warm phosphate buffer stock and then fixed in 4% paraformaldehyde for 15 min at 37°C for detection of extracellular antigen. For detection of intracellular antigens, cells were washed in cold methanol at 20°C for 15 min. Next, cells were blocked with a solution of 5% normal goat serum, 5% glycerol in phosphate buffer stock for 1 h at room temperature. Tissue was incubated with a primary antibody suspended in phosphate buffer stock, 0.1% Tween 20, 0.1% bovine serum albumin and Triton 0.1% for 1 h at room temperature. After incubation, cells were washed three times in phosphate buffer stock, followed by incubation with secondary antibody suspended in Tris buffered saline, with 0.1% Tween 20 (TBS-T) and 0.1% bovine serum albumin. Cells were washed three times with PBS, followed by addition of mounting media containing DAPI (VECTA shield) and examined by conventional and confocal fluorescence microscopy. We used the following antibodies: collagen (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, goat IgG), fibronectin (Santa Cruz Biotechnology, Inc., mouse IgG1), and α-SMA (R&D Biosystems, Mouse IgG1).
Statistical methods
The Mann-Whitney U test was performed to determine if the differences in protein expression between hypoxic and normoxic cells at 24, 48, and 72 hours. A P value of .05 or less was considered statistically significant. SAS version 9 (SAS Institute Inc., Cary, N.C.) was used for statistical analyses. Data are expressed as mean ± SD.
Results
HIF-1α and α-SMA expression by hypoxic and normoxic fibroblasts
We first determined the response of the fibroblasts to hypoxia by measuring the expression of HIF-1α at 24, 48, and 72 hours. At all time points, there was significantly increased amount of HIF-1α in hypoxic fibroblasts when compared to normoxic fibroblasts (Fig. 2, P<0.05). We next determined the expression of α-SMA production which is a marker for myofibroblasts. At all time points, α-SMA production was significantly higher by the hypoxic cells when compared to the normoxic cells. In addition, α-SMA production peaked by 48 hours and then began to decrease by 72 hours (Fig. 3, P<0.05). Overall, these results indicate that under hypoxic stimulus, fibroblasts will switch their phenotype to myofibroblasts with increased expression of α-SMA production and the peaked myofibroblast conversion occurs by 48 hours of hypoxic injury.
Figure 2.
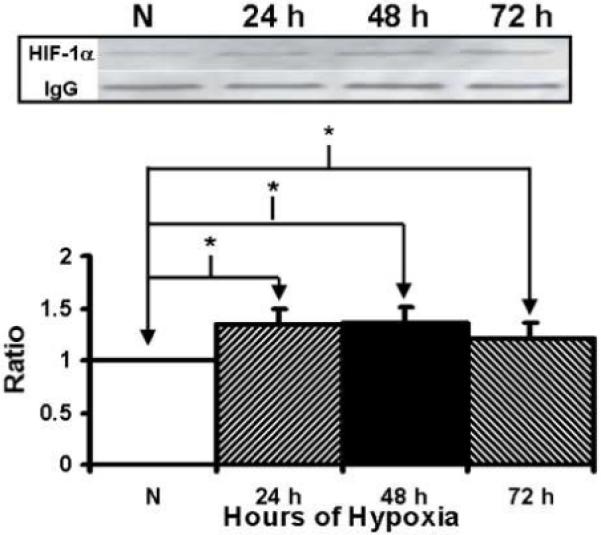
Immunoprecipitation followed by Western blot analysis of hypoxia inducible factor-1α (HIF-1α by hypoxic and normoxic fibroblasts at different time points. Upper panel is representative blots of HIF-1α by normoxic (N), 24 hour (24 h), 48 hour (48 h), and 72 hour (72 h) fibroblasts and controls with IgG loading. Lower panel shows pooled data for HIF-1α. The expression of HIF-1α (*) was significantly higher by the hypoxic fibroblasts than the normoxic fibroblasts at all time points (P<0.05). Data are presented as mean ± SD.
Figure 3.
