

Abstract
Mitochondria are key organelles that perform essential cellular functions and play pivotal roles in cell death and survival signaling. Hence, they represent an attractive target for drugs to treat metabolic, degenerative and hyperproliferative diseases. Targeting mitochondria with organelle-specific agents or prodrugs has proven to be an effective therapeutic strategy. More specifically, controling the cellular ROS balance via selective delivery of an antioxidant “payload” into mitochondria is an elegant emerging therapeutic concept. Herein, we review the recent medicinal chemistry and clinical data of these exploratory strategies which should point the way for future generations of therapeutics.
Keywords: mitochondria-targeted antioxidant, reactive oxygen species, oxidative stress, nitroxide, TEMPO, gramicidin S
1. Why targeting mitochondria?
The mitochondrion is a discreet organelle present in most eukaryotic cells. Its unusual structure is comprised of four distinct compartments that carry out specialized functions: the outer mitochondrial membrane (OMM), the intermembrane space (IMS), the inner mitochondrial membrane (IMM), and the mitochondrial matrix. The IMM is highly folded into cristae, which house the protein complexes of the electron transport chain (ETC) and F1F0-ATPase, controlling the fundamental rates of cellular metabolism. This essential role of the mitochondrion is responsible for its reference as the “power plant of the cell”. However, the function of mitochondria is not limited to supplying cellular energy (McBride et al. 2006). Adenosine triphosphate (ATP) production through the oxidative phosphorylation (OXPHOS) process requires a continuous flow of electrons. As such, mitochondria are the major source of reactive oxygen species (ROS, i.e. superoxide and H2O2), generated as byproducts of the ETC (Murphy 2009; Starkov 2008). ROS reflect the level of cellular oxidative stress, causing severe damage to macromolecules when overproduced. Consequently, according to the Harman’s oxidative stress theory, they have been linked to aging, age-related pathologies, and death (Balaban et al. 2005). However, when produced in a controlled amount, ROS may also play important signaling roles in various redox-dependent processes, including apoptosis (Bayir et al. 2006; Kagan et al. 2009b), cell proliferation (Fruehauf and Meyskens Jr. 2007) and hypoxia (Hamanaka and Chandel In Press). Furthermore, mitochondria are active players in cellular calcium homeostasis (Graier et al. 2007). Mitochondrial Ca2+ accumulation regulates functions as diverse as aerobic metabolism and induction of cell death (Celsi et al. 2009). Finally, mutations in mitochondrial DNA (mtDNA) are responsible for many mitochondrial metabolic disorders, and are thought to contribute to aging by promoting apoptosis (Kujoth et al. 2005; Reeve et al. 2008a). Thus, because of their pivotal role in controling cell life and death (Green and Kroemer 2004; Mattson and Kroemer 2003; Wasilewski and Scorrano 2009), mitochondria represent an attractive target for mitochondrial gene therapy (Koene and Smeitink 2009) as well as drugs treating either degenerative or hyperproliferative diseases (Fig. 1).
Fig 1.
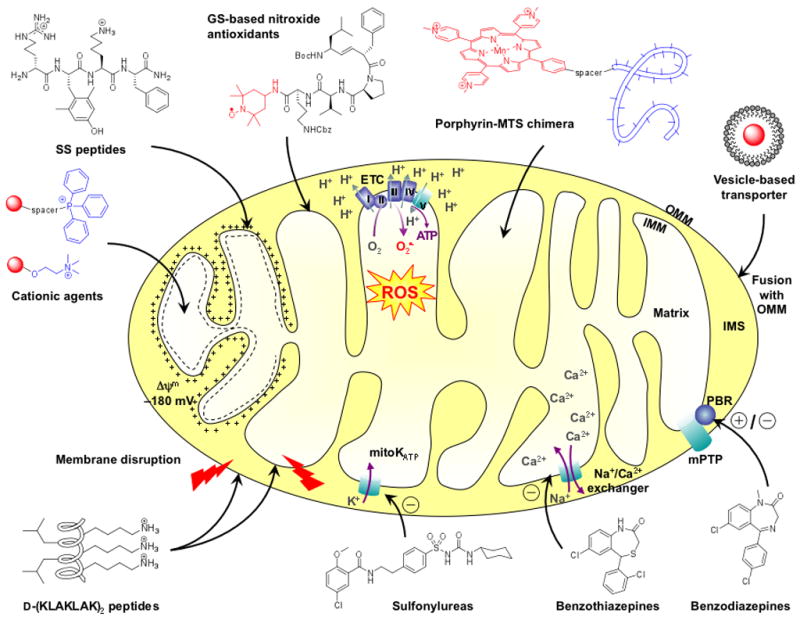
Schematic representation of a mitochondrion and the mode of action of representative mitochondria targeting compounds. Cationic compounds (triphenylphosphine (TPP)-based agents, choline esters, SS peptides) are attracted by the negative potential of the inner mitochondrial membrane (IMM). Driven by their high affinity for IMM-specific phospholipids, gramicidin S (GS)-based antioxidants deliver the nitroxide ROS (reactive oxygen species) scavenger into the matrix. Mitochondrial targeting sequences (MTS) can be utilized as vehicles to deliver metalloporphyrin superoxide dismutase (SOD)-mimics into the matrix. Alternatively, the mitochondrial agent can be encapsulated in a vesicle which undergoes fusion with the outer mitochondrial membrane (OMM). The filled circle represents the anti- or pro-oxidant payload. D-(KLAKLAK)2 and analogs are cationic amphipathic α-helical peptides able to disrupt mitochondrial membranes, hence triggering apoptosis. Other chemical agents target specific mitochondrial proteins. For instance, sulfonylureas block the mitochondrial ATP-regulated K+ channel (mitoKATP), benzothiazepines are inhibitors of the mitochondrial Na+-Ca2+ exchanger, and benzodiazepines are agonists or antagonists of the peripheral benzodiazepine receptor (PBR). ATP, adenosine triphosphate; ETC, electron transport chain; IMS, intermembrane space.
2. Therapeutic opportunities and challenges
2.1. Mitochondrial diseases1
Mitochondrial dysfunction triggers the cell death signaling cascade and results in organ failure and disease. Therapeutic intervention at the mitochondrial level can be envisioned for general cell-degenerative as well as hyperproliferative diseases, i.e. cancers (Gogvadze et al. 2009; Trachootham et al. 2009). Hyperproliferative cells are susceptible to pro-oxidant-induced apoptosis via an increase of their oxidative stress level. The redox status of many tumors is significantly altered compared with that of normal tissue, and pro-oxidant drugs can exploit this difference for treatment.
Conversely, aging and degenerative diseases are associated with an elevated oxidant state that may cause mitochondrial damage. In this case, antioxidants targeting mitochondria are expected to exert a mitigating effect. Several pathologies are found in this category, all sharing the common features of disturbances of mitochondrial Ca2+, ATP, or ROS metabolism. They include cardiovascular diseases (Lesnefsky et al. 2001), f. ex. atherosclerosis (Di Lisa et al. 2009), ischemia/reperfusion injury, heart failure, stroke (Dirnagl et al. 1999), and traumatic brain injury; aging (Balaban et al. 2005) and neurodegenerative diseases (Celsi et al. 2009; Reddy 2008; Reeve et al. 2008b), f. ex. Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and Friedreich’s ataxia (FRDA); chronic autoimmune inflammatory diseases, f. ex. rheumatoid arthritis (RA) (Gelderman et al. 2007); metabolic diseases, f. ex. diabetes (Friederich et al. 2009) and obesity (Rogge 2009); as well as ionizing radiation injury (Pearce et al. 2001).
2.2. Mode of action of drugs
Small molecule drugs or biologics can act on mitochondria through various pathways (Todesco et al. 2006). Some of these mechanisms will be discussed in greater detail in the following sections, and a detailed discussion would vastly exceed the scope of this review, but noteworthy current approaches include ETC inhibition, OXPHOS uncoupling, mitochondrial Ca2+ modulation, and control of oxidative stress via decrease or increase of mitochondrial ROS accumulation. The inhibition of the ETC can occur via direct inhibition of a protein subunit of one (or more) of the enzyme complexes or via acceptance of electrons flowing across the ETC instead of the natural acceptors ubiquinone or cytochrome c (cyt c) (Todesco et al. 2006). In the OXPHOS uncoupling event, protons are shifted from the mitochondrial matrix to the IMS and do not pass across the F1F0-ATPase back to the matrix, but instead migrate directly across the IMM. This bypass results in heat production, but lack of ATP formation. Typical examples for agents that promote OXPHOS uncoupling are weak acids and weak bases, which can be protonated in the IMS and carry protons across the IMM (Todesco et al. 2006). Interestingly, compounds affecting the activity of inner-membrane uncoupling proteins (UCPs) can prevent cell death (Mattson and Kroemer 2003).
An important event triggering the apoptotic cascade is the mitochondrial membrane permeabilization (MMP), which initiates the collapse of the mitochondrial potential, the loss of cyt c and the release of protease and nuclease activators. The prevention of this process can be achieved with inhibitors of the mitochondrial permeability transition pore (mPTP) complex (Zorov et al. 2009), openers of the mitochondrial ATP-regulated (mitoKATP) or Ca2+-activated (mitoKCa) potassium channels (Szewczyk et al. 2009), or inhibitors of the mitochondrial Na+-Ca2+ exchange (Mattson and Kroemer 2003). Modulation of mitochondrial Ca2+ can also be envisioned by interference with mitochondria-specific Ca2+ transporters.
