

Abstract
Eight new antimicrobial natural products named chrysophaentins A-H belonging to a new structural class have been isolated from the chrysophyte alga Chrysophaeum taylori. Their structures were determined by extensive 2D NMR and MS techniques and are characterized by the presence of two polyhalogenated, polyoxygenated ω,ω’-diarylbutene units connected by two ether bonds to form the suite of macrocyclic natural products. Chrysophaentin A, the most potent of these antibiotics, inhibited the growth of clinically relevant Gram-positive bacteria including methicillin-resistant Staphylococcus aureus (MIC50 1.5 ± 0.7 μg/mL), multiple drug resistant S. aureus (1.3 ± 0.4 μg/mL), and vancomycin-resistant Enterococcus faecium (MIC50 2.9 ± 0.8 μg/mL). In vitro enzyme assays and transmission electron microscopy showed chrysophaentin A to inhibit the GTPase activity of the bacterial cytoskeletal protein FtsZ with an IC50 value of 6.7 ± 1.7 μg/mL, as well as GTP-induced formation of FtsZ protofilaments. Saturation Transfer Difference (STD) NMR experiments further confirmed chrysophaentin A binds to FtsZ, and NMR competition experiments with GTPγS showed chrysophaentin A and GTP to bind competitively to FtsZ. Last, molecular docking showed chrysophaentin A to bind in and occlude a large portion of the GTP binding site of FtsZ in a manner that is consistent with the binding epitope determined by STD NMR.
Introduction
There is a continuous need to identify new lead compounds with novel mechanisms of action to treat infections caused by multi-drug resistant bacteria. Infectious diseases are the leading cause of death worldwide,1 and it has been estimated that in the United States more people die from methicillin-resistant Staphylococcus aureus (MRSA) related infections than from HIV.2 Infections involving drug resistant bacteria are more difficult to treat due to increased costs and decreased efficacies.3 In fact 90,000 people die from hospital-acquired bacterial infections in the United States each year in part due to the fact that clinically important bacteria are characterized by multiple antibiotic resistance to so-called drugs of last resort such as fluoroquinolones, vancomycin, and carbapemens.3,4
A relatively new target in antimicrobial drug discovery programs is the bacterial cell division protein FtsZ: not only is this protein essential for cell division but it is highly conserved among almost all bacteria making it an attractive antimicrobial target.5 At the early stages of cell division, FtsZ, a structural homolog of the eukaryotic cytoskeletal protein tubulin, undergoes guanosine 5-triphosphate (GTP)-dependent polymerization to form protofilaments that assemble into a dynamic and contractile structure known as the Z-ring, which marks the plane of cell division.6,7 Although efforts to identify inhibitors of FtsZ have increased rapidly in recent years, the target remains under exploited.5,8 Compounds reported to inhibit the function of FtsZ include phenolic natural products viriditoxin,9a berberine,9b cinnamaldehyde,9c and totarol,9d together with synthetic inhibitors such as PC190723,9e zantrins,9f and OTBA9g (reviewed in references 5 and 8). Marine natural products represent a potential source for new FtsZ inhibitors because of their unique structures and strong antimicrobial activities. Several recent, relevant examples include the marine actinomycete-derived marinomycins10a and lynamicins;10b the marine sponge-derived motualevic acids,10c and the red alga-derived neurymenolides.10d
Continuing with our search for new anti-infectives from marine organisms, we identified a methanol extract of the chrysophyte alga Chrysophaeum taylori that strongly inhibited the growth of Staphylococcus aureus, MRSA, Enterococcus faecium, and vancomycin-resistant E. faecium (VREF). Bio-assay and LC-MS guided fractionation led to the isolation of eight new polyhalogenated, polyoxygenated bisdiarylbutene ether macrocycles termed chrysophaentin A-H (1-8). Their planar structures were determined by extensive spectroscopic methods including NMR and MS. Here we describe the antimicrobial activities of compounds 1-8; and by using enzymatic assays, transmission electron microscopy (TEM), Saturation Transfer Difference (STD) NMR and molecular docking we show chrysophaentin A to inhibit the GTPase activity and polymerization of FtsZ and establish its mode of binding.

Structure Determination
HR-ESI-MS of chrysophaentin A (1) gave a molecular ion at m/z 675.0154 [M-H]− consistent with a molecular formula of C32H24Cl4O8, requiring nineteen degrees of unsaturation. The presence of four Cl atoms was confirmed by MS in-source fragmentation experiments where fragmentation was induced by increasing the cone voltage from 30 to 125 eV. Fragment ions at m/z 649 [M-H-HCl]−, 603 [M-H-2HCl]−, 567 [M-H-3HCl]−, and 531 [M-H-4HCl]− and their respective isotopic patterns clearly indicated the loss of four consecutive chlorine atoms. The IR spectrum of 1 showed bands at 3380 and 1680 cm−1, implying the existence of hydroxyl and aromatic functionalities, respectively. The downfield region of the 1H NMR spectrum of 1 in MeOH-d4 contained signals for eight aromatic protons including two doublets at δ 6.18 (1H, d, J = 2.8 Hz) and 6.30 (1H, d, J = 2.8 Hz) corresponding to a tetrasubstituted benzene ring, a broad signal at δ 6.16 (2H, br s) suggesting the presence of two nearly equivalent protons,11 and four singlets at δ 6.179 (1H, s), 6.28 (1H, s), 6.81 (1H, s), and 6.84 (1H, s); as well as signals for two olefinic protons at δ 5.99 (1H, t, J = 8.7 Hz) and 6.07 (1H, t, J = 8.1 Hz). Thirty-two resonances were observed in the 13C NMR spectrum of 1, and the HSQC spectrum contained cross peaks ascribable to eight aromatic methine carbons (δ 103.8, 107.9, 109.1×2, 116.0, 116.7, 117.1, and 177.3), four benzyl methylene signals (δ 30.6, 30.4, 33.7, and 40.6), and two olefinic methines (δ 127.7 and 127.9).
Analysis of the 2D NMR data (HSQC, HMBC, COSY, and ROESY) of 1 allowed us to identify two main fragments (I and II) shown in Figure 1 as follows. HSQC and HMBC correlations from the aromatic protons at δ 6.16-6.84 led to the construction of four independent tetrasubstituted benzene rings, A-D (Figure 1). Rings A and C possessed chloro and alkyl substituents at positions 2 and 5, respectively, and two oxy substituents at positions 1 and 4. Rings B and D each contained three oxy substituents, with ring B displaying ortho-coupled AB-type proton signals and ring C displaying AA’-type signals. COSY data then correlated the olefinic triplet at δ 5.99 (H-8) to the benzylic methylene at δ 3.23 (2H, d, J = 8.7 Hz, H-7) and in turn, diagnostic HMBC correlations observed from these methylene protons to the carbon resonances at δ 116.0 (C-6), 126.7 (C-5), 150.4 (C-4), and 134.7 (C-9), and from the benzylic methylene protons at δ 3.39 (2H, br s, H-10) to the carbon resonances at δ 107.9 (C-12), 133.0 (C-11), 135.9 (C-16), and 127.7 (C-8), linked rings A and B via a 2-butene chain. The partial structure I was completed by assigning the deshielded quaternary carbon at δ 134.7 (C-9) to a chlorinated alkene. The geometry of the C8/C9 double bond was established as E on the basis of strong ROEs between the methylene protons H-7 and H-10. Thus, the partial structure I was characterized by the presence of two aryl rings ω,ω’-linked to an (E)-2-chlorobut-2-ene moiety.
Figure 1.
