

Abstract
This paper presents aspects of the coordination chemistry of mono- and divalent manganese complexes supported by the anionic tris(phosphino)borate ligand, [PhBPiPr3] (where [PhBPiPr3] = [PhB(CH2PiPr2)3]−). The Mn(II) halide complexes, [PhBPiPr3]MnCl (1 and [PhBPiPr3]MnI (2), have been characterized by X-ray diffraction, SQUID magnetometry, and EPR spectroscopy. Compound 2 serves as a precursor to a series of Mn azide, alkyl, and amide species: [PhBPiPr3]Mn(N3) (3), [PhBPiPr3]Mn(CH2Ph) (4), [PhBPiPr3]Mn(Me) (5), [PhBPiPr3]Mn(NH(2,6-iPr2-C6H3)) (6), [PhBPiPr3]Mn(dbabh) (7), and [PhBPiPr3]Mn(1-Ph(isoindolate)) (8). The complexes 2 – 8 feature a divalent-metal center and are pseudotetrahedral. They collectively represent an uncommon structural motif for low-coordinate, polyphosphine-supported Mn complexes. Two Mn(I) species have also been prepared. These include the Tl-Mn adduct, [PhBPiPr3]Tl-MnBr(CO)4 (9), and the octahedral complex [PhBPiPr3]Mn(CNtBu)3 (10). Some of our initial synthetic efforts to generate [PhBPiPr3]Mn≡Nx species are briefly described, as are DFT studies that probe the electronic viability of these types of multiply bonded target structures.
I. Introduction
Transition metal complexes containing metal-ligand multiple bonds figure prominently in coordination chemistry, especially in atom- and group-transfer processes.1, 2 Metal species of the types LnM=E and LnM≡E, where E = O, NR, CR2, can act as intermediates in numerous catalytic reactions including epoxidation,3-6 aziridination,7-10 cyclopropanation,11-13 olefin metathesis,14-17 and C-H bond insertion.10, 18 Besides having broad synthetic utility, metal-to-ligand multiply bonded species also serve as biomimetic models of active sites in metalloenzymes.19-22 Until a few years ago, examples of stable complexes featuring metal-to-ligand multiple bonds were rare for the later metals,23-25 especially for those in the first-row.26 While the paucity of these compounds was typically attributed to an inherent incompatibility of high d-electron counts with multiply bonded π-donor substituents,1 low-coordinate geometries have provided one strategic path to circumvent this limitation. The recent rise in literature reports of well-defined LnM=E and LnM≡E systems for the metals M = Fe,27-33 Co,34-38 Ni,39-43 and Cu44, 45 strongly attests to the viability of these types of species for the first-row, mid-to-late metals.
Manganese compounds featuring multiply bonded terminal functionalities such as nitrides and imides are far more abundant than their later first-row counterparts.46 For example, Mn(V) nitrides are well known to be stabilized by various ligand auxiliaries, including porphyrins,47-51 macrocyclic amines,52, 53 cyanides,54 and Schiff bases.55-58 In contrast, Mn(VI) and Mn(VII) nitrides are rare. Wieghardt and coworkers prepared two examples of Mn(VI) nitrides in situ, but neither compound has been fully characterized.52, 54 Hursthouse and coworkers isolated the sole example of a Mn(VII) nitride, [Mn(N)(NtBu)3]2−, which is supported by imido ligands.59 Other relevant manganese imides from the Hursthouse group include species of the type MnVII(NtBu)3X, as well as the homoleptic compound [MnVI(NtBu)4]2−.60-62 Terminal Mn(V) imides are also stabilized by corroles, as recently reported by Abu-Omar and coworkers.63-65 Finally, elegant work from Groves and Carreira strongly implicates Mn(V) acylimides (generated in situ from the nitrides) as effective group-transfer catalysts to olefin and enol ether substrates.47, 55, 66
Our group has been investigating the tris(phosphino)borate (generally denoted as [BP3]) and hybrid bis(phosphino)pyrazolylborate ([BP2(pz)]) ligands as scaffolds for M≡E linkages. Our endeavors have led to the characterization of stable Co(III)36 and Fe(II/III) imides, 27, 29 as well as distinctive examples of Fe(IV) imides and nitrides.28, 67 Motivated by these results, we wish to extend this chemistry to other mid-to-late first-row metals such as Ni68 and Mn. In particular, we are interested in whether [BP3]Mn≡Nx species (d3 and d4) are electronically accessible, and in defining synthetic methods for their generation. In pursuit of this task, we began to study aspects of the fundamental coordination chemistry of previously unexplored [BP3]Mn-systems. In this first study, we report on the synthesis and characterization of a family of monovalent- and divalent-manganese compounds supported by the [PhBPiPr3] ligand (where [PhBPiPr3] = [PhB(CH2PiPr2)3]−), some of which might ultimately serve as precursors for the preparation of [BP3]Mn≡Nx species. Complementary theoretical studies are described for hypothetical imide [PhBPiPr3]Mn(NtBu) and nitride [PhBPiPr3]Mn(N) systems. These species appear to be plausible target structures based upon electronic considerations. While studies towards well-defined [BP3]Mn≡Nx systems are ongoing, the four-coordinate [PhBPiPr3]Mn(II) species that are described here represent structurally unusual examples of low-coordinate manganese complexes supported by polyphosphine ligands.
II. Results and Discussion
IIa. Synthesis and Characterization of [PhBPiPr3]Mn(II) Halides
The Mn(II) halides [PhBPiPr3]MnCl (1) and [PhBPiPr3]MnI (2) were readily prepared by mixing the thallium reagent [PhBPiPr3]Tl with MnX2 (where X = Cl or I) (Scheme 1).69 This methodology has been used previously by us to prepare related compounds of divalent Fe, Ru, Co, and Ni.68, 70, 71 The halides 1 and 2 are colorless in solution and can be crystallized from benzene/petroleum ether. Compound 1 provided light pink blocks in low yield (33 %), while 2 was obtained as colorless blocks in moderate yield (62 %). Because of its higher crystalline yield, compound 2 was used as the preferred starting material throughout the research described herein.
Scheme 1.
The halides 1 and 2 were characterized by various techniques including X-ray crystallography, SQUID magnetometry, and EPR spectroscopy. The solid-state structures of 1 and 2 are shown in Figures 1 and 2. The chloride complex 1 crystallizes as a mixture of its monomer (1a) and its dimer {[PhBPiPr3]Mn(μ-Cl)}2 (1b), whereas the iodide complex 2 crystallizes exclusively as a monomer. A similar monomer-dimer equilibrium was observed for [PhBP3]CoX systems in solution (where X = Cl or Br).71 Solid-state structures of monomeric 1a and 2 are pseudotetrahedral with approximate three-fold symmetry about the axis containing B, Mn, and X (I-Mn-B = 176.6°, Cl-Mn-B = 175.5°). The Mn-P bond distances are nearly equal, as well as the P-Mn-P bond angles. For example, monomer 1a has Mn-P distances of 2.53 Å and P-Mn-P angles that are near 90° (90.6 to 92.6°). Likewise, compound 2 exhibits a narrow spread in its Mn-P bond distances (2.524 to 2.538 Å) and P-Mn-P angles (90.7 to 91.4°). The Mn-P bond distances are not unusual for high-spin Mn(II),72 but are significantly longer than those of tris(phosphino)borate complexes of later first-row transition metal ions. The series of structurally characterized chloride complexes, [PhBPiPr3]MCl for M = Mn, Fe, Co, and Ni, show decreasing M-P bond distances, which reflect both their decreasing ionic radii across the series as well as their total spin S.