Additional strategies for drug-induced perturbation of mitochondrial biochemistry include the inhibition of the cyt c-catalyzed peroxidation of the mitochondria-specific phospholipid cardiolipin (CL) (Borisenko et al. 2008; Kagan et al. 2009a; Kagan et al. 2009b), and the targeting of other specific mitochondrial proteins via inhibition of kinases, F1F0-ATPase, enzymes of the Krebs cycle, or members of the anti-apoptotic Bcl-2 family (Armstrong 2007; Gogvadze et al. 2009). It has been known for a while that prevention of the oxidative cellular damage via a decrease of mitochondrial ROS accumulation can be achieved by the delivery of antioxidants acting as radical and/or electron scavengers. Several drugs are able to inhibit the β-oxidation of unsaturated fatty acids, causing cellular accumulation of fat (Todesco et al. 2006). Alternatively, anti-apoptotic agents could be designed via inhibition of the cyt c-catalyzed peroxidation of CL (Borisenko et al. 2008; Kagan et al. 2009a; Kagan et al. 2009b).
Finally, the mitochondrial biochemistry is also severely derailed by mtDNA binding/oxidation or inhibition of mtDNA synthesis, or modulation of mitochondrial fission/fusion. Chemical agents that bind to mtDNA often result in inhibition of DNA synthesis (Todesco et al. 2006). If sufficient selectivity in the binding process can be achieved, this mechanism of action may represent an interesting strategy to block the expression of mutated mtDNA responsible for genetic mitochondrial disorders. Recently, compounds that modulate mitochondrial fission/fusion have been proposed as a valuable alternative in treatment of neurodegenerative diseases (Lu 2009).
2.3. Strategies to target mitochondria
While the OMM is relatively permeable due to the abundance of the voltage-dependent anion channel (VDAC) protein, the IMM is highly impermeable and acts as a rigid barrier to the passive diffusion of all types of molecules. It is also rich in the unusual phospholipid CL, and maintains a strong negative internal potential of −180 mV required for the ETC function. A widely used strategy for targeting mitochondria takes advantage of this remarkable biophysical membrane property, since cationic molecules are attracted to and accumulate preferentially within the negatively charged mitochondrial matrix (Murphy and Smith 2007). Another strategy is based on the affinity of an agent to mitochondrial membrane components, particularly to the phospholipid CL which is exclusively found in the IMM (Hoye et al. 2008). In addition to the former more specific properties, sufficient lipophilicity is also required to achieve a significant enrichment in mitochondrial compartments.
An emerging approach to the selective delivery of bioactive cargo molecule into mitochondria uses a carrier of short peptide sequences with specific physicochemical properties (Yousif et al. 2009). For instance, Horton et al. recently reported such mitochondria-penetrating peptides with alternating cationic and hydrophobic residues (Horton et al. 2008). Other variants have been based on an oligomeric carbohydrate scaffold, always attaching key guanidinium moieties due to their delocalized cationic form (Yousif et al. 2009). Finally, the tethering of active molecules to mitochondrial targeting sequences (MTSs) has also been successively utilized (Yousif et al. 2009). MTSs are peptides used by cells for the delivery of nuclear-encoded mitochondrial proteins, containing structural motifs recognized by the mitochondrial import machinery.
Another class of mitochondrial delivery vectors, suitable for the import of large or impermeable molecules, is the vesicle-based transporter system (Yousif et al. 2009). The targeted agent is encapsulated in a cationic liposome, which undergoes cellular internalization and subsequent fusion with the OMM (Ko et al. 2009).
In summary, by the use of a broad range of diverse delivery systems, the targeting of mitochondria for therapeutic benefits can be utilized to enrich both antioxidants as well as pro-oxidants in mitochondrial compartments. Antioxidants are of primary interest for their antiaging properties, with some of the main applications centered around cardioprotection and neurodegenerative diseases, while pro-oxidant and cytotoxic agents are under investigation for cancer therapy. This review will describe recent advances in the development of mitochondria-targeted agents, with an emphasis on the selective delivery of an antioxidant “payload” into mitochondria (Fig. 2; use this Figure as a reference for compound structures described throughout the review). For a summary of selected major mitochondrial targeting agents and their current clinical status, if known, see Table 1.
Fig 2.

Chemical structures of featured mitochondria-targeting agents and clinical drug candidates. For chimera molecules, substructures highlighted in dashed boxes represent the targeted bioactive components, and substructures highlighted in dashed circles represent the mitochondria-targeting cationic entities. Ph, phenyl; Me, methyl; Et, ethyl; Boc, tert-butoxycarbonyl; Cbz, benzyloxycarbonyl; Ac, acetyl.
TABLE I.
Major mitochondria-targeting agents
| Class | Compound | Mechanism of Action—In Vitro/In Vivo Activity | Current Status |
|---|---|---|---|
| TPP-based conjugates | Mito-VitE | Antioxidant: radical scavenger; antiapoptotic agent | — |
| Mito-Q | Antioxidant: radical scavenger; antiapoptotic agent | Phase II: PD, liver damage associated with hepatitis C | |
| Mito-SOD | Antioxidant: SOD mimetic | — | |
| Mito-Peroxidase | Antioxidant: peroxidase mimetic | — | |
| Mito-PBN | Antioxidant: radical scavenger | — | |
| Mito-CP | Antioxidant: radical scavenger; SOD and catalase mimetic; antiapoptotic agent | — | |
| Mito-TEMPOL | Antioxidant: radical scavenger; SOD and catalase mimetic | — | |
| TPEY-TEMPO | Antioxidant: radical scavenger; SOD and catalase mimetic; antiapoptotic agent; radioprotective agent | — | |
| HVTP | NO donor; cyt c/CL peroxidase inhibition; antiapoptotic agent | — | |
| TPP-OA | Prevention of CL peroxidation; antiapoptotic agent | — | |
| Other delocalized lipophilic cations | Rhodamine 123 | Mitochondrial chaperone | — |
| Flupirtine | Antioxidant: radical scavenger (?);antiapoptotic agent | (Nonopioid analgesic) | |
| Sulfonylureas and related compounds | Glibenclamide | Mito KATP blocker | (Antidiabetic agent) |
| Diazoxide | Mito KATP opener | Antihypertensive agent | |
| Sulofenur | OXPHOS uncoupler | (Anticancer agent) | |
| Benzodiazepines and other PBR ligands | Ro5-4864 | PBR agonist; potential antiapoptotic agent | — |
| PK-11195 | PBR antagonist; potential anticancer agent | — | |
| Benzothiazepines | CGP-37157 | Mitochondrial Na+-Ca2+ exchanger inhibitor | — |
| Modified phospholipids | NBD-CL | Cyt c/CL peroxidase inhibition; antiapoptotic agent | — |
| Anthracyclines | Adriamycin Daunomycin |
Mitochondrial membranes disruption; oxidative damaging agent | Anticancer agents with mitochondrial toxicity |
| Nucleoside analogs | AZT | Mitochondrial enzymes inhibition; mtDNA damaging agent | Antiviral agent with mitochondrial toxicity |
| SS tetrapeptides | SS-02 | Antioxidants: radical scavengers; antiapoptotic agents; cardioprotective agents: myocardial infarction, ischemic brain injury; | — |
| SS-31 | neuroprotective agents: ALS, PD; renal protective agents | — | |
| Choline esters of GSH and NAC | Mito-GSH | Antioxidants: radical scavengers | — |
| Mito-NAC | — | ||
| GS-based conjugates | XJB-7-75 | Antioxidants: radical scavengers; SOD and catalase mimetics; | — |
| XJB-5-125 | antiapoptotic agents; | — | |
| XJB-5-131 | cardioprotective agents: hemorrhagic shock; | — | |
| jp4_039 | potential anti-inflammatory agents; radioprotective agents | Preclinical trials: radioprotection | |
| Clinical drug candidates | CoQ10 | Antioxidant: radical scavenger; dietary supplement (not mitochondria-targeted) | Clinical trials: cardiovascular and neurodegenerative diseases, bipolar disorder |
| Idebenone | Antioxidant: radical scavenger (not mitochondria-targeted) | Phase III: FRDA; clinical trials: DMD, LHON, primary progressive multiple sclerosis | |
| As2O3 | Sulfhydryl-based proteins inhibition | Marketed: acute promyelocytic leukemia | |
| Elesclomol | Pro-oxidant | Phase III suspended: metastatic melanoma | |
| Motexafin gadolinium | Pro-oxidant | Phase III: brain metastases (combination with radiation therapy), hematological malignancies (combination with chemotherapy) | |
| 2-Methoxy-estradiol | SOD inhibition | Phase II: solid tumors and metastatic cancers | |
| Imexon | Binding to thiols | Phase I/II: pancreatic and lung cancers |
Empty entries in the last column indicate that there is no recent information. Activities in parentheses are likely unrelated to the mitochondrial effects of these compounds. The question mark (?) indicates a speculative mechanism of action.
TPP, triphenylphosphonium; PBR, peripheral benzodiazepine receptor; GSH, glutathione; NAC, N-acetyl-L-cysteine; GS, gramicidin S; SOD, superoxide dismutase; cyt c, cytochrome c; CL, cardiolipin; Mito-KATP, ATP-regulated potassium channel; OXPHOS, oxidative phosphorylation; mtDNA, mitochondrial DNA; ALS, amyotrophic lateral sclerosis; PD, Parkinson’s disease; FRDA, Friedreich’s ataxia; DMD, Duchenne muscular dystrophy; LHON, Leber’s hereditary optic neuropathy.
3. Lead structures and medicinal chemistry progress
3.1. Non peptide-based mitochondria-targeted agents
Delocalized lipophilic cations
Taking advantage of the substantial negative electrochemical potential maintained across the IMM, the class of delocalized lipophilic cations is particularly effective at crossing the hydrophobic membranes and, hence, preferentially accumulates >1,000 fold within the mitochondrial matrix.