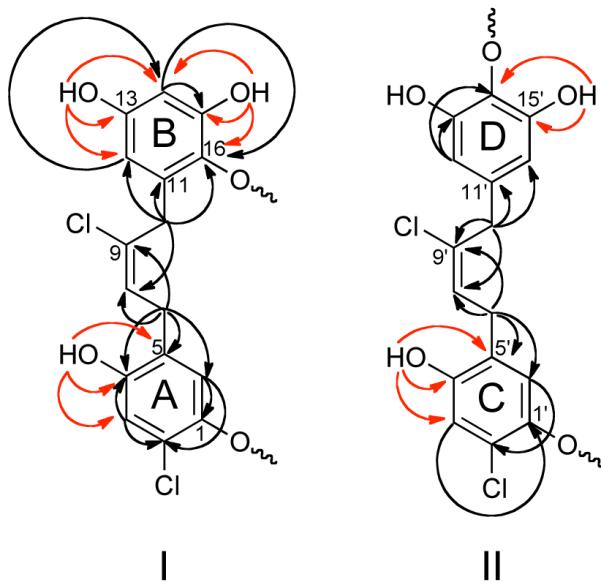
Key HMBC correlations used to establish partial structures corresponding to fragments I and II in compound 1. 1H–13C and OH–13C correlations are shown by black and red arrows, respectively.
2D NMR data corresponding to fragment II closely resembled those of I, and indicated that the partial fragment II also contained an ω,ω’-diarylalkene unit. In particular, a set of long-range correlations from the methylene protons at δ 3.57 (2H, br s, H-10′) to the aromatic carbons at δ 109.1 (C-12′ and C-16′), to the chlorinated olefinic quaternary carbon at δ 134.4 (C-9′), and to the olefinic methine carbon at δ 127.9 (C-8′) linked ring D to the 2-butene moiety as shown. Additional HMBC correlations from the remaining benzylic methylene at δ 3.28 (2H, d, J = 7.9 Hz, H-7′) to the aromatic carbons at δ 116.7 (C-6′) and 150.7 (C-4′), and to the olefinic carbons C-8′ and C-9′, linked ring C to the remainder of fragment II (Figure 1), and ROEs between the methylene protons H-7′ and H-10′ indicated an E geometry at the C-8′/C-9′ double bond of fragment II (see table S1).
Inspection of the partial fragments I and II revealed that together they contained eighteen of the required nineteen degrees of unsaturation, and only eighteen of the twenty-four protons were attached to carbons. Consequently there had to be six hydroxyl groups and fragments I and II had to be connected through two ether linkages to satisfy the unsaturation index and molecular formula of 1. This was further corroborated by acetylation of 1, which yielded the hexa-acetate 1a (Table S1).
The 1H NMR spectrum of 1 recorded in DMF-d7 displayed excellent line shape and resolution for the resonances ascribable to six hydroxyl protons at δ 9.40-10.1. (Spectra of 1 dissolved in DMSO-d6, CD3CN, and CDCl3 were also recorded but gave unsatisfactory results.) In fact, HMBC and ROESY correlations from these hydroxyl protons allowed unambiguous assignment of the positions of the hydroxyl groups and ether linkages connecting fragments I and II. Long-range correlations from the hydroxyl proton at δ 9.90 (1H, s, OH-4) to the carbon resonances at δ 116.1 (C-3), 125.6 (C-5), and 149.7 (C-4), and from the hydroxyl resonance at δ 10.1 (1H, s, OH-4′) to the carbon resonances at δ 116.0 (C-3′), 125.9 (C-5′), and 150.4 (C-4′), showed that the ether bonds must occur at C-1 and C-1′ in rings A and C, respectively. Similarly, HMBC correlations from the hydroxyl protons at δ 9.41 (1H, s, OH-13) and δ 9.58 (1H, s, OH-15) to carbons corresponding to C-12-C–14 (δC 106.7, 155.5, 103.1) and C-14–C-16 (δC 103.1, 150.7, 134.9), respectively (Table 1), provided clear evidence of their respective locations at C-13 and C-15, as well as positioning the ether bond at C-16 in ring B. The chemical equivalency of the aromatic protons H-12′ and H-16′ (δ 6.25, br s), and of the hydroxyl protons OH-13′ and OH-15′ (δ 9.44, s) suggested a symmetrical arrangement of the substituents on ring D and positioning the ether linkage at C-14. This was further corroborated by HMBC correlations between protons OH-13′/OH-15′ and carbon resonances at δ 151.2 (C-13′/C-15′) and 128.6 (C-14′). Finally, connectivities between fragments I and II were assigned from a ROESY spectrum. In particular, ROEs between OH-15′ and H-6 (δ 6.39) required C-14′ of ring D to be connected to C-1 of ring A via an ether bond, while an ROE between H-6′ (δ 6.51) and methylene protons H-10 (δ 3.57) suggested rings B and C were linked by the second ether bond at C-16 and C-1′, respectively. Therefore the structure of chrysophaentin A was established as a macrocyclic dimer composed of two ω,ω’-diaryl-2-chlorobut-2-ene moieties linked through two ether bonds in an assymetrical fashion.
Table 1.
NMR Spectroscopic Data for Chrysophaentin A (1)
| 1 (MeOH-d4) | 1 (DMF-d7) | |||||||
|---|---|---|---|---|---|---|---|---|
| position | δ Ca | δHb (J in Hz) | HMBCc | ROESYd | δ Ce | δHf (J in Hz) | HMBCa | ROESYd |
| 1 | 148.1 | 146.8 | ||||||
| 2 | 120.0 | 118.2 | ||||||
| 3 | 117.1 | 6.81 s | 1, 2, 4, 5 | 116.1 | 7.00 s | 1, 2, 4, 5 | OH-4 | |
| 4 | 150.4 | 149.7 | ||||||
| OH-4 | 9.90 s | 3, 4, 5 | 3, 8 | |||||
| 5 | 126.7 | 125.6 | ||||||
| 6 | 116.0 | 6.179 s | 1, 2, 4, 5, 7 | 115.2 | 6.39 s | 1, 2, 4, 5 | 7, 10, OH-15′ | |
| 7 | 30.6 | 3.23 d (8.7) | 4, 5, 6, 8, 9 | 10 | 29.8 | 3.36 d (8.8) | 4, 5, 8, 9 | 6, 8, 10 |
| 8 | 127.7 | 5.99 t (8.7) | 5, 7, 9, 10 | 126.3 | 6.05 t (8.8) | 7, 9, 10 | 7 | |
| 9 | 134.7 | 134.4 | ||||||
| 10 | 33.7 | 3.39 br s | 8, 9, 11, 12, 16 | 7, 12, 6′ | 32.3 | 3.57 | 8, 9, 11, 12 | 6, 7, 6′ |
| 11 | 133.0 | 131.7 | ||||||
| 12 | 107.9 | 6.18 d (2.8) | 10, 13, 14, 16 | 10 | 106.7 | 6.27 d (2.6) | 13, 14, 15, 16 | 10, OH-13 |
| 13 | 155.6 | 155.5 | ||||||
| OH-13 | 9.41 s | 12, 13, 14 | 12, 14 | |||||
| 14 | 103.8 | 6.30 d (2.8) | 12, 13, 15, 16 | 103.1 | 6.48 d (2.8) | 12, 16, 13, 15 | OH-14, OH-13 | |
| 15 | 151.3 | 150.7 | ||||||
| OH-15 | 9.58 s | 14, 15, 16 | 14 | |||||
| 16 | 135.9 | 134.9 | ||||||
| 1′ | 148.9 | 147.9 | ||||||
| 2′ | 121.1 | 120.1 | ||||||
| 3′ | 117.3 | 6.84 s | 1′, 2′, 4′, 5′ | 116.0 | 7.05 s | 1′, 2′, 4′, 5′ | OH-4′ | |
| 4′ | 150.7 | 150.4 | ||||||
| OH-4′ | 10.1 s | 3′, 4′, 5′ | 3′, 8′ | |||||
| 5′ | 127.2 | 125.9 | ||||||
| 6′ | 116.7 | 6.28 s | 1′, 2′, 4′, 5′, 7′ | 8′, 10′, 10 | 116.4 | 6.51 s | 1′, 2′, 4′, 5′, 7′ | |
| 7′ | 30.4 | 3.28 br d (8.1) | 4′, 5′, 6′, 9′ | 10′ | 30.3 | 3.36 br d (8.3) | 8′ | 6′, 8′ |
| 8′ | 127.9 | 6.07 t (8.1) | 5′, 7′, 9′, 10′ | 6′, 10′ | 127.1 | 6.23 t (8.3) | 9′ | 7′ |
| 9′ | 134.4 | 133.1 | ||||||
| 10′ | 40.6 | 3.57 br s | 8′, 9′, 11′, 12′ | 6′, 7′, 8′, 12′ | 39.2 | 3.72 | ||
| 11′ | 136.7 | 135.7 | ||||||
| 12′, 16′ | 109.1 | 6.16 br s | 12′, 13′, 14′ | 10′ | 107.6 | 6.25 | 11′ | OH-13′ |
| 13′, 15′ | 151.8 | 151.2 | ||||||
| OH-13′, OH-15′ |
9.44 s | 13′, 14′ | 6, 12′ | |||||
| 14′ | 129.7 | 128.6 | ||||||
Recorded at 125 MHz; referenced to residual MeOH-d4 at δ 49.1.