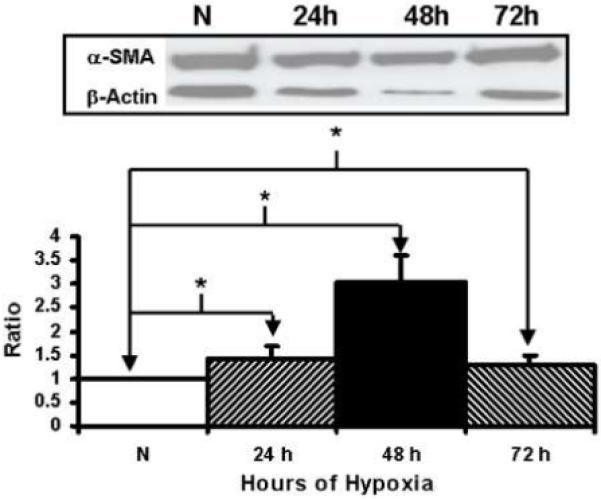
Western blot analysis of α-smooth muscle actin (α-SMA) by hypoxic and normoxic fibroblasts at different time points. Upper panel is representative blots of α-SMA by normoxic (N), 24 hour (24 h), 48 hour (48 h), and 72 hour (72 h) fibroblasts and controls with β-actin loading. Lower panel shows pooled data for α-SMA. The expression of α-SMA (*) was significantly by the hypoxic fibroblasts when compared to than the normoxic fibroblasts at all time points (P<0.05). Data are presented as mean ± SD.
Immunohistochemical analysis for α-SMA, collagen, and fibronectin in normoxic and hypoxic fibroblasts
Immunostaining for α-SMA, collagen, and fibronectin was performed in normoxic and hypoxic cells at 24, 48, and 72 hours.. These proteins are all markers of myofibroblasts. Cells staining for red are positive for α-SMA and fibronectin and those staining green are positive for collagen (Fig. 4). This demonstrated that there was increased expression of α-SMA, fibronectin, and collagen over time suggesting that fibroblasts were converting their phenotype to myofibroblasts under hypoxia.
Figure 4.
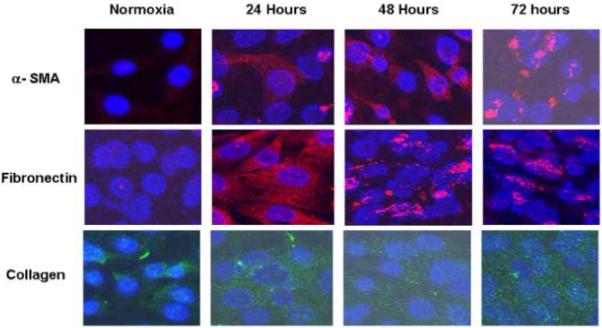
Immunostaining performed for HIF-1α, α-SMA, collagen, and fibronectin in normoxic and hypoxic fibroblasts. Cells staining for red are positive for α-SMA and fibronectin and those staining green are positive for collagen. This demonstrated that there was increased expression of α-SMA, fibronectin, and collagen over time suggesting that fibroblasts were converting their phenotype to myofibroblasts under hypoxia.
MMP-2 and MMP-9 expression by hypoxic and normoxic fibroblasts
MMP-2 production in hypoxic fibroblasts when compared to normoxic fibroblast was significantly increased by 48 to 72 hours (Fig. 5, P<0.05). There was no difference in the pro MMP-2. We did not observe any MMP-9 expression.
Figure 5.
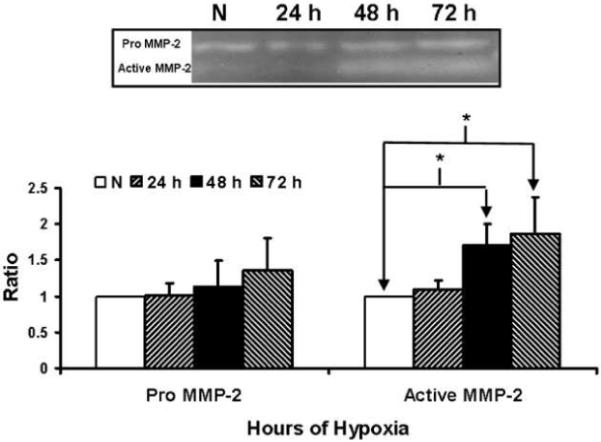
Matrix metalloproteinase-2 (MMP-2) analysis by Zymography by hypoxic and normoxic fibroblasts at different time points. Upper panel is a representative zymogram by normoxic (N), 24 hour (24 h), 48 hour (48 h), and 72 hour (72 h) fibroblasts. Lower panel shows pooled data for MMP-2. Data are presented as mean ± SD.