The most important class of molecules based on this approach has been developed by the tethering of antioxidants to a triphenylphosphonium (TPP) salt head piece (Murphy and Smith 2007). The vitamin E conjugate MitoVit E is effective at preventing lipid peroxidation, with the α-tocopherol component acting as a chain-terminating antioxidant (Smith et al. 1999). Similarly, MitoQ is an ubiquinone derivative attached to the TPP cation by a 10-carbon aliphatic chain (Kelso et al. 2001). Both compounds were found to be more potent than their untargeted analogs (Trolox and Idebenone, respectively) in preventing oxidative stress-induced apoptosis in cultured fibroblasts from FRDA patients (Jauslin et al. 2003). Further in vivo studies reported that MitoQ selectively protected mitochondria against cardiac ischemia/reperfusion injury in rats (Adlam et al. 2005). This compound is now under clinical development (cf. section 4). The TPP-based strategy was also applied to other antioxidants. MitoSOD is a mitochondria-targeted version of the superoxide dismutase (SOD) mimetic M40403, which degrades superoxide (Murphy and Smith 2007). MitoPeroxidase is a mitochondria-targeted version of Ebselen, whose peroxidase-mimetic activity is triggered by its selenium atom (Filipovska et al. 2005). MitoPBN is a mitochondria-targeted version of the spin trap α-phenyl-N-tert-butylnitrone (PBN), and has been shown to react with mitochondrial carbon-centered radicals (Murphy et al. 2003). Several TPP-nitroxide conjugates have also been designed (the mechanism of action of nitroxides will be discussed in the “Gramicidin S-based conjugates” section). Mito-CP (Mito-carboxy proxyl) was shown to inhibit peroxide-induced oxidative stress and apoptosis in cultured cells (Dhanasekaran et al. 2005). Similar TPP-TEMPO conjugates were reported, which differ only by the linker connecting both entities. While the ester-linked MitoTEMPOL was found not to be more active than its untargeted version (Dessolin et al. 2002), the imine-linked TPEY-TEMPO concentrated in mitochondria, exhibiting anti-apoptotic and radioprotective properties (Jiang et al. 2009, submitted). This suggests that the role of the linker connecting TEMPO and TPP is essential for their penetration into cells (Kagan et al. 2009c). In a different approach, precursors of nitric oxide (NO) donors were also used to target mitochondria by attachment to the TPP moiety. Indeed, NO readily inhibits peroxidases by forming iron-nitrosyl complexes or by quenching reactive peroxidase intermediates. Hence, the prodrug (2-hydroxyaminovinyl)-triphenylphosphonium (HVTP) was shown to protect cells against apoptosis via inhibition of the peroxidase activity of the cyt c/CL complex (see also the section “Modified phospholipids” for further details on the cyt c/CL complex) (Belikova et al. 2009; Stoyanovsky et al. 2008). Finally, manipulation of the unsaturation levels of CL species by mitochondria-targeted fatty acids has been proposed to modulate the sensitivity of cells to undergo apoptosis. The rationale was based on the observation that CL saturated and mono-unsaturated fatty acid chains are not oxidized by the cyt c/CL peroxidase. Thus, a TPP-conjugate with the mono-unsaturated oleic acid (TPP-OA) exhibited anti-apoptotic activity, indicating that the cell’s sensitivity to apoptosis may be modulated via the mitochondrial delivery of oxidizable or non oxidizable fatty acids that may be integrated into CL species (Kagan et al. 2009c).
Apart from the TPP-conjugates, other classes of lipophilic cationic mitochondria-selective molecules have been described as attractive candidates for therapeutic purposes. Historically, the first examples were the fluorescent lipophilic cation rhodamine 123 and other anticancer cyanine dyes (Murphy 1997). Notably, rhodamine 123 has been used as a chaperone to selectively deliver the anticancer drug cisplatin into the mitochondria of tumor cells. More recently, the triaminopyridine flupirtine, a non opioid analgesic, was shown to be an effective antioxidant in mitochondria, with potential applications as an anti-apoptotic agent (Schlüter et al. 2000). It was proposed to act as a free radical scavenger.
However, in spite of their effective in vitro prevention of mitochondrial damage, lipophilic cations suffer from a major drawback. Charge accumulation into the matrix results in mitochondrial membrane depolarization, which may account for the toxicity of these compounds at concentrations >10 μM (Murphy 1997).
Sulfonylureas and related compounds
The cardioprotective action of potassium channel openers (KCOs) appeared to be mediated by the interaction of these compounds with the mitoKATP channels (Szewczyk et al. 2009). MitoKATP can be blocked by antidiabetic sulfonylureas such as glibenclamide, or activated by KCOs such as diazoxide. Thus, therapeutic applications might be envisioned in the treatment of myocardial infarction and stroke (Szewczyk and Marbán 1999). Some diarylsulfonylureas, a distinct class of antitumor agents (f. ex. sulofenur), were shown to be also effective in uncoupling OXPHOS. However, the mechanism of antitumor activity could not be linked to this property (Szewczyk and Wojtczak 2002).
Benzodiazepines and other PBR ligands
In addition to the well known central benzodiazepine receptor GABAA, a second class of binding sites termed peripheral benzodiazepine receptor (PBR) is localized primarily in mitochondrial membranes (Veenman and Gavish 2006). This receptor is thought to be a component or a regulator of mPTP, a multiprotein complex involved in the regulation of programmed cell death (Zorov et al. 2009). Therefore, PBR agonists such as Ro5-4864 (4′-chlorodiazepam) and PBR antagonists such as the isoquinoline carboxamide PK-11195 may have potential utility as anti-apoptotic agents or pro-apoptotic antitumor agents, respectively (Galiegue et al. 2003).
Benzothiazepines
Mitochondria are involved in cellular Ca2+ homeostasis. An increase in intra-mitochondrial Ca2+ concentration has been shown to activate Ca2+-sensitive dehydrogenases in the Krebs cycle, thereby increasing ATP synthesis (Koene and Smeitink 2009). In a study with fibroblasts of children bearing a mutation responsible for the complex I (NADH:ubiquinone oxidoreductase) deficient Leigh syndrome, inhibition of the mitochondrial Na+-Ca2+ exchange by the benzothiazepine CGP-37157 resulted in an increase in mitochondrial Ca2+ concentration and subsequent ATP production (Visch et al. 2004). These findings suggest that complex I deficiency was associated with altered cytosolic Ca2+ homeostasis. Therefore, mitochondrial Ca2+ modulation may represent a new therapeutic approach towards complex I deficiency syndromes, such as encephalopathies and neurodegenerative diseases.
Furthermore, it has been proposed that inhibition of the mitochondrial Na+-Ca2+ exchanger could enhance glucose-stimulated insulin secretion in pancreatic β-cells (Pei et al. 2003). To this end, novel benzothiazepine derivatives have been reported as potential therapeutics for type II diabetes.
Modified phospholipids
During the apoptotic stage, the mitochondria-specific phospholipid CL interacts with cyt c to form a peroxidase complex that catalyzes CL oxidation by utilizing ROS. This process represents an essential mechanism for the induction of the cell death program before the point-of-no-return, caspase 3 activation. Kagan and coworkers have explored a new approach for inhibiting the cyt c peroxidase activity by utilizing modified CL bearing an oxidizable and fluorescent 7-nitro-2,1,3-benzoxadiazole moiety (NBD-CL) (Borisenko et al. 2008). They demonstrated that this conjugate formed high-affinity complexes with cyt c and blocked cyt c-catalyzed oxidation of peroxidase substrates. Thus, NBD-CL may represent a potential regulator of apoptosis.
Other drugs with mitochondrial toxicity
The anticancer activity of anthracyclines is mainly attributed to their DNA intercalation. However, the cytotoxic side effects of the widely used adriamycin and daunomycin have been associated with mitochondrial dysfunction (Jung and Reszka 2001). Interaction of these drugs with mitochondria appears to follow complex mechanisms, including major membrane disruption caused by the high affinity of anthracyclines to CL, and the redox activity of the quinone moiety which results in oxidative damage of proteins.
Nucleoside analogs used as antiviral drugs, such as AZT (zidovudine), can cause mitochondrial damage through two mechanisms (Szewczyk and Wojtczak 2002). The short-term mechanism directly affects the activity of mitochondrial enzymes, via the competitive inhibition of the ADP/ATP antiport and of the nucleoside diphosphate kinase. The long-term mechanism alters the mtDNA, via oxidative damage to mtDNA and inhibition of the polymerase responsible for mtDNA replication.
NSAIDs can inhibit the β-oxidation of fatty acids and cause the uncoupling of OXPHOS, due partly to induction of mPTP (Szewczyk and Wojtczak 2002). Local anesthetics uncouple OXPHOS and inhibit mitochondrial F1F0-ATPase and ETC enzymes (Szewczyk and Wojtczak 2002).
Hence, many late-stage adverse drug toxicity problems have prompted companies to screen for mitochondrial toxicity early in the drug discovery process, in order to reduce late-stage attrition (Dykens and Will 2007).
Other chemical classes
A variety of structurally diverse small molecules have been described to act on mitochondrial targets. For instance, a novel group of anticancer agents called “mitocans” has been reviewed by Neuzil et al. (Neuzil et al. 2007). These compounds usually cause the mitochondrial destabilization of tumor cells through the activation of mitochondrial mediators of apoptosis, including the Bcl-2 protein family, F1F0-ATPase, ETC proteins, and mPTP and its components. A recent review by Toogood also listed several small molecule drugs, most of them acting as pro-oxidants (Toogood 2008). These agents are chemically extremely diverse, ranging from natural polyheterocycles and macrolides to steroid, benzodiazepine, isoquinoline, polyphenol or guanidinone derivatives. We refer to these excellent references for further reading (Neuzil et al. 2007; Toogood 2008).