Recorded at 500 MHz; referenced to residual MeOH-d4 at δ 3.30.
Proton showing HMBC correlation to indicated carbon.
Proton showing ROESY correlation to indicated carbon.
Recorded at 125 MHz; referenced to residual DMF-d7 at δ 34.89.
Recorded at 500 MHz; referenced to residual DMF-d7 at δ 2.92.
To view the conformational features of chrysophaentin A, a 3D model was constructed via a full exploration of the conformational space of 1 using the AMBER force field12 (see Computational Details), with each of the obtained conformations minimized with the Polak-Ribier Conjugate Gradient (PRCG) algorithm. The global minimum energy conformer is shown in Figure 2 where the interproton distances observed are in good agreement with the ROESY data. In particular the distances measured between protons H-6/H-10 (2.5 Å), H-10/H-12 (2.8 Å), H-6′/H-8′ (2.6 Å), H-7′/H-10′ (2.3 Å), and H-7/H-10 (3.2 Å) were consistent with the strong ROEs observed for each of these proton pairs.
Figure 2.
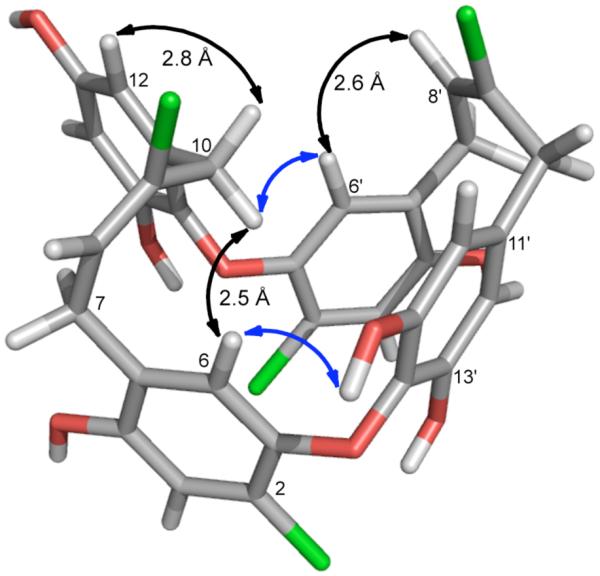
Global minimum energy conformer of 1 obtained by molecular dynamics and PRCG minimization. Selected strong ROE’s are indicated with double-sided black arrows together with corresponding interproton distances observed in the model. Blue arrows show key ROEs used to connect fragments I and II.
HR-ESI-MS and MS in-source experiments showed chrysophaentin B (2) and C (3) to possess the same molecular formula, C32H24BrCl3O8 (m/z 718.9655 [M-H]− and 718.9650 [M-H]−), while that of chrysophaentin D (4) was assigned as C32H24Br2Cl2O8 (m/z 762.9168 [M-H]−). The 2D NMR data (HSQC, HMBC, COSY, and ROESY) for compounds 2-4 were almost superimposable with those of 1. Indeed, analysis of the 1D and 2D NMR data showed 2 and 3 to be the 2- and 2′-brominated analogues of 1, while 4 was shown to be the 2,2′-dibrominated analogue (see Table S2 for NMR data for compounds 2-4).
The HR-ESI-MS of chrysophaentin E (5) showed a pseudomolecular ion peak at m/z 677.0317 [M-H]− corresponding to a molecular formula of C32H26Cl4O8 (calcd for C32H25Cl4O8, 677.0304) that differed from that of 1 by addition of two hydrogen atoms and required eighteen degrees of unsaturation, one less than compound 1. The 1H NMR spectrum of 5 in MeOH-d4 (see Table 2) showed signals corresponding to four benzyl methylenes at δ 3.32 (2H, d, J = 7.9 Hz), 3.41 (2H, d, J = 7.8 Hz), 3.52 (2H, br s), and 3.69 (2H, br s), and two olefinic triplets at δ 5.88 (1H, t, J = 7.8 Hz) and 5.73 (1H, t, J = 7.8 Hz), suggesting the presence of two sets of 2-butene chains. The aromatic region of the 1H NMR spectrum also revealed four downfield singlets at δ 6.44 (1H, s), 6.70 (1H, s), 6.74 (1H, s), and 6.84 (1H, s); three meta-coupled protons belonging to an AA’B spin system at δ 6.13 (1H, d, J = 1.8 Hz) and 6.16 (2H, d, J = 1.8 Hz) and corresponding to a 1,3,5-trisubstituted benzene ring; and two singlets belonging to an AA’ spin system at δ 6.41 (2H, s) corresponding to a symmetrical tetrasubstituted ring. The 2D NMR data of 5 were consistent with two sets of two aryl rings ω,ω’-linked to an (E)-2-chlorobut-2-ene chain and closely resembled that of 1; however there were some significant differences in the chemical shifts of protons and carbons of rings B and C. The most notable difference appeared at C-16 (ring B) where the oxygenated substitution in 1 was absent in 5. Moreover, the 1H NMR spectrum of 5 in DMF-d7 showed signals corresponding to seven hydroxyl protons (one more than 1) at δ 9.34 (2H, s), 9.52 (1H, s), 9.55 (1H, s), 9.60 (2H, s), and 9.82 (1H, s). Together these data suggested the two diarylalkene moieties in 5 were linked via one ether bond only. HMBC correlations of the hydroxylated protons located the seven hydroxyl groups at C-4, C-13, C-15, C-1′, C-4′, C-13′, and C-15′. Thus, an ether bond must link C-1 in ring A to C-14′ in ring D, completing the structure of chrysophaentin E as an acyclic bisdiarylbutene (5).
Table 2.