TIMP-1 and TIMP-2 expression by hypoxic and normoxic fibroblasts
TIMP-1 and TIMP-2 are naturally occurring inhibitor of MMP-9 and MMP-2 respectively. There was significantly increased production of TIMP-1 by hypoxic fibroblasts at 48-72 hours when compared to normoxic fibroblasts (Fig. 6, P<0.05). There was significantly increased production of TIMP-2 by hypoxic fibroblasts at 72 hours when compared to normoxic fibroblasts (Fig. 7, P<0.05).
Figure 6.
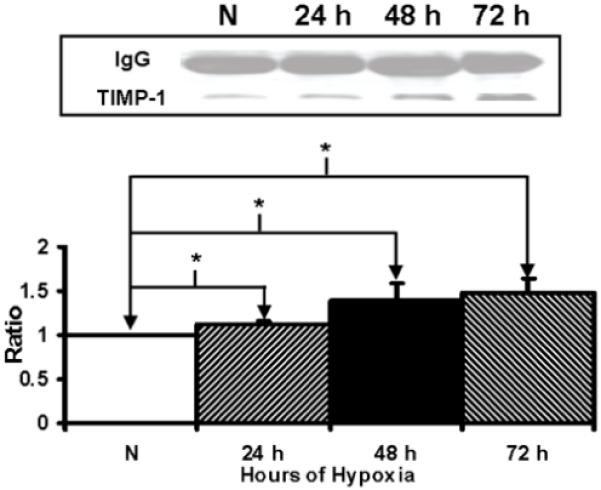
Immunoprecipitation followed by Western blot analysis of tissue inhibitor of matrix metalloprotease-1 (TIMP-1) by hypoxic and normoxic fibroblasts at different time points. Upper panel is representative blots of TIMP-1 by normoxic (N), 24 hour (24 h), 48 hour (48 h), and 72 hour (72 h) fibroblasts and controls with IgG loading. Lower panel shows pooled data for TIMP-1. The expression of TIMP-1 (*) was significantly higher by the hypoxic fibroblasts than the normoxic fibroblasts at all time points (P<0.05). Data are presented as mean ± SD.
Figure 7.
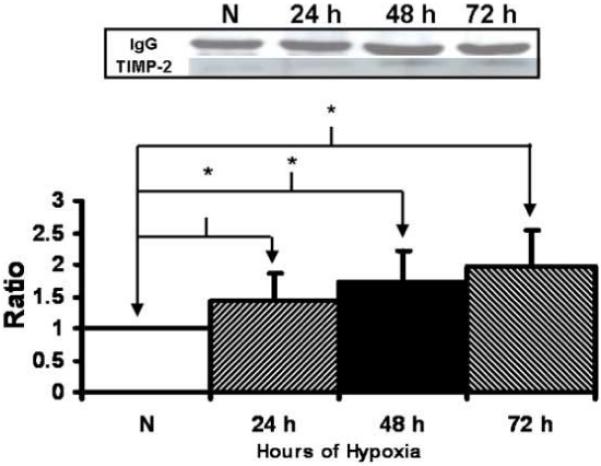
Immunoprecipitation followed by Western blot analysis of tissue inhibitor of matrix metalloprotease-2 (TIMP-2) by hypoxic and normoxic fibroblasts at different time points. Upper panel is representative blots of TIMP-2 by normoxic (N), 24 hour (24 h), 48 hour (48 h), and 72 hour (72 h) fibroblasts and controls with IgG loading. Lower panel shows pooled data for TIMP-2. The expression of TIMP-2 (*) was significantly higher by the hypoxic fibroblasts than the normoxic fibroblasts at all time points (P<0.05). Data are presented as mean ± SD.