3.2. Peptide- and amino acid-based mitochondria-targeted agents
D-(KLAKLAK)2 pro-apoptotic peptides and analogs
Mitochondria-disrupting peptides are capable of invading the mitochondria of mammalian cells and trigger apoptosis. They are typically derived from the sequences of membrane-disrupting antimicrobial peptides. Since they can be fused easily to tissue- or tumor-specific peptides or antibodies, they may be attractive as new targeted anticancer agents. However, they usually suffer from low potencies, thereby limiting their clinical utility (Ellerby et al. 1999). Recently, Horton and Kelley were able to improve the mitochondrial localization and membrane-disrupting activity of the widely used cationic amphipathic α-helical killer peptide D-(KLAKLAK)2 (Horton and Kelley 2009). By increasing its hydrophobicity via the exchange of the leucine residue for a cyclohexylalanine or a 6-carbon alkyl chain residue, a dramatic increase in potency could be achieved, with LC50 values of ca. 2 μM for the corresponding analogs.
Mn-porphyrin-oligopeptide conjugates
A new class of mitochondria-targeted SOD-mimics was reported recently, consisting of a manganese metalloporphyrin conjugated to a signal oligopeptide (Met-Leu-Ser-Leu-Arg-Gln-Ser-Ile-Arg-Phe-Lys-Gly-Cys-S-spacer-porphyrin) (Asayama et al. 2006), whose sequence was based on the leader sequence of the yeast cyt c oxidase subunit IV (Murphy 1997). The SOD activity of the resulting conjugate was confirmed by monitoring the rate of decomposition of the highly toxic peroxynitrite, a product of the reaction of superoxide with NO.
SS tetrapeptides
The Szeto-Schiller (SS) peptides feature a common structural motif of alternating aromatic (Phe, Tyr, Dmt (2′,6′-dimethyltyrosine)) and basic (Arg, Lys) residues (Szeto 2006; Szeto 2008). Despite their triple positive net charge at physiological pH values, their aromatic-cationic amino acid sequence allows them to freely penetrate cells in a potential-independent, non saturable manner (Zhao et al. 2003). Rapid uptake of the radiolabeled tetrapeptide [3H]SS-02 ([3H]Dmt-D-Arg-Phe-Lys-NH2) occurred with maximal levels reached by 2 min and an achievement of a 100-fold concentration in mitochondria (Zhao et al. 2004). Further experiments suggested that these peptides were predominantly targeted to the IMM, allowing only 20% to reach the mitochondrial matrix via potential-driven mechanisms, despite their cationic nature. Thus, contrary to small lipophilic cations which cause toxicity at >10 μM, their uptake is not self-limiting and they do not cause mitochondrial depolarization even at 1 mM concentrations.
The free radical scavenging abilities of these peptides are likely to originate from their Tyr or Dmt residues, with Dmt being the more effective reducing agent than Tyr (Winterbourn et al. 2004). The specific location of the Tyr or Dmt residue in the peptide sequence was inconsequential, since SS-02 and SS-31 (D-Arg-Dmt-Lys-Phe-NH2) were found to be equally effective in scavenging ROS and inhibiting linoleic acid oxidation. In agreement with this hypothesis, replacement of Dmt with Phe resulted in a loss of the scavenging activity of the corresponding analog SS-20 (Phe-D-Arg-Phe-Lys-NH2) (Zhao et al. 2004).
In vitro cellular experiments showed that SS-02 and SS-31 can reduce intracellular ROS production and apoptosis, and prevent mitochondrial depolarization, mitochondrial permeability transition (MPT), and Ca2+-induced swelling (Zhao et al. 2004). Much more potent than SS-02, the estimated 5,000-fold enrichment of SS-31 in mitochondria could explain its strong efficacy even at <1 nM concentrations (Zhao et al. 2005).
Furthermore, both SS-02 and SS-31 were shown to protect against the loss of contractile force induced by 30 min of global ischemia in the isolated perfused guinea pig heart (Zhao et al. 2004). The ability of SS-02 to prevent myocardial stunning has been confirmed in vivo in rats (Song et al. 2005), and pre-ischemic i.p. administration of SS-31 (and even SS-20) to rats significantly reduced infarct size (Cho et al. 2007a). These results support the hypothesis that ROS play a major role in reperfusion-induced myocardial stunning. Surprisingly, however, the non-scavenging peptide SS-20 exhibited a better activity than SS-31. This suggests that SS-20 can reduce ROS generation more effectively by improving mitochondrial bioenergetics and myocardial ATP content during ischemia. Moreover, in a mouse model of ischemic cerebral injury, SS-31 was shown to exert its antioxidant and neuroprotective activity through the inhibition of the scavenger receptor CD36 (Cho et al. 2007b). Although further studies are required to investigate the precise mode of action of the peptide on CD36 pathways, these data suggest that targeting CD36 with specific antioxidants might represent a new therapeutic strategy to treat ischemic brain stroke patients.
Significantly, an in vivo study in a mouse model of ALS reported that daily i.p. injections of SS-31 led to a significant improvement in survival and motor performance (Petri et al. 2006). In a mouse model of PD, both SS-31 and SS-20 demonstrated remarkable neuroprotective effects (Yang et al. 2009). These findings confirm the critical role for oxidative stress in the pathogenesis of neurodegenerative diseases and support the potential use of antioxidants as therapeutic agents.
An islet transplantation study in mice demonstrated that SS-31 could improve the viability of isolated pancreatic islet cells and graft function in recipients with type 1 diabetes (Thomas et al. 2007). The authors concluded that the peptide might be useful for optimizing islet transplantation and increasing the pool of eligible organ donors.
More recently, it was reported that SS-31 protected against renal damage in an unilateral ureteral obstruction (UUO) model, although the mechanism by which protection was afforded remains to be determined (Mizuguchi et al. 2008).
In summary, SS peptides have exhibited marked antioxidant properties in a range of in vivo studies, including myocardial infarction, ischemic brain injury, ALS, PD, islet transplantation, and UUO models. Hence, such mitochondria-targeted antioxidants clearly represent a promising approach to treating related pathologies.
Choline esters of glutathione and N-acetyl-L-cysteine
The tripeptide glutathione (L-γ-glutamyl-L-cysteinylglycine or GSH) plays an important role in protecting cells against oxidants and electrophiles (Pompella et al. 2003). Upon donating an electron to unstable molecules such as ROS, two GSH react together to form glutathione disulfide (GSSG). Increasing mitochondrial glutathione and other thiol-based antioxidants can be an effective strategy to prevent mitochondrial oxidative stress. Using a similar approach as with the TPP conjugated antioxidants, Sheu et al. prepared choline esters of glutathione (MitoGSH) and of its analog N-acetyl-L-cysteine (MitoNAC) for targeting to mitochondria (Sheu et al. 2006). Preliminary studies have shown that GSH and NAC can indeed protect against oxidative damage in cultured cells, but in vivo data have not yet been published.
Gramicidin S-based conjugates
An alternative concept using mitochondrial targeting of ROS scavengers was based on the affinity of certain natural antibiotics to microbial cell membranes. Due to the close evolutionary relationship between bacterial membranes and the IMM components, in particular their lipid composition (Prenner et al. 1999), the antibacterial membrane disruptor gramicidin S (GS) was hypothesized to serve as a template for mitochondrial targeting. Thus, a hemi-GS pentapeptide sequence was tethered to the stable free radical 4-amino-2,2,6,6-tetramethylpiperidine-N-oxyl (4-NH2-TEMPO, or 4-AT), the ROS scavenging “payload” (Fink et al. 2007a; Fink et al. 2007b; Hoye et al. 2008). The major advantage of sterically hindered free radicals such as 4-AT is their electron acceptor/donor nature depending on the redox potential of the environment (Samuni et al. 2002; Soule et al. 2007). By accepting one electron, they are reduced to hydroxylamines, which can act as direct radical scavengers and are then converted back into nitroxides (Mitchell et al. 2000). In other words, these compounds undergo redox recycling (Zhang et al. 1999). Nitroxides also possess SOD and catalase mimetic activities (Goldstein et al. 2003; Krishna et al. 1996), thus offering additional protective benefits against oxidative cellular damage. Another advantage of free radicals is the possibility to use electron spin resonance (ESR) to measure distribution of the spin label and detect oxidative stress in the local cellular environment (Mitchell et al. 2000). Thus, nitroxides combine several important features in a single functional moiety.
Based on these considerations, two different hemi-GS segments have been engineered to give, among other analogs, the conjugates XJB-7-75 (Boc-D-Phe-Pro-Val-Orn(Cbz)-Leu-4-AT), XJB-5-125 (Boc-Leu-D-Phe-Pro-Val-Orn(Cbz)-4-AT), and its isostere XJB-5-131, in which the Leu-D-Phe peptide bond has been replaced by a metabolically more stable isosteric (E)-alkene moiety. These compounds were shown to be effectively partitioning into the mitochondria of mouse embryonic cells, in their reduced form. They inhibited actinomycin D-induced superoxide production and CL peroxidation, and provided protection against apoptosis, at relatively low 10 μM concentrations (Jiang et al. 2007; Wipf et al. 2005). Furthermore, the gramicidin S-based 4-AT conjugates were devoid of any toxicity even at a higher concentration of 20 μM (Jiang et al. 2007).