Figure 1.
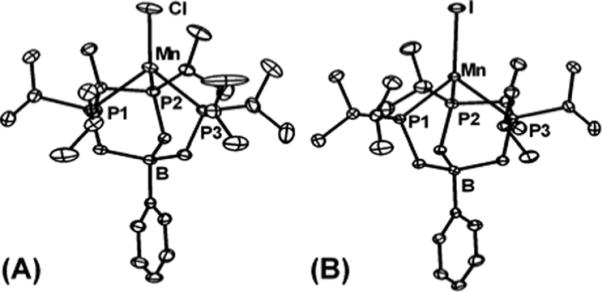
Displacement ellipsoid representations (50%) of (A) [PhBPiPr3]MnCl (1a) and (B)[PhBPiPr3]MnI (2). Hydrogen atoms and solvent molecules have been omitted for clarity. Selected bond distances (Å) and angles (deg) for 1a: Mn–Cl 2.2687(7), Mn–P1 2.5300(6), Cl–Mn–P1 125.38(2), Mn–P2 2.5298(6), Mn–P3 2.5289(6), Cl–Mn–P2 120.22(2), Cl–Mn–P3 126.14(2), P1–Mn–P2 90.55(2), P2–Mn–P3 92.64(2), P1–Mn–P3 92.22(2). For 2: Mn–I 2.6394(4), Mn–P1 2.5351(7), Mn–P2 2.5238(7), Mn–P3 2.5379(6), P1–Mn–I 127.19(2), P2–Mn–I 122.27(2), P3–Mn–I 124.09(2), P1–Mn–P2 91.39(2), P2–Mn–P3 90.68(2), P1–Mn–P3 90.95(2).
Figure 2.
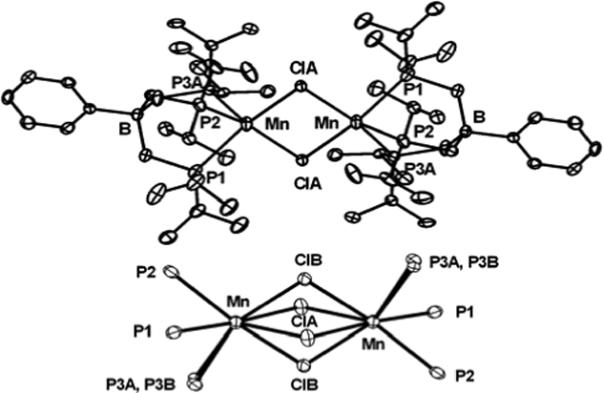
Displacement ellipsoid representations (50%) of {[PhBPiPr3]Mn(μ-Cl)}2 (1b) (top). Hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (deg) for 1b: Mn–ClA 2.4625(9), Mn–ClA 2.5327(9), Mn–ClB 2.4630(9), Mn–ClB 2.525(1), Mn–P1 2.6162(7), Mn–P2 2.6132(7), Mn–P3A 2.552(4), Mn–P3B 2.580(4), P1–Mn–P2 86.38(2), P3A–Mn–P1 87.13(7), P3B–Mn–P1 94.03(7), P3A–Mn–P2 92.55(8), P3B–Mn–P2 86.26(7). View of the core of 1b showing both positions of the disordered chloride and P3 atom (below). The angle between the Mn2(ClA)2 and Mn2(ClB)2 planes is 41°.
The dimer {[PhBPiPr3]Mn(μ-Cl)}2 (1b) crystallizes in the triclinic crystal system Pī and features a Mn2Cl2 core that contains an inversion center. The chloride and one Pi Pr2 unit are disordered, but both can be independently refined in two positions of nearly equal populations. The solid-state structure of one conformer is shown in Figure 2 (top). A simplified structure containing all of the disordered atoms in the core is shown below it. In the dimer, the Mn center is five-coordinate and roughly square pyramidal. Two phosphorus atoms and two chlorides comprise the base, with an axial phosphine at the apex of the pyramid. The Mn-P bond distances in the dimer are even more elongated relative to the monomer 1a (Mn-P = 2.613, 2.616, and 2.552 Å). One of these bonds (Mn-P3) is slightly shorter than the other two and corresponds to the Mn-Pax bond. The Mn-Cl bonds in the dimer are also significantly longer (2.463 and 2.533 Å) compared to the monomer (2.269 Å).
Magnetic susceptibility (SQUID) data were obtained on polycrystalline samples of 1 and 2. The μeff versus T plots are nearly identical and show very little temperature dependence (Figure 3A). From 60 to 300 K, the μeff values average 5.94 μB for 1 (calculated as a monomer) and 5.97 μB for 2. The μeff value for 1 is identical to its solution magnetic moment of 5.9(1) in benzene, where we presume the monomeric form is favored over the dimer form (Evans method, 3 runs).73, 74 These values are consistent with 5 unpaired electrons per Mn, verifying the high-spin configuration of these [PhBPiPr3]MnX species. The proximity of μeff to the spin-only value (5.92 μB) further suggests that the ground states of 1 and 2 are orbitally nondegenerate as expected for a ground-state electronic configuration where all the d-orbitals are singly populated (6S). The χMT versus T plots for 1 and 2 are also similar, but distinct at very low temperatures. From 4 to 75 K, χMT dips slightly for 2, perhaps due to intermolecular paramagnetic quenching (Figure 3B).75 The χMT versus T plot for 1, however, shows a gradual increase in χMT in the same temperature range. The small rise in χMT from 4.43 to 4.71 cm3 K/mol might be indicative of weak ferromagnetic coupling. Similarly weak coupling has been observed in another Mn(μ-Cl) dimer, and might thus arise from the presence of the dimer 1b.76
Figure 3.

(A) SQUID plot of μeff (μB) versus and (B) χMT (cm3K/mol) versus T for [PhBPiPr3]MnCl, 1, (■), and [PhBPiPr3]MnI, 2, (□). An expansion of the χMT vs. T plot at low temperatures (4 to 30 K) is shown in the inset.
EPR spectra were also recorded on the halides in methyl-THF glass at 8 K. The iodide 2 has an axial spectrum with transitions at ∼120 and 330 mT (Figure 4A). The observed g values, ∼ 6 and 2, match the calculated transitions for the ms = ± ½ states of a S = 5/2 paramagnet with E/D ∼ 0. Hyperfine structure due to the 55Mn nucleus (I = 5/2) is evident in the g = 2 signal, which shows a six-line splitting with A ∼ 63 G. The octahedral complexes MnX2(dmpe)2 (where X = Br, I) show nearly identical EPR signatures.77 Unlike 2, the chloride 1 has an EPR spectrum with numerous transitions occurring from 0 to 1000 mT (Figure 4B). The complicated spectrum is difficult to interpret and is tentatively attributed to a superposition of the monomer (∼ 130 and 360 mT) and dimer forms of 1. The dimer, an S = 5 paramagnet, could itself give rise to multiple transitions. For both 1 and 2, hyperfine coupling to the 31P nucleus (I = ½) is not observed, suggesting that the spin density is tightly localized on Mn and therefore indicative of attenuated covalent character at the Mn-P bond by comparison to other [BP3]M paramagnets we have studied.27, 71, 78
Figure 4.

EPR spectra of glassy methyl-THF solutions of (A) [PhBPiPr3]MnI (2) and (B) [PhBPiPr3]MnCl (1) at 8 K. Instrumental parameters: ν = 9.38 GHz, modulation frequency = 100 kHz, modulation amplitude = 5 G, microwave power = 0.064 mW, conversion time = 81.9 ms, time constant = 20.5 ms, sweep time = 83.9 ms, 3 scans.