Discussion
Venous injury and subsequent formation of venous neointimal hyperplasia are responsible for hemodialysis graft failure. Histologically, venous neointimal hyperplasia is characterized by a hyperplastic intima with increased cellular proliferation of α-SMA positive staining cells [4, 16-19]. There is substantial evidence that the fibroblasts play a significant role in the response to vascular injury [5, 20-25]. It is well established in experimental animal models of arterial stenosis and interposition of saphenous vein grafts used as arterial conduits that fibroblasts migrating from the adventitia contribute to stenosis formation [25, 26]. In an experimental hemodialysis graft porcine model, it has been observed that there is migration of adventitial and medial cells contributing to venous neointimal hyperplasia formation and these cells in part are derived from the fibroblasts which reside in the adventitia [5, 25, 26]. Experiments conducted in the present paper demonstrate that fibroblasts under hypoxic stimulus will change their phenotype to myofibroblast through a MMP-2/TIMP mediated pathway, offering a potential mechanism for venous neointimal hyperplasia formation [20, 27].
Venous neointimal hyperplasia formation is a dynamic and complicated process. The mechanisms that can potentially contribute to early venous injury with subsequent initiation of the signaling pathways that promote venous neointimal hyperplasia venous neointimal hyperplasia are multifactorial and felt to include hemodynamic factors such as high and low wall shear stress (WSS) [28-36] and local vessel wall hypoxia [7, 8]. There is increased expression of HIF-1α in venous neointimal hyperplasia removed from both clinical and experimental samples [7, 8]. Several lines of evidence prompted our investigation of HIF-1α including increased hypoxia within the vessel wall in regions of intimal hyperplasia in prosthetic vascular grafts used as arterial conduits in experimental animal models and atherosclerosis [37, 38]. Second, at a cellular level, hypoxia increases gene expression of several important matrix regulatory proteins through increased expression of HIF-1α which is known to regulate expression of vascular endothelial growth factor-A (VEGF-A), matrix metalloproteinases (MMPs), tissue inhibitors of matrix metalloproteinases (TIMPs), and others [39, 40]. Third, in patients with failed hemodialysis vascular access grafts, increased cellular proliferation with matrix deposition has been observed and MMPs have been shown to be involved in cellular proliferation and migration [5, 18, 19, 41]. These observations suggest that HIF-1α may play an important role in venous neointimal hyperplasia formation in hemodialysis grafts.
We hypothesized that hypoxia will induce fibroblasts to undergo a phenotypic switch to myofibroblasts through a MMP-2/TIMP mediated pathway. Several studies have demonstrated that hypoxia will stimulate fibroblasts from the pulmonary artery to differentiate into myofibroblast which is hypothesized to cause fibrosis associated with chronic inflammatory lung disease [42-45]. Furthermore, a recent study demonstrated that epithelial renal cells exposed to hypoxia will undergo epithelial mesenchymal transition with increased production of TIMP-1, MMPs, and other growth factors resulting in extracellular matrix production [46]. These findings are consistent with other studies which have demonstrated that PDGF, TGF-β, and recently MMPs are involved in the differentiation process of fibroblast to myofibroblast under hypoxic stimulus [42, 44, 47]. However, the role of hypoxia on converting venous fibroblasts to myofibroblasts and subsequent venous neointimal hyperplasia formation has not been studied.
In the present study, we used a murine embryonic fibroblast cell line (AKR-2B) to investigate the role of hypoxia on inducing fibroblast to undergo differentiation to myofibroblast which is the major cell found in venous neointimal hyperplasia formation. We elected to use a murine fibroblast cell line instead of fibroblasts isolated from veins because fibroblasts isolated from different vascular beds have been shown to be comprised of subpopulations of fibroblasts. These subpopulations have been shown to have differing abilities to proliferate and migrate in response to hypoxia [48]. Therefore, we elected for our experiments to use a fibroblast cell line. We observed using this cell line that myofibroblast production significantly increased as the length of hypoxia exposure increased. Furthermore, we observed a significant increase in active MMP-2, TIMP-1, and TIMP-2 at the same time points. These finding further extend our understanding of the pathophysiology of venous neointimal hyperplasia formation associated with hemodialysis graft failure. Recent experimental data from animal experiments suggests that MMP-2 may be responsible for the stenotic process, and clinical specimens removed from failed hemodialysis access specimens from both arteriovenous fistulas and grafts [5]. Furthermore, inhibition of MMPs by a non specific inhibitor in a pig model of a hemodialysis graft failure has resulted in reduction of intimal hyperplasia [15]. Finally, increased expression of HIF-1α has been observed in failed clinical specimens removed from patients with hemodialysis grafts and in early venous stenosis from a porcine experimental hemodialysis graft model.