NMR Spectroscopic Data for Chrysophaentin E (5)
| 5 (MeOH-d4) | 5 (DMF-d7) | ||||||
|---|---|---|---|---|---|---|---|
| position | δ Ca | δHb (J in Hz) | HMBCc | δ Ce | δHf (J in Hz) | HMBCc | ROESYd |
| 1 | 148.3 | 147.3 | |||||
| 2 | 120.6 | 119.4 | |||||
| 3 | 116.9 | 6.84 s | 1, 2, 4, 5 | 116.3 | 7.01 s | 1, 2, 4, 5 | OH-4 |
| 4 | 150.6 | 150.2 | |||||
| OH-4 | 9.82 s | 3, 4, 5 | 3 | ||||
| 5 | 126.5 | 125.6 | |||||
| 6 | 116.5 | 6.44 s | 1, 2, 4, 5, 7 | 115.8 | 6.58 s | 1, 2, 4, 5, 7 | 7 |
| 7 | 30.0 | 3.32 d (7.9) | 4, 5, 6, 8, 9 | 29.3 | 3.41 d (7.5) | 1, 4, 5, 8, 9 | 6, 8, 10 |
| 8 | 127.6 | 5.73 t (7.9) | 5, 7, 9, 10 | 127.3 | 5.74 t (7.5) | 5, 7, 9, 10 | 7 |
| 9 | 134.2 | 133.2 | |||||
| 10 | 40.5 | 3.52 br s | 8, 9, 11, 12 | 39.7 | 3.61 br s | 8, 9, 11, 12 | 7, 12 |
| 11 | 140.9 | 139.8 | |||||
| 12, 16 | 108.1 | 6.16 br d (1.9) | 10, 13, 14, 16 | 107.2 | 6.25 br s | 10, 13, 14, 16 | OH-13 |
| 13, 15 | 159.3 | 159.3 | |||||
| OH-13, OH-15 |
9.34 s | 12, 13, 14 | 12, 14 | ||||
| 14 | 101.6 | 6.13 br d (1.9) | 12, 13 | 101.2 | 6.26 d (1.8) | 12, 13 | OH-13 |
| 1′ | 146.8 | 146.6 | |||||
| OH-1′ | 9.52 s | 1′, 2′, 6′ | 6′ | ||||
| 2′ | 119.1 | 120.3 | |||||
| 3′ | 116.8 | 6.74 s | 1′, 2′, 4′, 5′ | 115.9 | 6.91 s | 1′, 2′, 5′ | OH-4′ |
| 4′ | 149.4 | 148.8 | |||||
| OH-4′ | 9.55 s | 3′, 4′, 5′ | 3′ | ||||
| 5′ | 127.1 | 126.1 | |||||
| 6′ | 118.4 | 6.70 s | 1′, 2′, 4′, 5′, 7′ | 117.8 | 6.90 s | 2′, 4′, 7′ | OH-1′, 7′, 8′ |
| 7′ | 29.9 | 3.41 d (7.9) | 4′, 5′, 6′, 8′, 9′ | 29.0 | 3.48 | 5′, 6′, 8′, 9′ | 6′, 8′, 10′ |
| 8′ | 128.3 | 5.88 t (7.9) | 5′, 7′, 9′, 10′ | 127.5 | 5.92 t (7.7) | 5′, 9′, 10′ | 6′, 7′ |
| 9′ | 133.4 | 133.1 | |||||
| 10′ | 40.3 | 3.69 br s | 8′, 9′, 11′, 12′ | 39.3 | 3.74 br s | 8′, 9′, 11′, 12′ | 7′, 12′ |
| 11′ | 136.4 | 135.1 | |||||
| 12′, 16′ | 109.3 | 6.41 s | 10′, 12′, 13′, 14′ | 108.3 | 6.49 | 11′, 12′, 13′, 14′ | 10′, OH-13′ |
| 13′, 15′ | 151.6 | 151.5 | |||||
| OH-13′, OH-15′ |
9.60 s | 12′, 13′, 14′ | 12′ | ||||
| 14′ | 130.3 | 129.4 | |||||
Recorded at 125 MHz; referenced to residual MeOH-d4 at δ 49.1.
Recorded at 500 MHz; referenced to residual MeOH-d4 at δ 3.30.
Proton showing HMBC correlation to indicated carbon.
Proton showing ROESY correlation to indicated carbon.
Recorded at 125 MHz; referenced to residual DMF-d7 at δ 34.89.
Recorded at 500 MHz; referenced to residual DMF-d7 at δ 2.92.
The total ion chromatogram (TIC) obtained from the LC-MS contained two additional compounds (6-7) with masses identical to 1-3 but eluting at considerably longer retention times. Among this group, chrysophaentin F (6) eluted from the C-12 HPLC column after chrysophaentin A (1 tR = 28.0 min; 6 tR = 41.3 min) and its molecular formula was determined to be C32H24Cl4O8 by HR-ESI-MS (675.0140 [M-H]−, calcd for C32H23Cl4O8, 675.0147), indicating 6 to be an isomer of 1. The 1H NMR and HSQC spectra of 6 displayed characteristic signals for a single ω,ω’-diarylalbutene unit, including two benzylmethylenes (δH 3.35, δC 31.0; δH 3.54, δC 39.8), one olefinic triplet (δH 5.89, δC 127.2), and four aromatic singlets (δH 6.23, δC 110.0 × 2; δH 6.42, δC 115.1; δH 6.90, δC 117.3; Table S3). HMBC and COSY experiments led to the identification of a 2-chlorobut-2-ene moiety linked in position 1 to a 2-chlorine-5-alkylbenzene-1,4-diol and in position 4 to a 5-alkylbenzene-1,2,3-triol. As these NMR data accounted for only half of the mass of 6, chrysophaentin F must be a symmetrical dimer comprising two identical diarylalkenes linked through two ether bonds. Chrysophaentin F differs from 1 in the location of the ether bond in ring B (para versus ortho), and connectivity was confirmed from an HMBC experiment (4JCH = 2 Hz) showing a correlation between H-6 and C-14′. Finally, the geometry of the double bond at C-8/C-9 was established as E on the basis of ROE correlations observed between the methylenes H-7 and H-10. Thus the structure of chrysophaentin F was established to be the symmetrical macrocyclic ether, 6.
Chrysophaentin G (7) displayed a major ion peak at m/z 718.9620 [M-H]− corresponding to a molecular formula of C32H24BrCl3O8, and differed from the molecular formula of 6 by the replacement of chlorine with a bromine atom. Successively, comparison of the 2D NMR data of 7 (Table S4) to that of 6 determined that the bromine atom was located at the C-2′ position. Chrysophaentin H (8) was the most hydrophobic compound of this group of antibiotics eluting from the C-12 HPLC column at even higher retention times than 6 (6 tR = 41.3 min; 8 tR = 50.5 min). Its molecular formula was assigned as C32H23BrCl4O8 (HR-ESI-MS m/z 752.9255 [M-H]−), which was 78 mu higher than 6. Once again, careful analysis of the 2D NMR data for 8 (Table S4) established chrysophaentin H to be the 12′-bromo-derivative of 6.
Antimicrobial Activity and Structure-Activity Relationships
Solid agar disk diffusion and microbroth dilution assays were used to evaluate the antimicrobial activities of compounds 1, hexa-acetate 1a, and 4-8 toward the drug susceptible bacteria Staphylococcus aureus (SA) and Enterococcus faecium, and drug resistant strains methicillin-resistant S. aureus (MRSA), multiple drug resistant SA (MDR-SA) and vancomycin-resistant E. faecium (VREF) (see Table 3). Our initial disk diffusion assays showed that only compounds 1, 4-6, and 8 inhibited the growth of all strains at loads ranging from 2-25 μg/disk. In both assay formats chrysophaentin A (1) was the most potent antibiotic giving respective minimum inhibitory concentrations (MIC50) of 1.8 ± 0.6, 1.5 ± 0.7, and 1.3 ± 0.4 μg/mL against SA, MRSA and MDR-SA; and 3.8 ± 1.9 and 2.9 ± 0.8 μg/mL toward E. faecium and VREF in microbroth dilution assays, and ~10 mm zones of inhibition at 2 μg/disk for all four strains. Chrysophaentins F and H (6 and 8) were the next most potent compounds with MIC50 values of 4-6 μg/mL toward S. aureus and MRSA, and ~9.5 μg/mL against VREF (see Table 3 for full profiles).
Table 3.