Compounds 1 and 2 were also studied by NMR spectroscopy and electrochemistry, but these data proved relatively uninformative. The 1H NMR spectra contained a few resolvable resonances and those that could be resolved were unusually broad by comparison to related [PhBPiPr3]FeX and [PhBPiPr3]CoX systems. The cyclic voltammograms of 1 and 2 did not reveal any reversible features (0.35 M [nBu4N][PF6] in THF). The lack of any reversible redox couples also contrasts the analogous halide complexes of Fe, Co, and Ni, all of which exhibit at least pseudo-reversible MII/MI redox couples.
Surprisingly, a survey of the CCDC revealed only three structures of monomeric four-coordinate Mn halide complexes with one or two phosphine donor(s).72, 79, 80 No such structures with three phosphine donors were found. Even monomeric five-coordinate polyphosphine-ligated Mn halides are few in number, with only three structurally characterized examples.81-83 Low-coordinate Mn halides are also known for the related tridentate ligand tris(pyrazoyl)borate (Tp). A handful of monomeric four-coordinate manganese structures featuring these ligands have been reported.84-88
IIb. Synthesis and Characterization of [PhBPiPr3]Mn(II) Azide, Alkyls, and Amides
A series of divalent-manganese complexes featuring other X-type ligands were readily prepared from the iodide 2 via metathesis (Scheme 2). For example, addition of excess sodium azide to 2 produced off-white [PhBPiPr3]Mn(N3) (3) in good yield (73 %). Compound 3 features a terminal azide ligand with a characteristic ν(N3) stretch at 2070 cm−1 (KBr, THF). Its solid-state structure is shown in Figure 5. Not surprisingly, the pseudohalide 3 exhibits a similar molecular geometry to the monomeric halides 1 and 2. In a CCDC search of all structurally characterized Mn complexes containing a terminal azide, the mean Mn-Nazide bond distance is 2.190 Å with a standard deviation of 0.096 Å. The Mn-N bond distance of 2.046(3) Å in 3 is therefore unusually short. Manganese complexes featuring similarly short Mn-N bond distances for terminal azides are typically in the oxidation state +3.53, 89-92
Scheme 2.
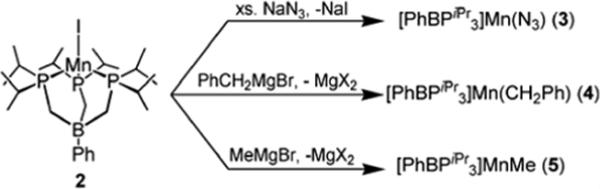
Figure 5.
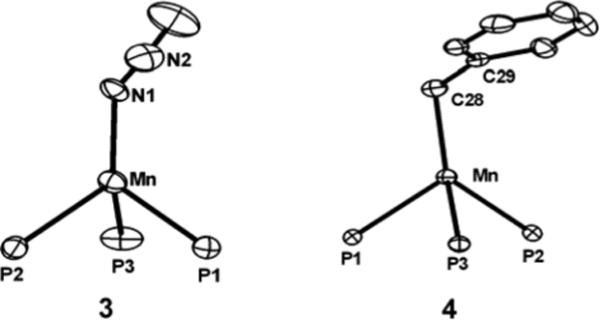
Displacement ellipsoid representations (50%) of [PhBPiPr3]Mn(N3) (3) and [PhBPiPr3]Mn(CH2Ph) (4). For clarity, hydrogen atoms have been omitted and only the phosphorous atoms of the ligand are shown. For 3, only one of the two molecules in the asymmetric unit is shown. Selected bond distances (Å) and angles (deg) for 3: Mn–N1 2.046(3), Mn–P1 2.534(1), Mn–P2 2.522(1), Mn–P3 2.521(2), N1–N2 1.112(5), N2–N3 1.202(6), N2–N1–Mn 128.8(3), P1–Mn–P2 92.33(4), P2–Mn–P3 93.21(5), P1–Mn–P3 91.04(5), N1–Mn–P1 123.1(1), N1–Mn–P2 125.7(1), N1–Mn–P3 122.2(1). For 4: Mn–C28 2.138(2), Mn–P1 2.5679(4), Mn–P2 2.5507(5), Mn–P3 2.5620(5), P1–Mn–P2 91.46(1), P2–Mn–P3 89.44(1), P1–Mn–P3 89.90(1), C28–Mn–P1 117.86(5), C28–Mn–P2 126.42(5), C28–Mn–P3 130.59(5), Mn–C28–C29 111.5(1).
Manganese alkyl compounds were also readily prepared from the addition of alkyl Gringards PhCH2MgBr and MeMgBr to 2 at −90° in THF. The bright yellow complex [PhBPiPr3]Mn(CH2Ph) (4) and pale yellow complex [PhBPiPr3]Mn(Me) (5) are soluble in hydrocarbons, and can be crystallized from cyclopentane at −30 °C. The benzyl complex 4 was characterized by X-ray diffraction, and its solid-state structure is shown in Figure 5. Related examples of low-coordinate monomeric organomanganese compounds are known and include trigonal (β-diketiminato)MnR and tetrahedral (P2)MnR2 species, where P2 represents bidentate phosphine or two PR3 ligands.93-95 Despite the low valence electron count of these Mn systems, rather little has been reported concerning their reactivity patterns. One interesting exception concerns tetrahedral Mn(II) dialkyl compounds supported by diimine ligands that readily undergo alkyl migration to the diimine ligand at ambient temperatures.96 We have found that the [PhBPiPr3]-supported manganese alkyls 4 and 5 are thermally stable up to 65 °C even under an atmosphere of dihydrogen or CO. This sharply contrasts the related iron(II) alkyls [PhBPiPr3]Fe-R, which lose alkane upon exposure to H2 and bind CO readily.97
Manganese amides were generated by the addition of lithium and potassium amides to 2 at −90° in THF (Scheme 3). In the metathesis reactions with lithium amides, a common side-product was the free ligand [PhBPiPr3][Li], easily identified by 31P NMR spectroscopy. Replacement of lithium by potassium minimizes the liberation of the [PhBPiPr3] anion. For example, 2,6-diispropylaniline can be deprotonated with KH and added to 2 to produce yellow [PhBPiPr3]Mn(NH(2,6-iPr2-C6H3)) (6) in high yield (90 %).
Scheme 3.