XJB-5-131 appeared to be the most effective in a series of GS-TEMPO conjugate analogs evaluated for their ability to protect against gut barrier dysfunction induced by hemorrhage in rats (Macias et al. 2007). More precisely, XJB-5-131 was able to ameliorate CL peroxidation and apoptosis induced by shock. Further in vivo studies demonstrated that i.v. infusion of XJB-5-131 could prolong survival in a rat model of lethal hemorrhagic shock (Macias et al. 2007). These findings suggest that treatment with XJB-5–131 may prolong the period of time that patients can survive after loosing large quantities of blood, thereby allowing transport to appropriate care facilities. These results also support the concept that targeting mitochondrial ROS scavengers is a reasonable therapeutic strategy for the managment of hemorrhagic shock and other related conditions, such as stroke or myocardial infarction.
Another in vivo study reported that i.v. administration of XJB-5-131 decreased nitric oxide (NO) production in mice treated with lipopolysaccharide (Fink et al. 2007a), revealing possible anti-inflammatory properties. Additional pharmacologic, pharmacokinetics and toxicologic studies are required to determine the full therapeutic potential of this compound series.
A further exploration into the utility of GS-TEMPO conjugates revealed their efficacy as radioprotectants. Ionizing radiation activates a variety of cytoplasmic transduction pathways and triggers apoptosis. It is assumed that radiolysis of water and subsequent generation of ROS induces damage of genomic DNA, followed by a mitochondria-dependent apoptotic response (Mitchell et al. 2000).
A recent study demonstrated that XJB-5-125 could effectively protect cells against superoxide generation, CL oxidation, and apoptosis induced by γ-irradiation, if given either 10 min before or 1 h after irradiation (Jiang et al. 2008). A previous experiment, using peptide conjugate analogs attached to the potent NO synthase (NOS) inhibitor AMT (2-amino-6-methyl-5,6-dihydro-4H-1,3-thiazine), revealed that the targeting of a NOS antagonist was even more radioprotective than the targeting of an ROS scavenger (Kanai et al. 2007).
Detailed structure-activity studies suggested that the effective partitioning of nitroxides was necessary but not sufficient for their antioxidant activity in mitochondria (Jiang et al. 2007). Monte Carlo simulations showed that active hemi-GS peptide-nitroxide conjugates with intact β-turn structure were positioned at the interface between polar and non polar regions of the lipid membrane. Thus, the optimized localization of the scavenger moiety inside the polar region of mitochondrial membranes was essential to allow successful competition with O2 for electrons from ETC, in order to prevent ROS formation.
In the course of improving the physicochemical properties of XJB-5-131, jp4_039 was identified as a small molecular weight analog with a marketed radioprotective/mitigative activity (Frantz, Pierce et al., in preparation; Gokhale et al., submitted). While jp4_039 only contains the alkene peptide isostere fragment of XJB-5-131, its physicochemical properties apparently allow it to effectively partition passively into mitochondria (Rajagopalan et al. 2009).
4. Recent clinical data
A number of non specific antioxidants have been evaluated as potential therapeutics. For instance, coenzyme Q10 (CoQ10) is a natural electron acceptor in the ETC. Because of its strong antioxidant properties, it is used as a dietary supplement and has been undergoing clinical trials for cardiovascular (Pepe et al. 2007) and neurodegenerative diseases (Kaufmann et al. 2009) as well as bipolar disorder. Idebenone (Catena®) is a CoQ10 analog currently under extensive development by Santhera Pharmaceuticals for the treatment of several neurological diseases. Its positive effect in cardiac hypertrophy and neurological symptoms associated with FRDA has been demonstrated (Meier and Buyse 2009), and two phase III trials have been initiated (Schulz et al. 2009). Clinical studies are ongoing for the treatment of Duchenne Muscular Dystrophy (DMD), Leber’s Hereditary Optic Neuropathy (LHON) and primary progressive multiple sclerosis.
Furthermore, phase II clinical trials of the mitochondria-targeted antioxidant MitoQ® for PD and liver damage associated with hepatitis C have been completed (Tauskela 2007), and initial positive results have been reported by Antipodean Pharmaceuticals.
In addition, many anticancer therapies currently under investigation aim at exploiting the increased oxidative stress of tumor cells to selectively kill them via pro-oxidant-induced apoptosis (Engel and Evens 2006; Toogood 2008; Trachootham et al. 2009).
Arsenic trioxide (Trisenox™) is currently marketed by Cell Therapeutics for the treatment of acute promyelocytic leukemia. As2O3 has undoubtedly multiple biological targets, but the common mechanism of action seems to be its interaction with sulphydryl groups, especially those of mPTP members, leading to the production of ROS and subsequent apoptosis (Carney 2008).
Elesclomol™, an injectable drug whose phase III clinical evaluation for the treatment of metastatic melanoma has recently been suspended (Synta Pharmaceuticals) (Kirshner et al. 2008), and Motexafin Gadolinium (MGd), a metalloporphyrin that enhances radiation therapy for brain cancer (Pharmacyclics) (Richards and Mehta 2007), selectively kill cancer cells through apoptosis as a result of an increased level of oxidative stress. The phase III clinical trial for Elesclomol™ was suspended due to safety concerns, including an imbalance in overall survival, with a greater number of deaths occurring in the combination arm (Elesclomol™ with paclitaxel) compared to the control arm (paclitaxel alone).
2-Methoxyestradiol (Panzem®) was shown to inhibit SOD, leading to ROS accumulation in cancer cells (Huang et al. 2000), and phase II clinical trials are ongoing for the treatment of solid tumors and metastatic cancers (Sutherland et al. 2007).
Imexon is an aziridine-containing iminopyrrolidone that binds to thiols (GSH, cysteine), causing an accumulation of ROS and mitochondrial swelling, and leading to apoptosis. It is being evaluated for the treatment of pancreatic and lung cancers (Dragovich et al. 2007).
Other redox-active anticancer candidates are at various stages of clinical development and have been reviewed previously (Engel and Evens 2006; Toogood 2008; Trachootham et al. 2009). These are mainly general modulators of the cellular redox system, and not specifically targeted to mitochondria.
5. Conclusion and future perspectives
Drug compartimentalization in different organelles within a cell has not yet been fully explored. The growing interest in mitochondrial targeting is revealing new avenues to identify both novel therapeutics as well as potential sources of toxicity for existing medicines.
Although the TPP-based compound class represents the majority of the nonpeptidic mitochondrial targeting agents synthesized to date, and in spite of the significant initial success of this approach, there are still controversies surrounding this approach. A recent instructive study conducted by Horobin et al. (Horobin et al. 2007) failed to establish simple correlations between mitochondria targeting capacity and physicochemical properties of mitochondria-specific agents. Indeed, upon analysis of >100 available “mitochondriotropics”, only a third were classified as lipophilic cations, contrary to the general prejudice. Acids and anions were represented in similar numbers as charge-neutral compounds. Two thirds of the known mitochondriotropics are lipophilic while one third is hydrophilic. In fact, amphiphilicity is not a general property of mitochondria-targeted molecules. Their mitochondria targeting capacity could not be attributed to the presence of a mitochondria-specific tag, but rather this feature was derived from their overall molecular properties.
Alternatively, the concept of dual function agents that use a vehicle to deliver a bioactive payload into mitochondria may be particularly promising. Initially developed with radical and electron scavengers, this approach might be ideally extended to any feasible anti- or pro-oxidant, or even to mitochondrial protein ligands.
Preclinical and clinical data have demonstrated the considerable potential of mitochondrial targeting approaches, and potential therapeutic applications span a broad range of pathological conditions. However, it is worth mentioning the controversies which have emerged with regard to the oxidative stress theory of aging (Pérez et al. 2009), the utility of antioxidants for cancer treatment (Gottlieb 2009), and the link between mitochondrial dysfunction and insulin resistance (Turner and Heilbronn 2008). Clearly, a better understanding of the mitochondrial biology is still needed to enable the design of the most beneficial therapeutic approach with respect to the modulation of the redox balance of the targeted cells. Nonetheless, the increasing prevalence of age-related disorders calls for innovative solutions, and mitochondrial drugs clearly have the potential to emerge as a key platform technology for the next generation of medicines.