Antimicrobial Data for 1, 1a, 4-8.
| Agar disk diffusion (μg/disk)a | MIC50 (μg/mL) | |||||||
|---|---|---|---|---|---|---|---|---|
| compound | S. aureus | MRSA | E. faecium | VREF | S. aureus | MRSA | E. faecium | VREF |
| 1 | 2 | 2 | 2 | 2 | 1.8 ± 0.6 | 1.5 ± 0.7 | 3.8 ± 1.9 | 2.9 ± 0.8 |
| 1a | NAb | NA | NA | NA | — c | — | — | — |
| 4 | 25 | 25 | 25 | 25 | > 25 | 20 ± 6.5 | > 50 | > 25 |
| 5 | 25 | 10 | 10 | 10 | 11 ± 3.8 | 8.9 ± 2.8 | > 25 | > 25 |
| 6 | 10 | 10 | 10 | 10 | 5.3 ± 2.0 | 4.2 ± 1.3 | > 25 | 9.5 ± 3.0 |
| 7 | NA | NA | NA | NA | 17 ± 5.4 | 12 ± 3.1 | > 50 | 25 ± 7.3 |
| 8 | 5 | 5 | 10 | 10 | 4.5 ± 1.4 | 4.7 ± 1.4 | > 25 | 9.4 ± 2.8 |
Lowest concentrations leading to 7-10 mm zones of inhibition
NA, no zone of inhibition observed at 25 μg/disk
—, not tested.
These screening results provided insight into structure-activity relationships for the chrysophaentins. The hexaacetate derivative of chrysophaentin A (1a) was inactive at loads as high as 25 μg/disk showing the importance of the hydroxyl groups for antimicrobial activity of 1. The remarkably weaker potency of chrysophaentin D (4) compared to A (1) showed the detrimental effect of bromine compared to chlorine on rings A and C as replacement with bromines results in an approximate 12-fold decrease in MIC50 values toward all four strains. The acyclic metabolite chrysophaentin E (5) was also found to be inactive toward E. faecium and VREF at concentrations as high as 25 μg/mL, and showed significantly higher MIC50 values towards S. aureus and MRSA when compared to the chlorinated cyclic bisdiarylbutene ethers 1 and 6 establishing the importance of the macrocyclic structure. Among the symmetrically linked dimers 6 and 7, the tetrachloro compound 6 was at least 3x more potent than compound 7, which differs only by replacement of a chlorine atom on ring C by bromine. With regard to the respective arrangements of the two diaryl butene ether units, comparison of the antibacterial activities of the tetrachlorinated macrocycles 1 and 6 shows chrysophaentin A to be 3-5 times more potent than chrysophaentin F indicating the position of the ether linkage relative to the 2-butene unit affects activity. Specifically, the ortho-linked chrysophaentin A is more potent than the para-linked chrysophaentin F.
To assess specificity as an antimicrobial, we tested chrysophaentin A for cytotoxicity against the human colon tumor cell line HCT-116, the murine leukemia cell line P388, and a control mammalian cell line BSC-1. Interestingly, chrysophaentin A did not inhibit the growth of any of the cancer cell lines at concentrations as high as 50 μg/mL, and did not show cytotoxicity toward the control cells at concentrations as high as 100 μg/mL.
Inhibition of GTPase activity and Polymerization of FtsZ in vitro
The bacterial cytoskeletal protein FtsZ is a GTPase that plays a central role in bacterial cell division. At the time of replication, FtsZ localizes to the mid cell and undergoes GTP-dependent polymerization to form a dynamic and contractile structure known as the Z-ring, which marks the future plane of cell division. Inhibition of proper FtsZ assembly can block cell division by preventing Z-ring formation, ultimately leading to bacterial cell death. A number of natural product and synthetic FtsZ inhibitors have been reported, some of which contain multiple phenolic or aromatic moieties. Owing to structural similarities between chrysophaentins A and some FtsZ inhibitors, and our observation that 1 inhibits SA, MRSA and MDR-SA with almost identical potencies – suggesting a mechanism of action other than those exhibited by commonly used antibiotics – we tested chrysophaentin A for its ability to inhibit recombinant FtsZ in vitro by two means. Using a colorimetric GTPase assay that measures production of inorganic phosphate upon FtsZ-mediated hydrolysis of GTP to GDP+Pi, we showed chysophaentin A inhibits FtsZ in a dose-dependent manner with an IC50 value of 6.7 ± 1.7 μg/mL. Next we used transmission electron microscopy (TEM) to visualize the effects of 1 on polymerization of FtsZ. In the presence of GTP, FtsZ undergoes polymerization to form protofilaments (Figure 3a), while in the presence of chrysophaentin A polymerization is inhibited (Figure 3b). In contrast, 1 had no effect on tubulin polymerization at concentrations as high as 150 μM (see Supporting Information). Together these results demonstrate that chrysophaentin A is an FtsZ inhibitor that exhibits at least a 15-fold selectivity for FtsZ over tubulin.
Figure 3.

Transmission electron micrographs of FtsZ in the presence of (A) GTP, and (B) GTP and chrysophaentin A. (A) Polymerization of recombinant E. coli FtsZ was induced with addition of GTP for 5 min at rt before transferring to a carbon grid, staining with 3% uranyl acetate, and visualizing by TEM, 44 000 x. (B) Incubation of FtsZ with 1 (50 μM) prior to addition of GTP inhibits polymerization and protofilament formation. No filaments were observed over the entire grid shown in panel B.
It is interesting to note that chrysophaentin A inhibits the growth of multiple bacterial strains with MIC50 values that are considerably lower than the IC50 values observed in in vitro GTP-ase assays. This phenomenon has been observed for several other FtsZ inhibitors, including unrelated compounds totarol9d and OTBA9g for example. Because inhibition of FtsZ polymerization can result in inhibition of multiple down stream pathways involved in cytoskeletal reorganization and bacterial cell division, this differential activity has been attributed to amplification of the effect of inhibiting the first step in this process. Similar effects are observed with microtubule inhibitors where micromolar concentrations are required for in vitro inhibition of tubulin polymerization, while nanomolar concentrations disrupt microtubule assembly in vivo.13
Characterization of Chrysophaentin A Binding to FtsZ by STD NMR
To identify the regions of chrysophaentin A involved in FtsZ binding, we recorded Saturation Transfer Difference (STD) NMR14 spectra of 1 in the presence of recombinant FtsZ using published conditions optimized for our instrumentation. Samples typically contained a 100-fold excess of chrysophaentin A relative to FtsZ with respective concentrations of 1.5 mM and 15 μM. An expansion of a representative difference spectrum (red) and control spectrum (black) is shown in Figure 4 to include strongly enhanced and non-overlapping aromatic and olefinic protons of 1. Normalization of the signal/s of greatest intensity (δ 6.85) in the difference spectrum to those of the reference spectrum showed signals for the aromatic protons H-3 (100%), H-14 (100%), H-12 (100%), and H-3′ (98%) to display the strongest enhancements, while the overlapped signals of the aromatic and olefinic protons H-6′ and H-8 showed a combined enhancement of ~50%. Thus, when bound to FtsZ, the face of chrysophaentin A displaying protons H-3, H-14, H-12, and H-3′ (rings A, B and C) is in closest proximity to the protein. It is worth mentioning that under the buffer conditions used to prepare these complexes we were unable to observe signals for the remaining two aromatic protons H-12′ and H-16′, both of which reside on ring D, in either 1H or STD NMR spectra.
Figure 4.

Reference (black) and STD NMR difference (red) spectra of chrysophaentin A in complex with FtsZ. Spectral expansion shows non-overlapped aromatic and olefinic signals of 1 displaying the strongest enhancements upon binding to FtsZ. Samples comprising 100:1 1:FtsZ were prepared in 20 mM NaPO4, 50 mM NaCl, 5 mM MgCl2, pH 6.8, and spectra recorded at 298 K. Overlaid spectra were normalized to the signal for H-3 (δ 6.85), which gave the strongest enhancement.