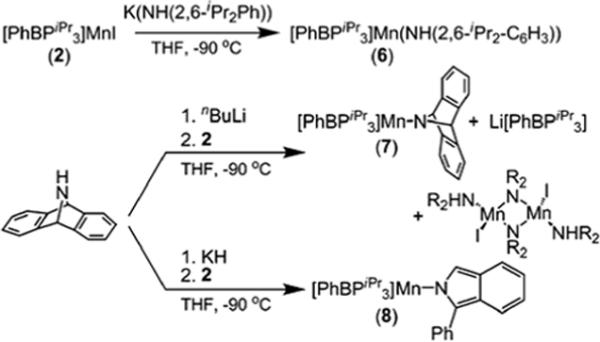
This strategy was also employed to install 2,3:5,6-dibenzo-7-azabicyclo[2.2.1]hepta-2,5-diene (Hdbabh) as the amide functionality. The lithium amide Li(dbabh) is a particularly interesting reagent to investigate because it has been successfully exploited as an N-atom transfer reagent to both Cr and Fe centers with concomitant loss of anthracene.28, 98 Methathesis of 2 and Li(dbabh) resulted in a mixture of the amide compound [PhBPiPr3]Mn(dbabh) (7), the bridged amide complex {(Hdbabh)MnI(μ-N-dbabh)}2 (see Supporting Information), and the ligand [PhBPiPr3][Li]. Though the yield of the desired compound 7 was quite low, yellow crystals of 7 could be cleanly chosen from the colorless crystals of the byproducts. Attempts to improve the yield of 7 by using K(dbabh) were unsuccessful. Deprotonation of Hdbabh with KH or benzyl potassium afforded a bright yellow-green solution; upon addition of 2 at −90°C, the reaction solution turned orange. Red-orange crystals were grown from this solution that were clearly indicative of a species different from 7. An X-ray diffraction study revealed the product to be a different Mn amide, [PhBPiPr3]Mn(1-Ph(isoindolate)) (8), an isomer of 7 wherein one of the five-membered rings has opened by an overall C–C bond cleavage. This decomposition most likely occurs during the deprotonation stage of the reaction since the solution turns brilliant yellow-green upon addition of the potassium base, whereas solutions of Li(dbabh) are pale pink.
The solid state structures of the Mn(II) amides 6, 7, and 8 are shown in Figure 6. Collectively, these amides are similar to the other pseudotetrahedral, high-spin [PhBPiPr3]Mn(II) species reported here in that they have typical Mn-P bond distances (ca. 2.55 Å) and P-Mn-P bond angles (ca. 90°). The apical N atom of 7 lies on the Mn-B axis, whereas compounds 6 and 8 exhibit a slightly canted N atom with N-Mn-B angles of 156° and 167° respectively. The Mn-N bond distances follow the order: 7 (1.948(1) Å) < 6 (1.997(2) Å) < 8 (2.054(2) Å). This trend likely reflects the relative steric repulsion between the amide groups and the isopropyl groups of the [PhBPiPr3] ligand.
Figure 6.
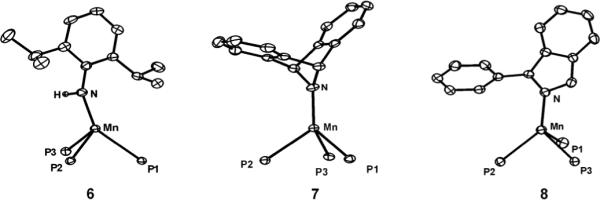
Displacement ellipsoid representations (50%) of [PhBPiPr3]Mn(NH(2,6-iPr2-C6H3)) (6), [PhBPiPr3]Mn(dbabh) (7), and [PhBPiPr3]Mn(1-Ph(isoindolate)) (8). For clarity, hydrogen atoms (except N-H) have been omitted and only the phosphorous atoms of the ligand are shown. Selected bond distances (Å) and angles (deg) for 6: Mn–N 1.997(2), Mn–P1 2.5877(6), Mn–P2 2.5813(6), Mn–P3 2.5637(6), P1–Mn–P2 87.96(2), P2–Mn–P3 89.95(2), P1–Mn–P3 93.11(2), N–Mn–P1 144.77(5), N–Mn–P2 105.73(5), N–Mn–P3 118.63(5). For 7: Mn–N 1.948(1), Mn–P1 2.5357(5), Mn–P2 2.5473(5), Mn–P3 2.5503(5), P1–Mn–P2 90.46(2), P2–Mn–P3 89.51(2), P1–Mn–P3 90.71(2), N–Mn–P1 126.19(4), N–Mn–P2 126.13(4), N–Mn–P3 122.92(4). For 8: Mn–N 2.054(2), Mn–P1 2.5308(7), Mn–P2 2.5550(7), Mn–P3 2.5718(6), P1–Mn–P2 95.13(2), P1–Mn–P3 89.54(2), P2–Mn–P3 90.09(2), N–Mn–P1 111.98(5), N–Mn–P2 136.31(5), N–Mn–P3 122.30(5).
The Mn(II) alkyls and amides were studied by cyclic voltammetry in THF with 0.35 M [nBu4N][PF6] as the supporting electrolyte and Ag/AgNO3 as the reference electrode. Interestingly, amide 7 is distinct amongst this series of complexes in that it exhibits a reversible 1-electron redox couple centered at −0.26 V (vs. Fc/Fc+, scan rate = 50 mV/s), indicative of a MnII/MnIII redox event (Figure 7). The amides 6 and 8 exhibit irreversible oxidative events near this same potential.
Figure 7.
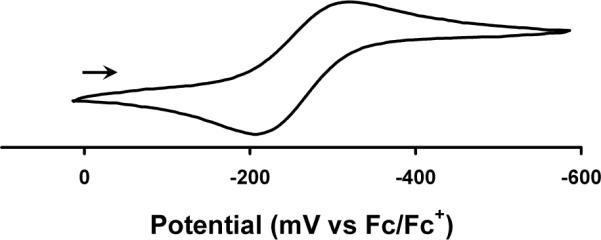
Cyclic voltammagram of [PhBPiPr3]Mn(dbabh) (7) recorded at a scan rate of 50 mV/s in THF with 0.35 M [nBu4][PF6] as the supporting electrolyte. Potentials are internally referenced to Fc/Fc+.
IIc. Synthesis and Characterization of [PhBPiPr3]Mn(I) Compounds
Monovalent-manganese compounds may prove to be valuable precursors to Mn≡NR multiply bonded species in future studies. Indeed, oxidative nitrene transfer to Fe(I) and Co(I) precursors has proven highly effective in the synthesis of related Fe≡NR, Co≡NR, and Co=NR species.29, 34-36, 99, 100 While monovalent-manganese complexes are numerous, they typically are octahedral 18-electron species stabilized by carbonyl ligands.101-105 Low-coordinate and unsaturated Mn(I) compounds, on the other hand, are uncommon. Notable examples of this latter class include a three-coordinate Mn(I) dimer supported by bulky β-diketiminate ligands and several five-coordinate Mn compounds of the type (P^P)2 Mn(CO)+ (where P^P is a bidentate phosphine).106-109
To pursue a low-coordinate Mn(I) precursor, [PhBPiPr3]Tl and MnBr(CO)5 were heated in benzene at 50 °C for several hours. The resulting yellow solution contained some TlBr precipitate, but to our surprise, the major product was a Tl-Mn adduct, [PhBPiPr3]Tl-MnBr(CO)4 (9), which was isolated in 39 % crystalline yield. Compound 9 is diamagnetic and exhibits a doublet in the 31P NMR spectrum (45.4 ppm, JTl-P 3 = 8060 Hz) that is shifted relative to [PhBPiPr3]Tl (24.3 ppm, JTl-P = 5913 Hz). The solid-state structure of 9 is shown in Figure 8. There are several crystal structures featuring a Tl-Mn bond, and the Tl-Mn bond of 2.6437(4) Å is similar to those reported previously.110, 111
Figure 8.