Acknowledgments
Grant sponsors: BARDA, CMCR, U.S. PHS NIH
Footnotes
Website of the Mitochondria Research Society: http://www.mitoresearch.org/
References
- Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RAJ, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- Armstrong JS. Mitochondrial Medicine: Pharmacological targeting of mitochondria in disease. Br J Pharmacol. 2007;151:1154–1165. doi: 10.1038/sj.bjp.0707288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asayama S, Kawamura E, Nagaoka S, Kawakami H. Design of Manganese Porphyrin Modified with Mitochondrial Signal Peptide for a New Antioxidant. Mol Pharmaceutics. 2006;3:468–470. doi: 10.1021/mp0500667. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, Oxidants, and Aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Bayir H, Fadeel B, Palladino MJ, Witasp E, Kurnikov IV, Tyurina YY, Tyurin VA, Amoscato AA, Jiang J, Kochanek PM, DeKosky ST, Greenberger JS, Shvedova AA, Kagan VE. Apoptotic interactions of cytochrome c: Redox flirting with anionic phospholipids within and outside of mitochondria. Biochim Biophys Acta Bioenergetics. 2006;1757:648–659. doi: 10.1016/j.bbabio.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Belikova NA, Jiang J, Stoyanovsky DA, Glumac A, Bayir H, Greenberger JS, Kagan VE. Mitochondria-targeted (2-hydroxyamino-vinyl)-triphenyl-phosphonium releases NO• and protects mouse embryonic cells against irradiation-induced apoptosis. FEBS Lett. 2009;583:1945–1950. doi: 10.1016/j.febslet.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisenko GG, Kapralov AA, Tyurin VA, Maeda A, Stoyanovsky DA, Kagan VE. Molecular Design of New Inhibitors of Peroxidase Activity of Cytochrome c/Cardiolipin Complexes: Fluorescent Oxadiazole-Derivatized Cardiolipin. Biochem. 2008;47:13699–13710. doi: 10.1021/bi801507s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney DA. Arsenic trioxide mechanisms of action - looking beyond acute promyelocytic leukemia. Leuk Lymphoma. 2008;49:1846–1851. doi: 10.1080/10428190802464745. [DOI] [PubMed] [Google Scholar]
- Celsi F, Pizzo P, Brini M, Leo S, Fotino C, Pinton P, Rizzuto R. Mitochondria, calcium and cell death: A deadly triad in neurodegeneration. Biochim Biophys Acta Bioenergetics. 2009;1787:335–344. doi: 10.1016/j.bbabio.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J, Won K, Wu D, Soong Y, Liu S, Szeto HH, Hong MK. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coronary Artery Dis. 2007a;18:215–220. doi: 10.1097/01.mca.0000236285.71683.b6. [DOI] [PubMed] [Google Scholar]
- Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT. A Novel Cell-permeable Antioxidant Peptide, SS31, Attenuates Ischemic Brain Injury by Down-regulating CD36. J Biol Chem. 2007b;282:4634–4642. doi: 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]
- Dessolin J, Schuler M, Quinart A, De Giorgi F, Ghosez L, Ichas F. Selective targeting of synthetic antioxidants to mitochondria: towards a mitochondrial medicine for neurodegenerative diseases? Eur J Pharmacol. 2002;447:155–161. doi: 10.1016/s0014-2999(02)01839-3. [DOI] [PubMed] [Google Scholar]
- Dhanasekaran A, Kotamraju S, Karunakaran C, Kalivendi SV, Thomas S, Joseph J, Kalyanaraman B. Mitochondria superoxide dismutase mimetic inhibits peroxide-induced oxidative damage and apoptosis: Role of mitochondrial superoxide. Free Radical Biol Med. 2005;39:567–583. doi: 10.1016/j.freeradbiomed.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Kaludercic N, Carpi A, Menabo R, Giorgio M. Mitochondria and vascular pathology. Pharmacol Rep. 2009;61:123–130. doi: 10.1016/s1734-1140(09)70014-3. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Dragovich T, Gordon M, Mendelson D, Wong L, Modiano M, Chow H-HS, Samulitis B, O’Day S, Grenier K, Hersh E, Dorr R. Phase I Trial of Imexon in Patients With Advanced Malignancy. J Clin Oncol. 2007;25:1779–1784. doi: 10.1200/JCO.2006.08.9672. [DOI] [PubMed] [Google Scholar]
- Dykens JA, Will Y. The significance of mitochondrial toxicity testing in drug development. Drug Discov Today. 2007;12:777–785. doi: 10.1016/j.drudis.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Ellerby HM, Arap W, Ellerby LM, Kain R, Andrusiak R, Del Rio G, Krajewski S, Lombardo CR, Rao R, Ruoslahti E, Bredesen DE, Pasqualini R. Anti-cancer activity of targeted pro-apoptotic peptides. Nat Med. 1999;5:1032–1038. doi: 10.1038/12469. [DOI] [PubMed] [Google Scholar]
- Engel RH, Evens AM. Oxidative stress and apoptosis: a new treatment paradigm in cancer. Front Biosci. 2006;11:300–312. doi: 10.2741/1798. [DOI] [PubMed] [Google Scholar]
- Filipovska A, Kelso GF, Brown SE, Beer SM, Smith RAJ, Murphy MP. Synthesis and Characterization of a Triphenylphosphonium-conjugated Peroxidase Mimetic: Insights into the Interaction of Ebselen with Mitochondria. J Biol Chem. 2005;280:24113–24126. doi: 10.1074/jbc.M501148200. [DOI] [PubMed] [Google Scholar]
- Fink MP, Macias CA, Xiao J, Tyurina YY, Delude RL, Greenberger JS, Kagan VE, Wipf P. Hemigramicidin-TEMPO conjugates: Novel mitochondria-targeted antioxidants. Crit Care Med. 2007a;35:S461–S467. doi: 10.1097/01.CCM.0000279192.96303.E7. [DOI] [PubMed] [Google Scholar]
- Fink MP, Macias CA, Xiao J, Tyurina YY, Jiang J, Belikova N, Delude RL, Greenberger JS, Kagan VE, Wipf P. Hemigramicidin-TEMPO conjugates: Novel mitochondria-targeted anti-oxidants. Biochem Pharmacol. 2007b;74:801–809. doi: 10.1016/j.bcp.2007.05.019. [DOI] [PubMed] [Google Scholar]
- Friederich M, Hansell P, Palm F. Diabetes, oxidative stress, nitric oxide and mitochondria function. Curr Diabetes Rev. 2009;5:120–144. doi: 10.2174/157339909788166800. [DOI] [PubMed] [Google Scholar]
- Fruehauf JP, Meyskens FL., Jr Reactive Oxygen Species: A Breath of Life or Death? Clin Cancer Res. 2007;13:789–794. doi: 10.1158/1078-0432.CCR-06-2082. [DOI] [PubMed] [Google Scholar]
- Galiegue S, Tinel N, Casellas P. The Peripheral Benzodiazepine Receptor: A Promising Therapeutic Drug Target. Curr Med Chem. 2003;10:1563–1572. doi: 10.2174/0929867033457223. [DOI] [PubMed] [Google Scholar]
- Gelderman KA, Hultqvist M, Olsson LM, Bauer K, Pizzolla A, Olofsson P, Holmdahl R. Rheumatoid Arthritis: The Role of Reactive Oxygen Species in Disease Development and Therapeutic Strategies. Antioxid Redox Signal. 2007;9:1541–1567. doi: 10.1089/ars.2007.1569. [DOI] [PubMed] [Google Scholar]
- Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria as targets for chemotherapy. Apoptosis. 2009;14:624–640. doi: 10.1007/s10495-009-0323-0. [DOI] [PubMed] [Google Scholar]
- Goldstein S, Merenyi G, Russo A, Samuni A. The Role of Oxoammonium Cation in the SOD-Mimic Activity of Cyclic Nitroxides. J Am Chem Soc. 2003;125:789–795. doi: 10.1021/ja028190w. [DOI] [PubMed] [Google Scholar]
- Gottlieb E. Cancer: The fat and the furious. Nature. 2009;461:44–45. doi: 10.1038/461044a. [DOI] [PubMed] [Google Scholar]
- Graier W, Frieden M, Malli R. Mitochondria and Ca2+ signaling: old guests, new functions. Pflügers Archiv Eur J Physiol. 2007;455:375–396. doi: 10.1007/s00424-007-0296-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Kroemer G. The Pathophysiology of Mitochondrial Cell Death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr Opin Cell Biol. doi: 10.1016/j.ceb.2009.08.005. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horobin RW, Trapp S, Weissig V. Mitochondriotropics: A review of their mode of action, and their applications for drug and DNA delivery to mammalian mitochondria. J Controlled Release. 2007;121:125–136. doi: 10.1016/j.jconrel.2007.05.040. [DOI] [PubMed] [Google Scholar]
- Horton KL, Kelley SO. Engineered Apoptosis-Inducing Peptides with Enhanced Mitochondrial Localization and Potency. J Med Chem. 2009;52:3293–3299. doi: 10.1021/jm900178n. [DOI] [PubMed] [Google Scholar]
- Horton KL, Stewart KM, Fonseca SB, Guo Q, Kelley SO. Mitochondria-Penetrating Peptides. Chem Biol. 2008;15:375–382. doi: 10.1016/j.chembiol.2008.03.015. [DOI] [PubMed] [Google Scholar]
- Hoye AT, Davoren JE, Wipf P, Fink MP, Kagan VE. Targeting Mitochondria. Acc Chem Res. 2008;41:87–97. doi: 10.1021/ar700135m. [DOI] [PubMed] [Google Scholar]
- Huang P, Feng L, Oldham EA, Keating MJ, Plunkett W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature. 2000;407:390–395. doi: 10.1038/35030140. [DOI] [PubMed] [Google Scholar]
- Jauslin ML, Meier T, Smith RAJ, Murphy MP. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003;17:1972–1974. doi: 10.1096/fj.03-0240fje. [DOI] [PubMed] [Google Scholar]