Chrysophaentin A Binds FtsZ in the GTP Binding Site
Among those FtsZ inhibitors reported to date, inhibition of GTPase activity and/or polymerization can occur through multiple modes of binding to FtsZ. To gain insight into the mode of binding of chrysophaentin A to FtsZ, we performed competition STD NMR experiments where increasing amounts of a non-hydrolyzable GTP analog, guanosine 5′-O-(3-thiotriphosphate) (GTPγS), known to bind to the GTP binding site of FtsZ with high affinity,15 was added to a 100:1 complex of chrysophaentin A:FtsZ. Spectra were recorded as described above on samples containing 1.25 mM 1 in the presence of 12.5 μM FtsZ. Difference spectra were monitored for a change in intensity of signals belonging to either chrysophaentin A or GTPγS during the titration. As seen in the spectral expansions showing the aromatic and olefinic region of the difference spectra (Figure 5), addition of 0.5 equivalents of GTPγS (625 μM) resulted in an ~50% uniform decrease in intensity for signals belonging to 1. Moreover, a new STD NMR signal appeared at δ 5.84 (H-1′-GTPγS) that was assigned to the anomeric proton of the ribose of GTPγS (Figure 5B). Step wise addition of another two equivalents of GTPγS to the complex further diminished the signal intensities of 1 concomitant with steady increases in signal intensities for the anomeric and guanosine protons of GTPγS, H-1′GTPγS and H-8GTPγS. As seen in Figure 5e, by addition of 3 equivalents of GTPγS relative to 1, signals for the natural product were imperceptible having been replaced by those of GTPγS. Thus chrysophaentin A and GTPγS bind the GTP binding site of FtsZ in a competitive manner.
Figure 5.

Competition of chrysophaentin A and GTPγS binding to FtsZ by STD NMR. (a) Expanded 1H STD NMR spectrum of chrysophaentin A (1.25 mM) in the presence of FtsZ (12.5 μM). (b-e) STD NMR spectra recorded on the same sample after addition of (b) 0.5, (c) 1.0, (d) 2.0 and (e) 3.0 eq GTPγS. Final concentrations of GTPγS were 625 μM, 1.25 mM, 2.50 mM and 3.75 mM, respectively.
Several crystal structures of FtsZ in complex with GTPγS or GDP have been solved, providing a detailed view of the GTP binding site, which resides in the N-terminal domain and includes the conserved motif GGGTGTG that forms a large portion of the nucleotide binding site.16 To further evaluate the mode of binding of 1 to FtsZ, we performed molecular docking studies using the program Autodock Vina 1.0.317 (see Computational Details). Since a crystal structure of E. coli FtsZ is not available, we generated for docking a homology model using the 2.1 Å crystal structure of Pseudomonas aeruginosa FtsZ,18 which displays the highest degree of conservation to E. coli FtsZ, as template. Docking simulations placed chrysophaentin A in the GTP binding site, consistent with our experimental data. Moreover, the docked conformation and protein-ligand interactions observed for the fourth lowest energy binding mode (−6.6 kcal/mol vs −7.0 kcal/mol for the global minimum energy binding mode) was entirely consistent with our STD NMR results (see Figure 6).
Figure 6.

Molecular docking of 1 to FtsZ with chrysophaentin A bound in the GTP binding site. (A) Docked model of 1 bound to an E. coli FtsZ homology model; FtsZ is displayed as a white surface where atoms within 5 Å of docked 1 are colored yellow; chrysophaentin A is shown in ball and stick representation with chlorine and oxygen atoms colored green and red, respectively. Protons displaying the strongest enhancements are labeled in italics. (B) Superposition of the docked model shown in panel A with GDP bound FtsZ (pdb accession number 1ofu.pdb17). GDP (blue) is shown in a ball and stick representation, and surfaces of residues comprising the GTP binding site of FtsZ are colored blue. Docking was performed using the program Autodock Vina 1.0.3.
In this docked model, chrysophaentin A occupies the triphosphate region of the GTP binding site and partially occludes the guanine-binding site as well. The docked model places 1 within hydrogen bonding distances of the side chain or back bone N and O atoms of Arg142, Gly20, Ala70, Asn43 Thr108, and Asn24. Furthermore, protons H-3, H-14, and H-12 which displayed the strongest STD enhancements (100%) are within van der Waal’s distances of Arg142 (2.9 Å), Thr108 (2.4 Å), and Ala48 (2.2 Å), respectively; while H-3′ of ring C (98% enhancement) is in close proximity to Gly20 (3.5 Å) and Gly21 (3.7 Å). Hydrophobic interactions are also apparent for olefinic protons H-8 and Gly71 (3.5 Å), and H-8′ and Ala48 (2.2 Å). Finally, the aromatic protons H-6 and H-6′ that displayed much weaker enhancements (~ 50%) are positioned 4.5 Å or greater from the protein surface suggesting a smaller contribution to FtsZ binding. It is interesting to note that sequence alignments with other FtsZ proteins whose x-ray structures are available show all residues in contact with chrysophaentin A in the E. coli model to be predicted to form a portion of the GTP binding site. For instance, sequence alignments would indicate Gly20, Gly21, and Asn24 to be involved in base recognition; Asn43, Ala48, Ala70, and Thr108 to be involved in phosphate binding; and Arg142 to contribute to ribose recognition,17 and these same residues are also in closest contact with 1. These results support the notion that chrysophaentin A inhibits the GTPase activity and polymerization of FtsZ through binding to the nucleotide binding site in a competitive manner with GTP.
Conclusions
In recent years, the bacterial cytoskeletal protein FtsZ, a tubulin homolog that helps to initiate bacterial cell division, has been the focus of numerous antimicrobial discovery efforts.9a,d,e,19 Here we have found a suite of marine natural products occurring in a chrysophyte alga, some of which exceed the potencies of other natural product FtsZ inhibitors. The structures of chrysophaentins A-H represent a new natural product scaffold comprising two polyhydroxylated, polyhalogenated ω,ω’-diarylbutene units connected through two ether bonds to form symmetrical or asymmetrical macrocycles. In multiple biological, biochemical and structural studies presented here, we have shown chrysophaentin A to inhibit the function of FtsZ in vitro, to be a competitive inhibitor with the natural substrate GTP, and to exhibit specificity over the eukaryotic homolog tubulin by at least 15-fold. These results provide a comprehensive approach to identifying and determining the mode of action of novel FtsZ inhibitors, and emphasize the value of marine natural products in discovery efforts.
Experimental Section
NMR Spectroscopy
NMR spectra of natural products were recorded in CD3OD or DMF-d7 at 600 MHz with an x,y,z-shielded gradient triple resonance probe, or at 500 MHz with a cryogenically cooled z-shielded gradient triple resonance probe. DQF-COSY, 2D-HOHAHA, HSQC, HMBC, and ROESY experiments were recorded using standard pulse programs with water suppression (Watergate). HSQC experiments were recorded with dwell times of 1.724 ms (1JC-H 145 Hz), and HMBC spectra with dwell times of 31.25 and 50 ms (2,3JC-H = 8 Hz and 5 Hz).
Saturation transfer difference (STD) NMR experiments were recorded with the carrier set at −1 or 12 ppm for on-resonance irradiation and 40 ppm for off-resonance irradiation. Control spectra were recorded under identical conditions on samples containing free compound 1 to test for artifacts. Selective protein saturation (2 s) was accomplished using a train of 50 ms Gauss-shaped pulses, each separated by a 1 ms delay, at an experimentally determined optimal power (49 dB on our probe); a T1ρ filter (30 ms) was incorporated to suppress protein resonances. Experiments were recorded using a minimum of 1024 scans and 32K points. On- and off-resonance spectra were processed independently, and subtracted to provide a difference spectrum.