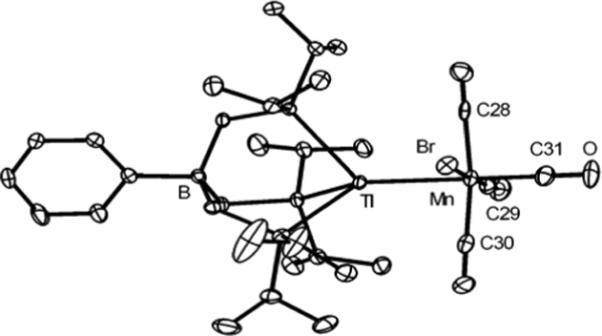
Displacement ellipsoid representations (50%) of [PhBPiPr3]Tl–MnBr(CO)4 (9). For clarity, hydrogen atoms have been omitted. Selected bond distances (Å) and angles (deg) for 9: Mn–Tl 2.6437(4), Mn–C28 1.913(3), Mn–C29 1.799(3), Mn–C30 1.851(3), Mn–C31 1.823(3), Mn–Br 2.5299(4), C31–Mn–Tl 172.74(8), Br–Mn–Tl 86.11(1), C29–Mn–C28 92.6(1), C29–Mn–C30 91.1(1), C28–Mn–Br 85.39(7), C29–Mn–Br 177.98(8), C30–Mn–Br 90.94(8), C31–Mn–Br 86.73(8).
Prolonged heating of a benzene solution of 9 resulted in its decomposition. By 31P NMR spectroscopy, one major product forms upon thermolysis, characterized by a broad peak at ∼50 ppm. An IR spectrum revealed two sharp stretches at 1999 and 1906 cm−1 (KBr/THF). We propose this species to be the Mn(I) tricarbonyl compound, [PhBPiPr3]Mn(CO)3 (Scheme 4). Indeed, the isoelectronic compound, [PhBPiPr3]Mn(CNtBu)3 (10), can be prepared by the reduction of [PhBPiPr3]MnI with sodium napthalenide followed by addition of excess CNtBu. Compound 10 is isolated in pure form but in low yield (22 %) due in part to the competitive formation of [PhBPiPr3]Na.112 Similarly, reduction of [PhBPiPr3]MnI with sodium napthalenide under CO produces the presumed degradation product of 9, [PhBPiPr3]Mn(CO)3. Unfortunately, this species has not been isolated in pure form. The solid-state structure of 10 confirms its assignment as a monovalent-Mn complex with three isocyanides (Figure 9). Relative to the high-spin Mn(II) species, the Mn-P bonds (2.36 to 2.38 Å) are more than 0.15 Å shorter. The P-Mn-P bond angles are also slightly contracted (86.5 − 89.2°). Compound 10 is also characterized by a reversible redox process at −0.66 V in its cyclic voltammagram, shown in Figure 10 (vs. Fc/Fc+, scan rate = 100 mV/s).
Scheme 4.
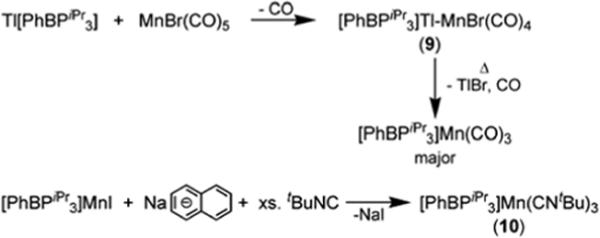
Figure 9.
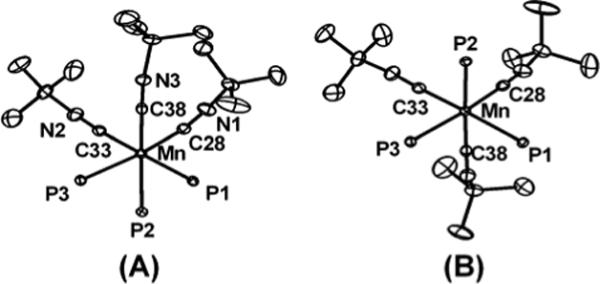
Displacement ellipsoid representations (50%) of [PhBPiPr3]Mn(CNtBu)3 (10) from the side (A) and top (B) views. For clarity, hydrogen atoms have been omitted and only the phosphorous atoms of the ligand are shown. Selected bond distances (Å) and angles (deg) for 10: Mn–C28 1.870(1), Mn–C33 1.864(1), Mn–C38 1.835(1), N1–C28 1.176(2), N2–C33 1.176(2), N3–C38 1.185(2), Mn–P1 2.3597(4), Mn–P2 2.3831(4), Mn–P3 2.3655(3), P1–Mn–P2 88.59(1), P2–Mn–P3 86.49(1), P1–Mn–P3 89.17(1), C33–Mn–C28 83.75(5), C38–Mn–C28 85.26(5), C38–Mn–C33 89.12(5).
Figure 10.
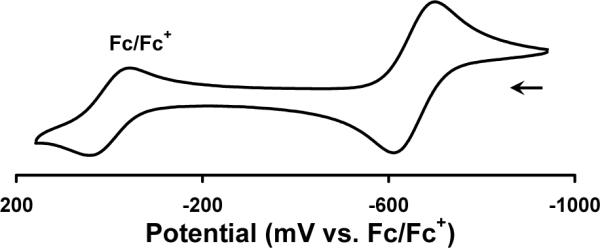
Cyclic voltammagram of [PhBPiPr3]Mn(CNtBu)3 (10) recorded at a scan rate of 100 mV/s in THF with 0.35 M [nBu4][PF6] as the supporting electrolyte. Potentials are internally referenced to Fc/Fc+.
IId. DFT Models of Mn=NtBu and Mn≡N species
DFT studies were undertaken to obtain some insight on the electronic viability of two species, [PhBPiPr3]MnIII≡NtBu and [PhBPiPr3]MnIV≡N. We have previously used DFT methods to probe the electronic structures of [BP3]M species with terminally bound Nx-functionalities that are strongly π-donating, for example [PhBP3]CoIII≡N(p-tolyl), [PhBP3]FeIII≡NtBu, and [PhBPiPr3]FeIV≡N.29, 36, 28
For the low-spin Fe and Co imides, a general d-orbital splitting diagram has been forwarded based upon these DFT studies. The d-orbitals, which transform as a1 + 2 e under idealized three-fold symmetry, split into two high-lying and three lower lying orbitals. The higher energy e-set, which in simplest terms can be thought of as comprising dxz and dyz, is destabilized by σ* and π* interactions with the phosphines and imide functionality, respectively. The remaining orbitals, of predominant dz2 (a1) and dxy and dx2-y2 (e) character, lie at lower energy and are predominantly non-bonding in nature.27
If we apply this qualitative d-orbital picture to the hypothetical d4 imide, [PhBPiPr3]Mn≡NtBu, the two ground states low-spin (S = 0) and intermediate-spin (S = 1) need to be considered. Recently, we have reported the characterization of an unusual FeIV imide that exhibits a triplet ground state.67 Because this species is isoelectronic to the hypothetical Mn(III) imide under consideration here, we might expect the latter to be a triplet as well. To test this from a theoretical standpoint, the imide [PhBPiPr3]Mn≡NtBu was optimized as a singlet and a triplet using the Jaguar computational program (B3LYP/LACVP**). The input geometry was derived from the crystal structure coordinates of [PhBPiPr3]Fe(NAd), where the metal and Nx-functionality were changed to Mn and NtBu respectively. A geometry optimization was then performed using a spin-unrestricted calculation and conducted without any symmetry constraints. (See Supporting Information)
The predicted structures for the singlet and triplet states of [PhBPiPr3]Mn≡NtBu differ quite remarkably from one another (Figure 11). The optimized singlet structure has a bent Mn-N-C angle (145°) with expanded P-Mn-P angles (97−98°), whereas the optimized triplet structure features a linear Mn-N-C angle (179°) with characteristic P-Mn-P angles (90−92°). The triplet state exhibits what appears to be a more likely geometry, in accord with the fact that it is predicted by DFT to be more energetically stable by 0.74 eV than the singlet state.
Figure 11.