There are several limitations of our study which must be discussed. The fibroblasts we used are an immortalized cell line which may behave different than fibroblasts found in veins. We did not perform any mechanistic experiments which examined the role of MMP-2, TIMP-1, and TIMP-2 in the phenotypic switch of fibroblasts to myofibroblasts under hypoxia which will be the focus of future studies. Finally, it is possible that there are other growth factors and cytokines which are responsible which were not evaluated in the present study.
In summary, we observed that in a murine fibroblast AKR-2B cell line, hypoxia induced conversion of fibroblasts to myofibroblasts with significantly increased expression of α-SMA. We also observed significant increase in active MMP-2, TIMP-1, and TIMP-2 production at the same time points. Understanding the role of hypoxia on converting fibroblasts to myofibroblasts through MMP-2/TIMP mediated pathway may help improve outcomes in hemodialysis graft failure and will be investigated in future studies.
Acknowledgments
The project described was supported by Award Number R01HL098967 (SM) from the National Heart, Lung, And Blood Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, And Blood Institute or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This work was presented at the 35Th Annual meeting of the Society of Interventional Radiology in Tampa, Florida.
References
- [1].Collins AJ, Kasiske B, Herzog C, et al. Excerpts from the United States Renal Data System 2003 Annual Data Report: atlas of end-stage renal disease in the United States. Am J Kidney Dis. 2003;42:A5–A7. [PubMed] [Google Scholar]
- [2].Miller PE, Carlton D, Deierhoi MH, Redden DT, Allon M. Natural history of arteriovenous grafts in hemodialysis patients. Am J Kidney Dis. 2000;36:68–74. doi: 10.1053/ajkd.2000.8269. [DOI] [PubMed] [Google Scholar]
- [3].Misra S, Bonan R, Pflederer T, Roy-Chaudhury P. BRAVO I: A pilot study of vascular brachytherapy in polytetrafluoroethylene dialysis access grafts. Kidney Int. 70:2006–2013. doi: 10.1038/sj.ki.5001869. [DOI] [PubMed] [Google Scholar]
- [4].Rekhter M, Nicholls S, Ferguson M, Gordon D. Cell proliferation in human arteriovenous fistulas used for hemodialysis. Arterioscler Thromb. 1993;13:609–617. doi: 10.1161/01.atv.13.4.609. [DOI] [PubMed] [Google Scholar]
- [5].Misra S, Doherty MG, Woodrum D, et al. Adventitial remodeling with increased matrix metalloproteinase-2 activity in a porcine arteriovenous polytetrafluoroethylene grafts. Kidney Int. 2005;68:2890–2900. doi: 10.1111/j.1523-1755.2005.00763.x. [DOI] [PubMed] [Google Scholar]
- [6].Li L, Terry CM, Blumenthal DK, et al. Cellular and morphological changes during neointimal hyperplasia development in a porcine arteriovenous graft model. Nephrol Dial Transplant. 2007;22:3139–3146. doi: 10.1093/ndt/gfm415. [DOI] [PubMed] [Google Scholar]
- [7].Misra S, Fu A, Rajan D, Juncos L, et al. Expression of hypoxia inducible factor-1 alpha, macrophage migration inhibition factor, matrix metalloproteinase-2 and -9, and their inhibitors in hemodialysis grafts and arteriovenous fistulas. J Vasc Interv Radiol. 2008;19:252–259. doi: 10.1016/j.jvir.2007.10.031. [DOI] [PubMed] [Google Scholar]
- [8].Misra S, Fu A, Pugionni A, et al. Increased expression of Hypoxia inducible factor-1α in a porcine model of chronic renal insufficiency with arteriovenous polytetrafluoroethylene grafts. J Vasc Interv Radiol. 2008;19:260–265. doi: 10.1016/j.jvir.2007.10.029. [DOI] [PubMed] [Google Scholar]