Computational Details
To allow full exploration of the conformational space of chrysophaentin A, molecular dynamics (MD) calculations were performed at three different temperatures (300 K, 500 K, 700 K/50 ns) using the AMBER force field (MacroModel software package)12 to give 100 structures, each of which was minimized using the Polak-Ribier Conjugate Gradient algorithm (PRCG, 1000 steps, maximum derivative less than 0.05 kcal/mol). These calculations provided the lowest energy minimum conformer for chrysophaentin A. A parallel analysis was performed using the MonteCarlo Multiple Minimum (MCMM) method (50K steps) of the MacroModel package leading to the same results obtained by MD calculations. Docking of the lowest energy structure of 1 to an E. coli homology model of FtsZ (built using SWISS-MODEL20 routines starting from the coordinates of Pseudomonas aeruginosa FtsZ, pdb accession code 1ofu.pdb18) was carried out with the program Autodock Vina 1.0.3. After initial docking runs showed all reasonable models to place 1 in or near to the GTP binding site, the grid was narrowed to an area slightly larger than the GTP binding site (28×20×24 Å). Docking was carried out with an exhaustiveness value of 512 and a maximum output of 25 structures. Agreement with the STD NMR data and calculated energies were used to arrive at the best docked-model. This approach provided a model that displayed the fourth lowest energy of the group, and its position within the GTP binding site was in full agreement with the measured STD NMR enhancements.
Biological Material
Samples of the chrysophyte alga Chrysophaeum taylori Lewis and Bryan21 were collected at −20 ft from Round Bay on the Island of St. John during the summer months of 2007 and 2009. The alga, found growing as fluffy colonies on course sand or coral rubble substrate, appears sulfur yellow in color, and when disturbed can turn to a rusty brown color within seconds. The multicellular structure of C. taylorii is extremely fragile and the mucilaginous cells do not preserve well. However, when freshly collected or freshly preserved (2.5% glutaraldehyde in sea water) samples are viewed by light microscopy, stalk like structures made up of branching mucilaginous ‘streamers’ of pear shaped, invaginated cells were visible. In addition, algal collections contained the known styrylchromone hormothamnione, and bore an uncanny resemblance at all stages of handling to field notes published by Gerwick in 1989.22
Isolation
The lyophilized material (200 g) was sequentially extracted with hexanes, chloroform, and methanol. The methanol extract (13 g) was partitioned between n-BuOH-H2O (1:1) and the organic layer (1.1 g) was fractionated on Sephadex LH-20. Fractions containing diarylalkene ethers (75.1 mg) were purified by reverse-phase HPLC (Jupiter Proteo C12, 250 × 10 mm, 4μ, DAD at 220 and 280 nm, flow rate 2.5 mL/min) eluting with a linear gradient of 50–80% MeOH in 0.05% TFA in 50 min to afford compounds 1 (3.5 mg, tR = 28.0 min), 2 (0.4 mg, tR = 28.6 min), 3 (0.8 mg, tR = 29.4 min), 4 (1.8 mg, tR = 32.0 min), 5 (2.6 mg, tR = 33.3 min), 6 (1.5 mg, tR = 41.3 min), 7 (1.4 mg, tR = 41.8 min), 8 (0.7 mg, tR = 50.5 min).
Chrysophaentin A (1)
colorless amorphous powder; non-optically active; UV (MeOH) λmax (log ε) 210 (4.2), 225 (3.9), 290 (3.4); IR (film) νmax 3384, 1675, 1449, 1203, 1143, 846, 802, 727; 1H and 13C NMR data, see Table 11; HR-ESI-MS 675.0154 [M-H]− corresponding to a molecular formula of C32H24Cl4O8 (calcd for C32H23Cl4O8, 675.0147).
Chrysophaentin B (2)
colorless amorphous powder; non-optically active; UV (MeOH) λmax (log ε) 210 (4.2), 225 (3.8), 290 (3.4); IR (film) νmax 3381, 1681, 1608, 1447, 1207, 1143, 846, 802, 723; 1H and 13C NMR data, see Table S1; HR-ESI-MS 718.9655 [M-H]− corresponding to a molecular formula of C32H24BrCl3O8 (calcd for C32H23BrCl3O8, 718.9642).
Chryophaentin C (3)
colorless amorphous powder; non-optically active; UV (MeOH) λmax (log ε) 210 (4.2), 225 (3.9), 290 (3.4); IR (film) νmax 3387, 1680, 1608, 1444, 1209, 1145, 846, 802, 722; 1H and 13C NMR data, see Table S1; HR-ESI-MS 718.9650 [M-H]− corresponding to a molecular formula of C32H24BrCl3O8 (calcd for C32H23BrCl3O8, 718.9642).
Chrysophaentin D (4)
colorless amorphous powder; non-optically active; UV (MeOH) λmax (log ε) 210 (4.1), 225 (3.9), 290 (3.4); IR (film) νmax 3397, 1681, 1608, 1447, 1207, 1144, 1016, 844, 802, 723; 1H and 13C NMR data, see Table S1; HR-ESI-MS 762.9168 [M-H]− corresponding to a molecular formula of C32H24Br2Cl2O8 (calcd for C32H23Br2Cl2O8, 762.9137).
Chrysophaentin E (5)
colorless amorphous powder; non-optically active; UV (MeOH) λmax (log ε) 210 (4.2), 225 (3.8), 290 (3.4); IR (film) νmax 3386, 1686, 1608, 1451, 1201, 1148, 849, 808, 730; 1 H and 13C NMR data, see Table 2; HR-ESI-MS 677.0317 [M-H]− corresponding to a molecular formula of C32H26Cl4O8 (calcd for C32H25Cl4O8, 677.00304).
Chrysophaentin F (6)
colorless amorphous powder; non-optically active; UV (MeOH) λmax (log ε) 210 (4.2), 225 (3.8), 290 (3.4); IR (film) νmax 3385, 1675, 1606, 1453, 1201, 1144, 846, 802, 733; 1H and 13C NMR data, see Table S2; HR-ESI-MS 675.0140 [M-H]− corresponding to a molecular formula of C32H24Cl4O8 (calcd for C32H23Cl4O8, 675.0147).
Chrysophaentin G (7)
colorless amorphous powder; non-optically active; UV (MeOH) λmax (log ε) 210 (4.2), 225 (3.8), 290 (3.4); IR (film) νmax 3381, 1677, 1601, 1435, 1210, 1148, 842, 812, 721; 1H and 13C NMR data, see Table S3; HR-ESI-MS 718.9620 [M-H]− corresponding to a molecular formula of C32H24BrCl3O8 (calcd for C32H23BrCl3O8, 718.9642).
Chrysophaentin H (8)
colorless amorphous powder; non-optically active; UV (MeOH) λmax (log ε) 210 (4.2), 225 (3.8), 290 (3.4); IR (film) νmax 3381, 1678, 1605, 1447, 1207, 1144, 840, 802, 725; 1H and 13C NMR data, see Table S3; HR-ESI-MS 752.9255 [M-H]− corresponding to a molecular formula of C32H23BrCl4O8 (calcd for C32H22BrCl4O8, 752.9252).
Antimicrobial activity
Antimicrobial assays, including agar disk diffusion and microbroth dilution formats, were performed as described in the CLSI guidelines where the solid agar assays were performed using plates seeded with overnight cultures of Staphylococcus aureus (ATCC 25923), methicillin-resistant S. aureus (ATCC BAA-41), Enterococcus faecium (ATCC 49032), vancomycin-resistant E. faecium (ATCC 70022), or Bacillus subtilis (ATCC 49343). Pure compounds or antibiotic standards were added to the plates on paper disks, and zones of inhibition measured after incubation for 18 h at 37 °C. Minimum inhibitory concentrations (MIC50) were determined using 96-well microbroth dilution assays. Antimicrobial activity of pure compounds or antibiotic standards were tested by adding serial dilutions to wells containing S. aureus, MRSA, or B. subtilis in MHII broth, or E. faecium or VRE in wells containing 10% Brain Heart Infusion broth. Plates were incubated at 37 °C overnight and absorbance at 600 nm measured.