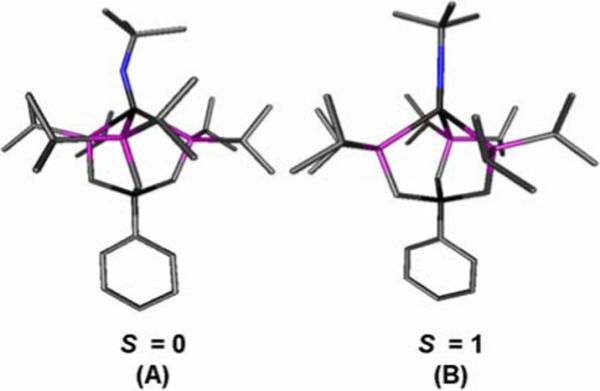
DFT-optimized structures (Jaguar 5.0, B3LYP/LACVP**) for (A) singlet [PhBPiPr3]Mn(NtBu) and (B) triplet [PhBPiPr3]Mn(NtBu). Selected bond distances (Å) and angles (deg) for (A): Mn–N 1.63; Mn–P 2.28, 2.36, 2.36; Mn–N–C 144.8; P–Mn–P 96.8, 97.2, 97.7; N–Mn–P 104.7, 126.2, 127.2. For (B): Mn–N 1.66; Mn–P 2.40, 2.44, 2.45; Mn–N–C 179.2; P–Mn–P 89.5, 89.8, 91.5; N–Mn–P 121.9, 126.2, 127.0.
Based on DFT studies, the known nitride [PhBPiPr3]FeIV≡N has a splitting diagram that differs from the structurally analogous imides in that the a1 orbital of dz2 parentage is more appreciably destabilized by sigma antibonding interactions with the nitride functionality and the phosphine donors.28 This situation leaves only two orbitals at very low energy, giving rise to the singlet ground state of [PhBPiPr3]FeIV≡N. Based on this splitting scheme, the hypothetical Mn nitride [PhBPiPr3]Mn≡N might be expected to populate a doublet ground state. To ascertain whether this would be the case, the Mn nitride [PhBPiPr3]Mn(N) was modeled as both an S = 1/2 and S = 3/2 system. In the optimized quartet structure, the nitride atom is severely tilted off the Mn-B axis by 30° (Figure 12A) in what appears to be an unfavorable geometry. The P-Mn-P bond angles (90 to 91°) and Mn-P bond distances (2.48 to 2.60 Å) are, however, typical of the pseudotetrahedral [PhBPiPr3]Mn(II) complexes described here. The Mn-N bond length of 1.62 Å seems excessively long for known Mn≡N species, which typically have Mn-N bond lengths in the range of 1.50 to 1.55 Å. These distortions in the optimized quartet structure presumably arise from population of the antibonding dz2 orbital.
Figure 12.
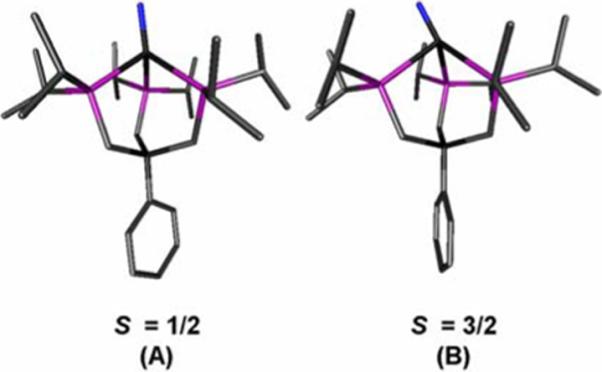
DFT-optimized structures (Jaguar 5.0, B3LYP/LACVP**) for (A) doublet [PhBPiPr3]Mn(N) and (B) quartet [PhBPiPr3]Mn(N). Selected bond distances (Å) and angles (deg) for (A): Mn–N 1.50; Mn–P 2.35, 2.41, 2.45; N–Mn–P 112.1, 122.3, 127.5; P–Mn–P 94.8, 95.6, 97.2. For (B): Mn–N 1.62; Mn–P 2.48, 2.49, 2.60; N–Mn–P 103.9, 109.3, 155.3; P–Mn–P 89.9, 90.5, 91.2.
Relative to the quartet state, the doublet state is energetically preferred by 0.41 eV and exhibits what appears to be a more reasonable geometry (Figure 12B). The nitride atom is canted off the Mn-B axis by only 9° and binds tightly to Mn with a Mn-N bond length of 1.50 Å. The P-Mn-P bond angles are slightly expanded (95 to 97°), approaching a more tetrahedral geometry. Similar parameters were found in the DFT optimized structure of [PhBPiPr3]FeIV≡N, which features expanded P-Fe-P bond angles (99−101°) and an Fe-N bond length of 1.49 Å. It is the combination of these two structural parameters that renders the a1 (dz2) orbital more antibonding for the metal nitrides than in the case of the related metal imides.
III. Concluding Remarks
With this report, we have extended our study of the coordination chemistry of first-row mid-to-late transistion metals supported by electron-releasing [BP3] ligands. As with Fe, Co, and Ni, the [PhBPiPr3] scaffold readily supports Mn(II) complexes in a pseudotetrahedral geometry. The preparative significance of the systems described herein is best underscored by noting how uncommon low-coordinate manganese complexes supported by polyphosphines are. The high donor strength and anionic charge of the [PhBPiPr3] ligand may be advantageous over polyphosphine ligands in general by helping to attenuate problematic phosphine dissociation. These two ligand properties are not always sufficient, however, as evident from the observed propensity to substitute manganese by an alkali metal cation or by thallium in the [PhBPiPr3] binding pocket under certain metathetical exchange reactions. Fortunately, the halide [PhBPiPr3]MnI does serve as a sufficiently useful reagent for most metathetical reactions, allowing the preparation of [PhBPiPr3]Mn(II) azide, alkyl, and amide species.
DFT calculations of the hypothetical Mn(III) imide [PhBPiPr3]Mn(NtBu) and nitride [PhBPiPr3]Mn(N) structures suggests that their preferred ground states will be triplet and doublet, respectively. While the preparation of Mn≡Nx species of these types has, at this stage, only been cursorily canvassed it is worth noting a few experimental observations. For instance, we have explored whether the Mn(I) complex 10 might undergo facile oxidative group transfer in the presence of organic aryl and alkylazides. We have also canvassed conditions that might have effected the oxidative deprotonation of the anilide species 6.42 Finally, attempts to generate a Mn(IV) nitride species, including thermolysis and photolysis of the amide complex [PhBPiPr3]Mn(dbabh) 7, as well as photolysis of the Mn azide 3, have been explored. As yet, none of these straightforward approaches have provided a tractable Mn≡Nx species. Therefore, while the preparation of well-defined low-valent Mn complexes with multiply bonded linkages remains a challenge, it is one that should be surmountable. Our hope is that manganese imides and nitrides of these types will benefit from synthetic access to unsaturated Mn(I) precursors, such as a four-coordinate [PhBPiPr3]Mn-PR3 species. Oxidative nitrene or nitride group transfer might then prove synthetically more fruitful. Efforts along these lines are part of ongoing studies in our labs.