- [9].Hughes D, Fu AA, Puggioni A, et al. Adventitial transplantation of blood outgrowth endothelial cells in porcine haemodialysis grafts alleviates hypoxia and decreases neointimal proliferation through a matrix metalloproteinase-9-mediated pathway--a pilot study. Nephrol Dial Transplant. 2009;24:85–96. doi: 10.1093/ndt/gfn433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dean RA, Butler GS, Hamma-Kourbali Y, et al. Identification of candidate angiogenic inhibitors processed by matrix metalloproteinase 2 (MMP-2) in cell-based proteomic screens: disruption of vascular endothelial growth factor (VEGF)/heparin affinity regulatory peptide (pleiotrophin) and VEGF/Connective tissue growth factor angiogenic inhibitory complexes by MMP-2 proteolysis. Mol Cell Biol. 2007;27:8454–8465. doi: 10.1128/MCB.00821-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ito TK, Ishii G, Chiba H, Ochiai A. The VEGF angiogenic switch of fibroblasts is regulated by MMP-7 from cancer cells. Oncogene. 2007;26:7194–7203. doi: 10.1038/sj.onc.1210535. [DOI] [PubMed] [Google Scholar]
- [12].Zhong J, Gencay MM, Bubendorf L, et al. ERK1/2 and p38 MAP kinase control MMP-2, MT1-MMP, and TIMP action and affect cell migration: a comparison between mesothelioma and mesothelial cells. Journal of cellular physiology. 2006;207:540–552. doi: 10.1002/jcp.20605. [DOI] [PubMed] [Google Scholar]
- [13].Misra S, Fu A, Anderson J, Glockner J, et al. The rat femoral arteriovenous fistula model: Increased expression of MMP-2 and MMP-9 at the venous stenosis. J Vasc Interv Radiol. 2008;19:587–594. doi: 10.1016/j.jvir.2008.01.005. [DOI] [PubMed] [Google Scholar]
- [14].Misra S, Fu AA, Puggioni A, Karimi KM, et al. Increased shear stress with up regulation of VEGF-A and its receptors and MMP-2, MMP-9, and TIMP-1 in venous stenosis of hemodialysis grafts. Am J Physiol Heart Circ Physiol. 2008;294:H2219–H2230. doi: 10.1152/ajpheart.00650.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rotmans JI, Velema E, Verhagen HJ, et al. Matrix metalloproteinase inhibition reduces intimal hyperplasia in a porcine arteriovenous-graft model. J Vasc Surg. 2004;39:432–439. doi: 10.1016/j.jvs.2003.07.009. [DOI] [PubMed] [Google Scholar]
- [16].Swedberg SH, Brown BG, Sigley R, Wight TN, Gordon D, Nicholls SC. Intimal fibromuscular hyperplasia at the venous anastomosis of PTFE grafts in hemodialysis patients. Clinical, immunocytochemical, light and electron microscopic assessment. Circulation. 1989;80:1726–1736. doi: 10.1161/01.cir.80.6.1726. [DOI] [PubMed] [Google Scholar]
- [17].Hofstra L, Tordoir JH, Kitslaar PJ, Hoeks AP, Daemen MJ. Enhanced cellular proliferation in intact stenotic lesions derived from human arteriovenous fistulas and peripheral bypass grafts. Does it correlate with flow parameters? Circulation. 1996;94:1283–1290. doi: 10.1161/01.cir.94.6.1283. [DOI] [PubMed] [Google Scholar]
- [18].Roy-Chaudhury P, Kelly BS, Miller MA, et al. Venous neointimal hyperplasia in polytetrafluoroethylene dialysis grafts. Kidney Int. 2001;59:2325–2334. doi: 10.1046/j.1523-1755.2001.00750.x. [DOI] [PubMed] [Google Scholar]
- [19].Roy-Chaudhury P, Sukhatme VP, Cheung AK. Hemodialysis vascular access dysfunction: a cellular and molecular viewpoint. J Am Soc Nephrol. 2006;17:1112–1127. doi: 10.1681/ASN.2005050615. [DOI] [PubMed] [Google Scholar]