GTPase activitiy of FtsZ in vitro
GTPase activity of recombinant E. coli FtsZ was measured in 96-well plate format using a colorimetric assay that measures inorganic phosphate production. Solutions containing FtsZ (2 μM) in MES buffer (50 mM MES, pH 6.5, 50 mM KCl, 5 mM MgCl2) were treated with serial dilutions of 1 (final concentrations 0–50 μg/mL in 5% DMSO), followed by addition of GTP added to a final concentration of 0.25 mM. Reactions were quenched 20 min later by addition of malachite green/ammonium molybdate and the color developed for 30 min. Inorganic phosphate was quantified by absorbance at 625 nm.
Protofilament formation of E. coli FtsZ was assessed by polymerization assays. Solutions containing recombinant E. coli FtsZ (6 μM) in MES buffer were treated with 5% DMSO, or 50 μM 1 in 5% DMSO, for 2 min at rt, followed by addition of GTP. Following an additional 5 min incubation period at rt, aliquots (5 μL) were adsorbed onto carbon films on lacey carbon supports on 400 mesh copper grids, rinsed with H2O, and exposed to 3% uranyl acetate for 5 min for negative staining. Images were acquired with an FEI Morgani transmission electron microscope, operating at 80kV, and equipped with an AMT Advantage camera, at 44,000x magnification.
Supplementary Material
Acknowledgement
We thank K. Thurber and R. Tycko for assistance with and use of the Morgani TEM, M. Bewley and M. Clore for help with collections, and W. Fenical and W. Gerwick for helpful discussions on chrysophyte algae. This work was supported by the NIH Intramural Research Program (NIDDK), and the Intramural AIDS Targeted Antiviral Program, Office of the Director, NIH (C.A.B.).
Footnotes
Supporting Information Available: Complete References 9d and 19; general experimental section; 1H and 13C NMR data and spectra for compounds 1a, 2-4, 6-8; 1H NMR and STD NMR spectra for complex of 1:FtsZ in aqueous buffer; and coordinates for the docked model of the global minimum energy structure of 1 to a homology model of E. coli FtsZ. This material is available free of charge on the internet at http://pubs.acs.org.
References
- 1.Rasko DA, Sperandio V. Nat. Rev. Drug Discovery. 2010;9:117. doi: 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- 2.Payne DJ. Science. 2008;321:1644. doi: 10.1126/science.1164586. [DOI] [PubMed] [Google Scholar]
- 3.Levy SB, Marshall B. Nat. Med. 2004;10:S122. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 4.Breukink E, de Kruijff B. Nat. Rev. Drug. Discov. 2006;5:321. doi: 10.1038/nrd2004. [DOI] [PubMed] [Google Scholar]
- 5.Lock RL, Harry EJ. Nat. Rev. Drug Discovery. 2008;7:324. doi: 10.1038/nrd2510. [DOI] [PubMed] [Google Scholar]
- 6.Oliva MA, Cordell SC, Löwe J. Nat. Struct. Mol. Biol. 2004;11:1243. doi: 10.1038/nsmb855. [DOI] [PubMed] [Google Scholar]
- 7.Bi EF, Lutkenhaus J. Nature. 1991;354:161. doi: 10.1038/354161a0. [DOI] [PubMed] [Google Scholar]
- 8.Kapoor S, Panda D. Expert Opin. Ther. Targets. 2009;13:1037. doi: 10.1517/14728220903173257. [DOI] [PubMed] [Google Scholar]
- 9.a) Wang J, Galgoci A, Srinivas K, Herath KK, Jayasuriya H, Dorso K, Vicente F, Gonzalez DC, Bramhill D, Singh S. J. Biol. Chem. 2003;278:44424. doi: 10.1074/jbc.M307625200. [DOI] [PubMed] [Google Scholar]; b) Domadia PN, Bhunia A, Sivaraman J, Swarup S, Dasgupta D. Biochemistry. 2008;47:3225. doi: 10.1021/bi7018546. [DOI] [PubMed] [Google Scholar]; (c) Domadia P, Swarup S, Bhunia A, Sivaraman J, Dasgupta D. Biochem. Pharmacol. 2007;74:831. doi: 10.1016/j.bcp.2007.06.029. [DOI] [PubMed] [Google Scholar]; d) Jaiswal R, Beuria TK, Mohan R, Mahajan SK, Panda D. Biochemistry. 2007;46:4211. doi: 10.1021/bi602573e. [DOI] [PubMed] [Google Scholar]; (e) Haydon DJ, et al. Science. 2008;321:1673. doi: 10.1126/science.1159961. [DOI] [PubMed] [Google Scholar]; (f) Margalit DN, Romberg L, Mets RB, Hebert AM, Mitchison TJ, Kiirschner MW, RayChaudhuri D. Proc. Natl. Acad. Sci. 2004;101:11821. doi: 10.1073/pnas.0404439101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Beuria TK, Singh P, Surolia A, Panda D. Biochem. J. 2009;423:61. doi: 10.1042/BJ20090817. [DOI] [PubMed] [Google Scholar]
- 10.a) Kwon HC, Kauffman CA, Jensen PR, Fenical W. J. Am. Chem. Soc. 2006;128:1622. doi: 10.1021/ja0558948. [DOI] [PubMed] [Google Scholar]; b) McArthur KA, Mitchell SS, Tsueng G, Rheingold A, White DJ, Grodberg J, Lam KS, Potts BCM. J. Nat. Prod. 2008;71:1732. doi: 10.1021/np800286d. [DOI] [PubMed] [Google Scholar]; (c) Keffer JL, Plaza A, Bewley CA. Org. Lett. 2009;11:1087. doi: 10.1021/ol802890b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Stout EP, Hasemeyer AP, Lane AL, Davenport TM, Engel S, Hay ME, Fairchild CR, Prudhomme J, Le Roch K, Aalbersberg W, Kubanek J. Org. Lett. 2009;11:225. doi: 10.1021/ol8024814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamazaki M, Maebayashi Y. Chem. Pharm. Bull. 1982;30:514. [Google Scholar]
- 12.Mohamadi F, Richards NG, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. J. Comput. Chem. 1990;11:440. [Google Scholar]
- 13.Jordan MA, Wilson L. Nat. Rev. Cancer. 2004;4:253. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 14.Mayer M, Meyer B. J. Am. Chem. Soc. 2001;123:6108. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- 15.Scheffers D–J, den Blaauwen T, Driessen AJM. Mol. Microbiol. 2000;35:1211. doi: 10.1046/j.1365-2958.2000.01791.x. [DOI] [PubMed] [Google Scholar]
- 16.Löwe J, Amos LA. Nature. 1998;391:203. doi: 10.1038/34472. [DOI] [PubMed] [Google Scholar]
- 17.Trott Oleg, Olson AJ. J. Comput. Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cordell SC, Robinson EJH, Löwe J. Proc. Natl. Acad. Sci. 2003;101:11821. [Google Scholar]
- 19.Czaplewski LG, et al. Bioorg. Med. Chem. Lett. 2009;19:524. doi: 10.1016/j.bmcl.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 20.Arnold K, Bordoli L, Kopp J, Schwede T. Bioinformatics. 2006;22:195. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 21.Lewis IF, Bryan HF. Am. J. Bot. 1941;28:343. [Google Scholar]
- 22.Gerwick WH. J. Nat. Prod. 1989;52:252. doi: 10.1021/np50062a005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.