Experimental Section
All manipulations were carried out using standard Schlenk or glovebox techniques under a dinitrogen atmosphere. Unless otherwise noted, solvents were deoxygenated and dried by thorough sparging with N2 gas followed by passage through an activated alumina column. Nonhalogenated solvents were tested with a standard purple solution of benzophenone ketyl in tetrahydrofuran to confirm effective oxygen and moisture removal. Deuterated solvents were purchased from Cambridge Isotopes Laboratories, Inc. and were degassed and stored over activated 3-Å molecular sieves prior to use. The compounds [PhBPiPr3]Tl,70 benzyl potassium,113 and Li(dbabh)98 were prepared according to literature procedures. The reagents 2,6-diisopropyl aniline and tBuNC were dried over sieves prior to use. Sodium azide was carefully dried by heating at 50 °C under vacuum for two days. (CAUTION: Azides are potentially explosive and should generally be stored away from heat and ignition sources.) Potassium hydride was crushed into a fine powder, washed liberally with petroleum ether, and stored at −30 °C. All other reagents were purchased from commercial vendors and used without further purification. Elemental analyses were performed by Desert Analytics, Tucson, AZ. Varian 300 MHz spectrometers were used to record the 1H NMR and 31P NMR spectra at ambient temperature. 1H chemical shifts were referenced to residual solvent, while 31P NMR chemical shifts were referenced to 85% H3PO4 at δ0 ppm. IR measurements were obtained with a KBr solution cell using a Bio-Rad Excalibur FTS 3000 spectrometer controlled by Bio-Rad Merlin Software (v. 2.97) set at 4 cm−1 resolution. Electrochemical measurements were recorded in a glovebox under a dinitrogen atmosphere using a BAS CV 100W (Bioanalytical Systems Inc., West Lafayette, IN). A glassy carbon working electrode, a platinum wire auxiliary-electrode, and an Ag/AgNO3 non-aqueous reference electrode were used in the electrochemical studies. X-ray diffraction studies were carried out in the Beckman Institute Crystallographic Facility on a Bruker Smart 1000 CCD diffractometer. Crystals were mounted on a glass fiber with Paratone-N oil.
EPR Measurements
X-band EPR measurements were recorded using a Bruker EMX spectrometer (controlled by Bruker Win EPR software version 3.0) equipped with a rectangular cavity working in the TE102 mode. Low temperature measurements were conducted with an Oxford continuous-flow helium cryostat. Accurate frequency values were recorded from a frequency counter on the microwave bridge. Solution spectra were acquired in 1-methyltetrahydrofuran. Samples were prepared in a glovebox under dinitrogen in quartz EPR tubes equipped with ground glass joints.
Magnetic Measurements
Magnetic measurements were recorded using a Quantum Designs SQUID magnetometer running Magnetic Property Measurement System Rev. 2 software. Data were recorded at 5000 G. The sample was suspended in the magnetometer in a plastic straw sealed under nitrogen with Lilly No. 4 gel caps. Loaded samples were centered within the magnetometer using the DC centering scan at 35 K and 5000 G. Data were acquired at 4−30 K (one data point every 2 K) and 30−300 K (one data point every 5 K). The magnetic susceptibility was adjusted for diamagnetic contributions using the constitutive corrections of Pascal's constants. The molar magnetic susceptibility (χm) was calculated by converting the calculated magnetic susceptibility (χ) obtained from the magnetometer to a molar susceptibility (using the multiplication factor {(molecular weight)/[(sample weight)(field strength)]}). Effective magnetic moments were calculated using eqn (1).
Computational Methods
All calculations were performed using the Jaguar 5.0 program package (Jaguar 5.0, Schrodinger, LLC, Portland, OR, 2002). The calculation employed the hybrid DFT functional B3LYP and the basis set LACVP**. Input coordinates for the geometry optimizations were derived from the solid state structure of [PhBPiPr3]Fe≡NAd, where Fe was replaced by Mn.99 For the Mn imide, the adamantyl group was truncated to a tert-butyl group; and for the nitride, the adamantyl group was removed completely. Spin states and molecular charges were imposed as described in the text. The calculations were spin unrestricted, and no symmetry constraints were used. The default values for geometry and SCF iteraction cutoffs were used, and all structures converged under these criteria.
Synthesis of [PhBPiPr3]MnCl (1)
A THF solution (18 mL) of [PhBPiPr3]Tl (250.0 mg, 36.46 mmol) was added dropwise to a stirring suspension of MnCl2 (46.3 mg, 36.36 mmol). The reaction mixture was stirred for 24 h, and filtered through a Celite pad. The colorless solution was concentrated under reduced pressure to dryness to yield an off-white solid. After washing the solid liberally with petroleum ether, the product was extracted into benzene. Vapor diffusion of petroleum ether into the benzene solution resulted in pale pink blocks (80 mg, 33% yield). 1H NMR (C6D6, 300 MHz) δ ∼20 (v. br), 10.3 (br), 8.1 (br). Anal. Calcd for C27H53BClMnP3: C 56.71; H 9.34; N 0. Found: C 56.43; H 9.44; N 0.19.
[PhBPiPr3]MnI (2)
A THF solution (20 mL) of [PhBPiPr3]Tl (284.7 mg, 41.52 mmol) was added dropwise to a stirring suspension of MnI2 (129.5 mg, 41.52 mmol). The reaction mixture was stirred for 12 h, and filtered through a Celite pad. The colorless solution was concentrated under reduced pressure to dryness to yield an off-white solid. After washing the solid liberally with petroleum ether, the product was extracted into hot benzene. Vapor diffusion of petroleum ether into the benzene solution resulted in colorless crystals (172 mg, 62% yield). 1H-NMR (C6D6, 300 MHz) δ 21 (v. br), 8.8 (br), 6.6 (br). Anal. Calcd for C27H53BIMnP3: C 48.89; H 8.05; N 0. Found: C 49.12; H 7.54; N < 0.05.
[PhBPiPr3]Mn(N3) (3)
A THF solution (15 mL) of [PhBPiPr3]MnI (406.0 mg, 0.612 mmol) was added to a suspension of NaN3 (203.0 mg, 3.06 mmol). The reaction mixture was stirred for 12 h, and filtered through a Celite pad. The cream-colored filtrate was concentrated under reduced pressure to dryness and extracted with benzene (258 mg, 73 % yield). Single crystals suitable for X-ray diffraction were grown from vapor diffusion of petroleum ether into the benzene solution. 1H NMR (C6D6, 300 MHz) δ 11 (v. br), 8.3 (br). Anal. Calcd for C27H53BMnN3P3: C 56.07; H 9.24; N 7.26. Found: C 55.67; H 8.85; N 7.34.
[PhBPiPr3]Mn(CH2Ph) (4)
An Et2O solution of benzylmagnesium bromide (107 μL, 1.41 M) was added dropwise to a THF solution of [PhBPiPr3]MnI (100.0 mg, 0.151 mmol) chilled to −90 °C. The solution was allowed to warm slowly to rt. The yellow solution was filtered through a Celite pad, concentrated under reduced pressure to dryness, and extracted with petroleum ether (83.4 mg, 88 % yield). Yellow block crystals suitable for X-ray diffraction were grown from cyclopentane at −30 °C. 1H-NMR (C6D6, 300 MHz) δ 16 (v. br), 10.3 (br), 8.3 (br). Anal. Calcd for C34H60BMnP3: C 65.08; H 9.64; N 0. Found: C 64.98; H 9.32; N <0.05.