- [20].Shi Y, O’Brien JE, Fard A, Mannion JD, Wang D, Zalewski A. Adventitial myofibroblasts contribute to neointimal formation in injured porcine coronary arteries. Circulation. 1996;94:1655–1664. doi: 10.1161/01.cir.94.7.1655. [DOI] [PubMed] [Google Scholar]
- [21].Shi Y, Patel S, Niculescu R, Chung W, Desrochers P, Zalewski A. Role of matrix metalloproteinases and their tissue inhibitors in the regulation of coronary cell migration. Arterioscler Thromb Vasc Biol. 1999;19:1150–1155. doi: 10.1161/01.atv.19.5.1150. [DOI] [PubMed] [Google Scholar]
- [22].Shi Y, Pieniek M, Fard A, O’Brien J, Mannion JD, Zalewski A. Adventitial remodeling after coronary arterial injury. Circulation. 1996;93:340–348. doi: 10.1161/01.cir.93.2.340. [DOI] [PubMed] [Google Scholar]
- [23].Scott NA, Cipolla GD, Ross CE, et al. Identification of a potential role for the adventitia in vascular lesion formation after balloon overstretch injury of porcine coronary arteries. Circulation. 1996;93:2178–2187. doi: 10.1161/01.cir.93.12.2178. [DOI] [PubMed] [Google Scholar]
- [24].Zalewski A, Shi Y. Vascular myofibroblasts. Lessons from coronary repair and remodeling. Arterioscler Thromb Vasc Biol. 1997;17:417–422. doi: 10.1161/01.atv.17.3.417. [DOI] [PubMed] [Google Scholar]
- [25].Kalra M, Miller VM. Early remodeling of saphenous vein grafts: proliferation, migration and apoptosis of adventitial and medial cells occur simultaneously with changes in graft diameter and blood flow. J Vasc Res. 2000;37:576–584. doi: 10.1159/000054091. [DOI] [PubMed] [Google Scholar]
- [26].Shi Y, O’Brien JE, Jr., Mannion JD, et al. Remodeling of autologous saphenous vein grafts. The role of perivascular myofibroblasts. Circulation. 1997;95:2684–2693. doi: 10.1161/01.cir.95.12.2684. [DOI] [PubMed] [Google Scholar]
- [27].Cai WJ, Koltai S, Kocsis E, et al. Remodeling of the adventitia during coronary arteriogenesis. Am J Physiol Heart Circ Physiol. 2003;284:H31–H40. doi: 10.1152/ajpheart.00478.2002. [DOI] [PubMed] [Google Scholar]
- [28].Fillinger M, Reinitz E, Schwartz R, Resetarits D, Paskanik A, Bredenberg C. Beneficial effects of banding on venous intimal-medial hyperplasia in arteriovenous loop grafts. Am J Surg. 1989;158:87–94. doi: 10.1016/0002-9610(89)90353-x. [DOI] [PubMed] [Google Scholar]
- [29].Fillinger M, Reinitz E, Schwartz R, et al. Graft geometry and venous intimal-medial hyperplasia in arteriovenous loop grafts. J Vasc Surg. 1990;11:556–566. [PubMed] [Google Scholar]
- [30].Haruguchi H, Teraoka S. Intimal hyperplasia and hemodynamic factors in arterial bypass and arteriovenous grafts: a review. J Artif Organs. 2003;6:227–235. doi: 10.1007/s10047-003-0232-x. [DOI] [PubMed] [Google Scholar]
- [31].Heise M, Schmidt S, Kruger U, et al. Local Haemodynamics and Shear Stress in Cuffed and Straight PTFE-venous Anastomoses: an in-vitro Comparison using Particle Image Velocimetry. Euro J Vasc Endovasc Surg. 2003;26:367–373. doi: 10.1016/s1078-5884(03)00243-0. [DOI] [PubMed] [Google Scholar]
- [32].Hofstra L, Bergmans DC, Hoeks AP, Kitslaar PJ, Leunissen KM, Tordoir JH. Mismatch in elastic properties around anastomoses of interposition grafts for hemodialysis access. J Am Soc Nephrol. 1994;5:1243–1250. doi: 10.1681/ASN.V551243. [DOI] [PubMed] [Google Scholar]
- [33].Keynton R, Evancho M, Sims R, Rodway N, Gobin A, Rittgers S. Intimal hyperplasia and wall shear in arterial bypass graft distal anastomoses: an in vivo model study. J Biomech Eng. 2001;123:464–473. doi: 10.1115/1.1389461. [DOI] [PubMed] [Google Scholar]