- Jiang J, Belikova NA, Hoye AT, Zhao Q, Epperly MW, Greenberger JS, Wipf P, Kagan VE. A Mitochondria-Targeted Nitroxide/Hemigramicidin S Conjugate Protects Mouse Embryonic Cells Against Gamma Irradiation. Int J Radiat Oncol Biol Phys. 2008;70:816–825. doi: 10.1016/j.ijrobp.2007.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Kurnikov I, Belikova NA, Xiao J, Zhao Q, Amoscato AA, Braslau R, Studer A, Fink MP, Greenberger JS, Wipf P, Kagan VE. Structural Requirements for Optimized Delivery, Inhibition of Oxidative Stress, and Antiapoptotic Activity of Targeted Nitroxides. J Pharmacol Exp Ther. 2007;320:1050–1060. doi: 10.1124/jpet.106.114769. [DOI] [PubMed] [Google Scholar]
- Jung K, Reszka R. Mitochondria as subcellular targets for clinically useful anthracyclines. Adv Drug Deliv Rev. 2001;49:87–105. doi: 10.1016/s0169-409x(01)00128-4. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Bayir A, Bayir H, Stoyanovsky D, Borisenko GG, Tyurina YY, Wipf P, Atkinson J, Greenberger JS, Chapkin RS, Belikova NA. Mitochondria-targeted disruptors and inhibitors of cytochrome c/cardiolipin peroxidase complexes: A new strategy in anti-apoptotic drug discovery. Mol Nutr Food Res. 2009a;53:104–114. doi: 10.1002/mnfr.200700402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, Tyurin VA, Jiang J, Stoyanovsky DA, Wipf P, Kochanek PM, Greenberger JS, Pitt B, Shvedova AA, Borisenko G. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radical Biol Med. 2009b;46:1439–1453. doi: 10.1016/j.freeradbiomed.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Wipf P, Stoyanovsky D, Greenberger JS, Borisenko G, Belikova NA, Yanamala N, Samhan Arias AK, Tungekar MA, Jiang J, Tyurina YY, Ji J, Klein-Seetharaman J, Pitt BR, Shvedova AA, Bayir H. Mitochondrial targeting of electron scavenging antioxidants: Regulation of selective oxidation vs random chain reactions. Adv Drug Deliv Rev. 2009c;61:1375–1385. doi: 10.1016/j.addr.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai A, Zabbarova I, Amoscato A, Epperly M, Xiao J, Wipf P. Mitochondrial targeting of radioprotectants using peptidyl conjugates. Org Biomol Chem. 2007;5:307–309. doi: 10.1039/b613334g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann P, Thompson JLP, Levy G, Buchsbaum R, Shefner J, Krivickas LS, Katz J, Rollins Y, Barohn RJ, Jackson CE, Tiryaki E, Lomen-Hoerth C, Armon C, Tandan R, Rudnicki SA, Rezania K, Sufit R, Pestronk A, Novella SP, Heiman-Patterson T, Kasarskis EJ, Pioro EP, Montes J, Arbing R, Vecchio D, Barsdorf A, Mitsumoto H, Levin B. Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann Neurol. 2009;66:235–244. doi: 10.1002/ana.21743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EC, Smith RAJ, Murphy MP. Selective Targeting of a Redox-active Ubiquinone to Mitochondria within Cells: Antioxidant and Antiapoptotic Properties. J Biol Chem. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- Kirshner JR, He S, Balasubramanyam V, Kepros J, Yang C-Y, Zhang M, Du Z, Barsoum J, Bertin J. Elesclomol induces cancer cell apoptosis through oxidative stress. Mol Cancer Ther. 2008;7:2319–2327. doi: 10.1158/1535-7163.MCT-08-0298. [DOI] [PubMed] [Google Scholar]
- Ko YT, Falcao C, Torchilin VP. Cationic Liposomes Loaded with Proapoptotic Peptide D-(KLAKLAK)2 and Bcl-2 Antisense Oligodeoxynucleotide G3139 for Enhanced Anticancer Therapy. Mol Pharmaceutics. 2009;6:971–977. doi: 10.1021/mp900006h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koene S, Smeitink J. Mitochondrial medicine: entering the era of treatment. J Intern Med. 2009;265:193–209. doi: 10.1111/j.1365-2796.2008.02058.x. [DOI] [PubMed] [Google Scholar]
- Krishna MC, Samuni A, Taira J, Goldstein S, Mitchell JB, Russo A. Stimulation by Nitroxides of Catalase-like Activity of Hemeproteins: Kinetics and Mechanism. J Biol Chem. 1996;271:26018–26025. doi: 10.1074/jbc.271.42.26018. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial Dysfunction in Cardiac Disease: Ischemia-Reperfusion, Aging, and Heart Failure. J Mol Cell Cardiol. 2001;33:1065–1089. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- Lu B. Mitochondrial dynamics and neurodegeneration. Curr Neurol Neurosci Rep. 2009;9:212–219. doi: 10.1007/s11910-009-0032-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias CA, Chiao JW, Xiao J, Arora DS, Tyurina YY, Delude RL, Wipf P, Kagan VE, Fink MP. Treatment With a Novel Hemigramicidin-TEMPO Conjugate Prolongs Survival in a Rat Model of Lethal Hemorrhagic Shock. Ann Surg. 2007;245:305–314. doi: 10.1097/01.sla.0000236626.57752.8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Kroemer G. Mitochondria in cell death: novel targets for neuroprotection and cardioprotection. Trends Mol Med. 2003;9:196–205. doi: 10.1016/s1471-4914(03)00046-7. [DOI] [PubMed] [Google Scholar]
- McBride HM, Neuspiel M, Wasiak S. Mitochondria: More Than Just a Powerhouse. Curr Biol. 2006;16:R551–R560. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- Meier T, Buyse G. Idebenone: An emerging therapy for Friedreich ataxia. J Neurol. 2009;256:25–30. doi: 10.1007/s00415-009-1005-0. [DOI] [PubMed] [Google Scholar]
- Mitchell JB, Russo A, Kuppusamy P, Krishna MC. Radiation, Radicals, and Images. Ann NY Acad Sci. 2000;899:28–43. doi: 10.1111/j.1749-6632.2000.tb06174.x. [DOI] [PubMed] [Google Scholar]
- Mizuguchi Y, Chen J, Seshan SV, Poppas DP, Szeto HH, Felsen D. A novel cell-permeable antioxidant peptide decreases renal tubular apoptosis and damage in unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2008;295:F1545–1553. doi: 10.1152/ajprenal.00395.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP. Selective targeting of bioactive compounds to mitochondria. Trends Biotechnol. 1997;15:326–330. doi: 10.1016/S0167-7799(97)01068-8. [DOI] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP, Echtay KS, Blaikie FH, Asin-Cayuela J, Cochemé HM, Green K, Buckingham JA, Taylor ER, Hurrell F, Hughes G, Miwa S, Cooper CE, Svistunenko DA, Smith RAJ, Brand MD. Superoxide Activates Uncoupling Proteins by Generating Carbon-centered Radicals and Initiating Lipid Peroxidation: Studies using a Mitochondria-targeted Spin Trap derived from α-Phenyl-N-tert-butylnitrone. J Biol Chem. 2003;278:48534–48545. doi: 10.1074/jbc.M308529200. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Smith RAJ. Targeting Antioxidants to Mitochondria by Conjugation to Lipophilic Cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- Neuzil J, Dong L-F, Ramanathapuram L, Hahn T, Chladova M, Wang X-F, Zobalova R, Prochazka L, Gold M, Freeman R, Turanek J, Akporiaye ET, Dyason JC, Ralph SJ. Vitamin E analogues as a novel group of mitocans: Anti-cancer agents that act by targeting mitochondria. Mol Aspects Med. 2007;28:607–645. doi: 10.1016/j.mam.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Pearce LL, Epperly MW, Greenberger JS, Pitt BR, Peterson J. Identification of Respiratory Complexes I and III as Mitochondrial Sites of Damage Following Exposure to Ionizing Radiation and Nitric Oxide. Nitric Oxide. 2001;5:128–136. doi: 10.1006/niox.2001.0338. [DOI] [PubMed] [Google Scholar]
- Pei Y, Lilly MJ, Owen DJ, D’Souza LJ, Tang X-Q, Yu J, Nazarbaghi R, Hunter A, Anderson CM, Glasco S, Ede NJ, James IW, Maitra U, Chandrasekaran S, Moos WH, Ghosh SS. Efficient Syntheses of Benzothiazepines as Antagonists for the Mitochondrial Sodium-Calcium Exchanger: Potential Therapeutics for Type II Diabetes. J Org Chem. 2003;68:92–103. doi: 10.1021/jo020446t. [DOI] [PubMed] [Google Scholar]
- Pepe S, Marasco SF, Haas SJ, Sheeran FL, Krum H, Rosenfeldt FL. Coenzyme Q10 in cardiovascular disease. Mitochondrion. 2007;7:S154–S167. doi: 10.1016/j.mito.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Pérez VI, Bokov A, Remmen HV, Mele J, Ran Q, Ikeno Y, Richardson A. Is the oxidative stress theory of aging dead? Biochim Biophys Acta. 2009;1790:1005–1014. doi: 10.1016/j.bbagen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petri S, Kiaei M, Damiano M, Hiller A, Wille E, Manfredi G, Calingasan NY, Szeto HH, Beal MF. Cell-permeable peptide antioxidants as a novel therapeutic approach in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2006;98:1141–1148. doi: 10.1111/j.1471-4159.2006.04018.x. [DOI] [PubMed] [Google Scholar]
- Pompella A, Visvikis A, Paolicchi A, De Tata V, Casini AF. The changing faces of glutathione, a cellular protagonist. Biochem Pharmacol. 2003;66:1499–1503. doi: 10.1016/s0006-2952(03)00504-5. [DOI] [PubMed] [Google Scholar]
- Prenner EJ, Lewis RNAH, McElhaney RN. The interaction of the antimicrobial peptide gramicidin S with lipid bilayer model and biological membranes. Biochim Biophys Acta Biomembr. 1999;1462:201–221. doi: 10.1016/s0005-2736(99)00207-2. [DOI] [PubMed] [Google Scholar]