- [34].Krueger U, Zanow J, Scholz H. Computational Fluid Dynamics and Vascular Access. Artificial Organs. 2002;26:571–575. doi: 10.1046/j.1525-1594.2002.07078.x. [DOI] [PubMed] [Google Scholar]
- [35].Meyerson SL, Skelly CL, Curi MA, et al. The effects of extremely low shear stress on cellular proliferation and neointimal thickening in the failing bypass graft. J of Vasc Surg. 2001;34:90–97. doi: 10.1067/mva.2001.114819. [DOI] [PubMed] [Google Scholar]
- [36].Van Tricht I, De Wachter D, Tordoir J, Verdonck P. Comparison of the hemodynamics in 6 mm and 4-7 mm hemodialysis grafts by means of CFD. Journal of Biomechanics. 2006;39:226–236. doi: 10.1016/j.jbiomech.2004.12.003. [DOI] [PubMed] [Google Scholar]
- [37].Nakata T, Shionoya S. Vascular lesions due to obstruction of the vasa vasorum. Nature. 1966;212:1258–1259. doi: 10.1038/2121258a0. [DOI] [PubMed] [Google Scholar]
- [38].Santilli SM, Tretinayak AS, Lee ES. Transarterial wall oxygen gradients at the deployment site of an intra-arterial stent in the rabbit. Am J Physiol Heart Circ Physiol. 2000;279:H1518–H1525. doi: 10.1152/ajpheart.2000.279.4.H1518. [DOI] [PubMed] [Google Scholar]
- [39].Koong AC, Denko NC, Hudson KM, et al. Candidate Genes for the Hypoxic Tumor Phenotype. Cancer Res. 2000;60:883–887. [PubMed] [Google Scholar]
- [40].Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- [41].Wang Y, Krishnamoorthy M, Banerjee R, et al. Venous stenosis in a pig arteriovenous fistula model anatomy, mechanisms and cellular phenotypes. Nephrol Dial Transplant. 2007;22:3139–3146. doi: 10.1093/ndt/gfm547. [DOI] [PubMed] [Google Scholar]
- [42].Karakiulakis G, Papakonstantinou E, Aletras AJ, Tamm M, Roth M. Cell type-specific effect of hypoxia and platelet-derived growth factor-BB on extracellular matrix turnover and its consequences for lung remodeling. The J Biol Chem. 2007;282:908–915. doi: 10.1074/jbc.M602178200. [DOI] [PubMed] [Google Scholar]
- [43].Leufgen H, Bihl MP, Rudiger JJ, et al. Collagenase expression and activity is modulated by the interaction of collagen types, hypoxia, and nutrition in human lung cells. Journal of Cell Physiol. 2005;204:146–154. doi: 10.1002/jcp.20289. [DOI] [PubMed] [Google Scholar]
- [44].Papakonstantinou E, Aletras AJ, Roth M, Tamm M, Karakiulakis G. Hypoxia modulates the effects of transforming growth factor-beta isoforms on matrix-formation by primary human lung fibroblasts. Cytokine. 2003;24:25–35. doi: 10.1016/s1043-4666(03)00253-9. [DOI] [PubMed] [Google Scholar]
- [45].Short M, Nemenoff RA, Zawada WM, Stenmark KR, Das M. Hypoxia induces differentiation of pulmonary artery adventitial fibroblasts into myofibroblasts. Am J Physiol Cell Physiol. 2004;286:C416–C425. doi: 10.1152/ajpcell.00169.2003. [DOI] [PubMed] [Google Scholar]
- [46].Higgins DF, Kimura K, Bernhardt WM, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. The Journal of Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Stenmark KR, Gerasimovskaya E, Nemenoff RA, Das M. Hypoxic activation of adventitial fibroblasts: role in vascular remodeling. Chest. 2002;122:326S–334S. doi: 10.1378/chest.122.6_suppl.326s. [DOI] [PubMed] [Google Scholar]
- [48].Das M, Dempsey EC, Reeves JT, Stenmark KR. Selective expansion of fibroblast subpopulations from pulmonary artery adventitia in response to hypoxia. Am J Physiol Lung Cell Mol Physiol. 2002;282:L976–L986. doi: 10.1152/ajplung.00382.2001. [DOI] [PubMed] [Google Scholar]