- Rajagopalan MS, Gupta K, Epperly MW, Franicola D, Zhang X, Wang H, Zhao H, Tyurin VA, Pierce JG, Kagan VE, Wipf P, Kanai AJ, Greenberger JS. The Mitochondria-targeted Nitroxide JP4-039 Augments Potentially Lethal Irradiation Damage Repair. In Vivo. 2009;23:717–726. [PMC free article] [PubMed] [Google Scholar]
- Reddy PH. Mitochondrial Medicine for Aging and Neurodegenerative Diseases. Neuromol Med. 2008;10:291–315. doi: 10.1007/s12017-008-8044-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve AK, Krishnan KJ, Turnbull D. Mitochondrial DNA Mutations in Disease, Aging, and Neurodegeneration. Ann NY Acad Sci. 2008a;1147:21–29. doi: 10.1196/annals.1427.016. [DOI] [PubMed] [Google Scholar]
- Reeve AK, Krishnan KJ, Turnbull DM. Age related mitochondrial degenerative disorders in humans. Biotechnol J. 2008b;3:750–756. doi: 10.1002/biot.200800066. [DOI] [PubMed] [Google Scholar]
- Richards GM, Mehta MP. Motexafin gadolinium in the treatment of brain metastases. Exp Opin Pharmacother. 2007;8:351–359. doi: 10.1517/14656566.8.3.351. [DOI] [PubMed] [Google Scholar]
- Rogge MM. The Role of Impaired Mitochondrial Lipid Oxidation in Obesity. Biol Res Nurs. 2009;10:356–373. doi: 10.1177/1099800408329408. [DOI] [PubMed] [Google Scholar]
- Samuni A, Goldstein S, Russo A, Mitchell JB, Krishna MC, Neta P. Kinetics and Mechanism of Hydroxyl Radical and OH-Adduct Radical Reactions with Nitroxides and with Their Hydroxylamines. J Am Chem Soc. 2002;124:8719–8724. doi: 10.1021/ja017587h. [DOI] [PubMed] [Google Scholar]
- Schlüter T, Struy H, Schönfeld P. Protection of mitochondrial integrity from oxidative stress by the triaminopyridine derivative flupirtine. FEBS Lett. 2000;481:42–46. doi: 10.1016/s0014-5793(00)01923-2. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Di Prospero NA, Fischbeck K. Clinical experience with high-dose idebenone in Friedreich ataxia. J Neurol. 2009;256:42–45. doi: 10.1007/s00415-009-1008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu S-S, Nauduri D, Anders MW. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochim Biophys Acta Mol Basis Dis. 2006;1762:256–265. doi: 10.1016/j.bbadis.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Smith RAJ, Porteous CM, Coulter CV, Murphy MP. Selective targeting of an antioxidant to mitochondria. Eur J Biochem. 1999;263:709–716. doi: 10.1046/j.1432-1327.1999.00543.x. [DOI] [PubMed] [Google Scholar]
- Song W, Shin J, Lee J, Kim H, Oh D, Edelberg JM, Wong SC, Szeto H, Hong MK. A potent opiate agonist protects against myocardial stunning during myocardial ischemia and reperfusion in rats. Coronary Artery Dis. 2005;16:407–410. doi: 10.1097/00019501-200509000-00011. [DOI] [PubMed] [Google Scholar]
- Soule BP, Hyodo F, Matsumoto K-i, Simone NL, Cook JA, Krishna MC, Mitchell JB. The chemistry and biology of nitroxide compounds. Free Radical Biol Med. 2007;42:1632–1650. doi: 10.1016/j.freeradbiomed.2007.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkov AA. The Role of Mitochondria in Reactive Oxygen Species Metabolism and Signaling. Ann NY Acad Sci. 2008;1147:37–52. doi: 10.1196/annals.1427.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoyanovsky DA, Vlasova II, Belikova NA, Kapralov A, Tyurin V, Greenberger JS, Kagan VE. Activation of NO donors in mitochondria: Peroxidase metabolism of (2-hydroxyamino-vinyl)-triphenyl-phosphonium by cytochrome c releases NO and protects cells against apoptosis. FEBS Lett. 2008;582:725–728. doi: 10.1016/j.febslet.2008.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland TE, Anderson RL, Hughes RA, Altmann E, Schuliga M, Ziogas J, Stewart AG. 2-Methoxyestradiol - a unique blend of activities generating a new class of anti-tumour/anti-inflammatory agents. Drug Discov Today. 2007;12:577–584. doi: 10.1016/j.drudis.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Szeto HH. Mitochondria-targeted peptide antioxidants: Novel neuroprotective agents. APPS J. 2006;8:E521–E531. doi: 10.1208/aapsj080362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto HH. Mitochondria-Targeted Cytoprotective Peptides for Ischemia-Reperfusion Injury. Antioxid Redox Signal. 2008;10:601–620. doi: 10.1089/ars.2007.1892. [DOI] [PubMed] [Google Scholar]
- Szewczyk A, Jarmuszkiewicz W, Kunz WS. Mitochondrial potassium channels. IUBMB Life. 2009;61:134–143. doi: 10.1002/iub.155. [DOI] [PubMed] [Google Scholar]
- Szewczyk A, Marbán E. Mitochondria: a new target for K+ channel openers? Trends Pharmacol Sci. 1999;20:157–161. doi: 10.1016/s0165-6147(99)01301-2. [DOI] [PubMed] [Google Scholar]
- Szewczyk A, Wojtczak L. Mitochondria as a Pharmacological Target. Pharmacol Rev. 2002;54:101–127. doi: 10.1124/pr.54.1.101. [DOI] [PubMed] [Google Scholar]
- Tauskela J. MitoQ - a mitochondria-targeted antioxidant. IDrugs. 2007;10:399–412. [PubMed] [Google Scholar]
- Thomas DA, Stauffer C, Zhao K, Yang H, Sharma VK, Szeto HH, Suthanthiran M. Mitochondrial Targeting with Antioxidant Peptide SS-31 Prevents Mitochondrial Depolarization, Reduces Islet Cell Apoptosis, Increases Islet Cell Yield, and Improves Posttransplantation Function. J Am Soc Nephrol. 2007;18:213–222. doi: 10.1681/ASN.2006080825. [DOI] [PubMed] [Google Scholar]
- Todesco L, Waldhauser K, Krähenbühl S. Mitochondrial Toxicity of Drugs. Chimia. 2006;60:37–39. [Google Scholar]
- Toogood PL. Mitochondrial drugs. Curr Opin Chem Biol. 2008;12:457–463. doi: 10.1016/j.cbpa.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- Turner N, Heilbronn LK. Is mitochondrial dysfunction a cause of insulin resistance? Trends Endocrinol Metab. 2008;19:324–330. doi: 10.1016/j.tem.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Veenman L, Gavish M. The peripheral-type benzodiazepine receptor and the cardiovascular system. Implications for drug development. Pharmacol Ther. 2006;110:503–524. doi: 10.1016/j.pharmthera.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Visch H-J, Rutter GA, Koopman WJH, Koenderink JB, Verkaart S, de Groot T, Varadi A, Mitchell KJ, van den Heuvel LP, Smeitink JAM, Willems PHGM. Inhibition of Mitochondrial Na+-Ca2+ Exchange Restores Agonist-induced ATP Production and Ca2+ Handling in Human Complex I Deficiency. J Biol Chem. 2004;279:40328–40336. doi: 10.1074/jbc.M408068200. [DOI] [PubMed] [Google Scholar]
- Wasilewski M, Scorrano L. The changing shape of mitochondrial apoptosis. Trends Endocrinol Metab. 2009;20:287–294. doi: 10.1016/j.tem.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC, Parsons-Mair HN, Gebicki S, Gebicki JM, Davies MJ. Requirements for superoxide-dependent tyrosine hydroperoxide formation in peptides. Biochem J. 2004;381:241–248. doi: 10.1042/BJ20040259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wipf P, Xiao J, Jiang J, Belikova NA, Tyurin VA, Fink MP, Kagan VE. Mitochondrial Targeting of Selective Electron Scavengers: Synthesis and Biological Analysis of Hemigramicidin-TEMPO Conjugates. J Am Chem Soc. 2005;127:12460–12461. doi: 10.1021/ja053679l. [DOI] [PubMed] [Google Scholar]
- Yang L, Zhao K, Calingasan NY, Luo G, Szeto HH, Beal MF. Mitochondria Targeted Peptides Protect Against 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Neurotoxicity. Antioxid Redox Signal. 2009;11:2095–2104. doi: 10.1089/ars.2009.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousif LF, Stewart KM, Kelley SO. Targeting Mitochondria with Organelle-Specific Compounds: Strategies and Applications. ChemBioChem. 2009;10:1939–1950. doi: 10.1002/cbic.200900185. [DOI] [PubMed] [Google Scholar]
- Zhang R, Goldstein S, Samuni A. Kinetics of superoxide-induced exchange among nitroxide antioxidants and their oxidized and reduced forms. Free Radical Biol Med. 1999;26:1245–1252. doi: 10.1016/s0891-5849(98)00328-1. [DOI] [PubMed] [Google Scholar]
- Zhao K, Luo G, Giannelli S, Szeto HH. Mitochondria-targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert-butyl hydroperoxide in neuronal cell lines. Biochem Pharmacol. 2005;70:1796–1806. doi: 10.1016/j.bcp.2005.08.022. [DOI] [PubMed] [Google Scholar]
- Zhao K, Luo G, Zhao G-M, Schiller PW, Szeto HH. Transcellular Transport of a Highly Polar 3+ Net Charge Opioid Tetrapeptide. J Pharmacol Exp Ther. 2003;304:425–432. doi: 10.1124/jpet.102.040147. [DOI] [PubMed] [Google Scholar]
- Zhao K, Zhao G-M, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH. Cell-permeable Peptide Antioxidants Targeted to Inner Mitochondrial Membrane inhibit Mitochondrial Swelling, Oxidative Cell Death, and Reperfusion Injury. J Biol Chem. 2004;279:34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovasc Res. 2009;83:213–225. doi: 10.1093/cvr/cvp151. [DOI] [PMC free article] [PubMed] [Google Scholar]