[PhBPiPr3]Mn(Me) (5)
An Et2O solution of methylmagnesium bromide (53.5 μL, 2.82 M) was added dropwise to a THF solution of [PhBPiPr3]MnI (100.0 mg, 0.151 mmol) chilled to −90 °C. The solution was allowed to warm slowly to rt. The pale yellow solution was filtered through a Celite pad, concentrated under reduced pressure to dryness, and extracted with petroleum ether (74.1 mg, 89 % crude yield). Crystals were grown from cyclopentane at −30 °C (42.3 mg, 51 % yield). 1H NMR (C6D6, 300 MHz) δ ∼ 17 (v. br), 10.1 (br), 8.3 (br). Anal. Calcd for C28H56BMnP3: C 60.99; H 10.24; N 0. Found: C 60.85; H 9.94; N < 0.05.
[PhBPiPr3]Mn(NH(2,6-iPr2-C6H3)) (6)
A THF solution (8 mL) of 2,6-diisopropylaniline (25.1 mg, 0.142 mmol) was added dropwise to a suspension of KH (5.7 mg, 0.142 mmol) chilled to −90 °C. The solution was allowed to warm slowly to rt. After stirring for 12 h, the solution was rechilled to −90 °C and a THF solution (8 mL) of [PhBPiPr3]MnI (93.9 mg, 0.142 mmol) was added dropwise. After slowly warming to rt, the bright yellow reaction was concentrated under reduced pressure to dryness. The product can be extracted with petroleum ether, filtered through a Celite pad to remove salts, and concentrated under reduced pressure to afford yellow solids (90.9 mg, 90 % yield). Single crystals suitable for X-ray diffraction were grown from TMS2O/petroleum ether at −30 °C. 1H NMR (C6D6, 300 MHz) δ 16.3 (v. br), 10.7 (br), 8.1 (br). Anal. Calcd for C39H71BMnNP3: C 65.73; H 10.04; N 1.97. Found: C 65.81; H 9.71; N 1.88.
[PhBPiPr3]Mn(dbabh) (7)
A THF solution (5 mL) of [PhBPiPr3]MnI (142.2 mg, 0.214 mmol) was added dropwise to a Et2O suspension (10 mL) of Li(dbabh) (42.7 mg, 0.214 mmol) chilled to −90 °C. The solution was allowed to warm slowly to rt. The yellow solution was filtered through a Celite pad, concentrated under reduced pressure to dryness, and extracted with hot Et2O. Cooling the ether solution to −30 °C afforded yellow blocks (49.7 mg, 32 % yield). 1H NMR (C6D6, 300 MHz) δ 21 (v. br), 10.3 (br), 8.1 (br). Anal. Calcd for C41H63BMnNP3: C 67.59; H 8.72; N 1.92. Found: C 66.28; H 8.40; N 1.77.
[PhBPiPr3]Mn(1-Ph(isoindolate)) (8)
A THF solution (10 mL) of benzyl potassium (40.0 mg, 0.307 mmol) was added to a solution of Hdbabh (59.4 mg, 0.307 mmol) chilled to −90 °C. The solution was allowed to warm slowly to rt. After stirring for 24 h, the solution was rechilled to −90 °C and a THF solution (10 mL) of [PhBPiPr3]MnI (203.9 mg, 0.307 mmol) was added dropwise. After slowly warming to rt, the orange solution was filtered through a Celite pad, concentrated under reduced pressure to dryness, and washed with petroleum ether, and extracted with hot Et2O. Cooling the ether solution to − 30 °C afforded red-orange crystals (71.8 mg, 35 % yield). 1H NMR (C6D6, 300 MHz) δ 11 (v. br), 8.2 (br). Anal. Calcd for C41H63BMnNP3: C 67.59; H 8.72; N 1.92. Found: C 66.34; H 9.83; N 1.82.
[PhBPiPr3]Tl-MnBr(CO)4 (9)
[PhBPiPr3]Tl (150.0 mg, 0.219 mmol) and MnBr(CO)5 (73.6 mg, 0.262 mmol) were mixed together in 10 mL of benzene. The reaction mixture was heated at 50 °C for 7 h. After cooling, the solution was concentrated under reduced pressure to dryness, extracted with benzene, filtered through a glass wool pipette, and concentrated to dryness. The yellow solids were washed liberally with petroleum ether and extracted with benzene. Vapor diffusion of petroleum ether into the solution afforded yellow-orange crystals (79.2 mg, 39 % yield). 1H NMR (C6D6, 300 MHz) δ 7.76 (d, J = 6.9 Hz, 2H, Ho of Ph), 7.53 (t, J = 6.9 Hz, 2H, Hm of Ph), 7.30 (t, J = 6.9 Hz, 1H, Hp of Ph), 2.00 (m, 6H, CH2P), 1.26 (m, 6H, CHMeMe′), 1.09 (d, J = 6.9 Hz, 18H, CHMeMe′), 1.01 (d, J = 6.9 Hz, 18H, CHMeMe′). 31P NMR (C6D6, 121 MHz) δ 45.4 (d, JTl-P = 8060 Hz). Anal. Calcd for C31H53BBrMnO4P3Tl: C 39.92; H 5.73; N 0. Found: C 40.35; H 5.83; N 0.06.
[PhBPiPr3]Mn(CNtBu)3 (10)
A THF solution (5 mL) of sodium naphthalenide (0.237 mmol, prepared by stirring excess Na and stoichiometric naphthalene in THF for 2 h, filter prior to use) was added to a stirring solution of [PhBPiPr3]MnI (150.0 mg, 0.226 mmol). tButylisocyanide (43.7 μL, 0.724 mmol) was then quickly added to the mixture. After 11 h, the reaction mixture was concentrated under reduced pressure to dryness, extracted with Et2O, and filtered through a glass wool pipette. The solution was again concentrated under reduced pressure to dryness, extracted with petroleum ether, and filtered through a glass wool pipette. Cooling of the solution to −30 °C afforded pale yellow crystals (39.4 mg, 22 % yield). 1H NMR (C6D6, 500 MHz) δ 8.15 (br, 2H), 7.63 (br, 2H), 7.34 (br, 1H), 2.19 (br, 6H), 1.54 (br, 18H), 1.39 (br, 18H), 1.27 (s, 27H), 0.94 (br, 6H). 31P NMR (C6D6, 121 MHz) δ 55.6 (br). Anal. Calcd for C42H80BMnN3P3: C 64.20; H 10.26; N 5.35. Found: C 63.84; H 10.10; N 5.22. IR (KBr/THF): ν(cm−1) 2048 (br).
Supplementary Material
Table 1.
X-ray Diffraction Experimental Details for compounds 1a, 1b, 2−4, and 6−10.
| 1a | 1b | 2 | 3 | 4 | |
|---|---|---|---|---|---|
| chemical formula | 0.98C27H53BP3Mn Cl·0.02 Tl C27H53BP3 | C54H106B2Cl2Mn2P6 | C27H53BP3MnI·C6H6 | 0.77C27H53BP3Mn N3·0.05 TlC27H53BP3·0.18 C27H53BP3MnCl | C34H60BMnP3 |
| formula weight | 573.66 | 1143.61 | 741.36 | 1165.21 | 627.48 |
| T (K) | 100 | 100 | 100 | 100 | 100 |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| a (Å) | 14.493(2) | 10.950(2) | 9.5424(8) | 11.946(2) | 9.4120(6) |
| b (Å) | 12.928(2) | 11.718(2) | 9.9006(8) | 34.586(7) | 11.8746(8) |
| c (Å) | 34.850(5) | 13.432(3) | 19.668(2) | 15.423(3) | 16.736(1) |
| α (°) | 90 | 68.000(3) | 89.780(1) | 90 | 90 |
| β (°) | 100.344(3) | 76.424(4) | 89.920(1) | 90 | 105.605(1) |
| γ (°) | 90 | 78.528(3) | 82.662(1) | 90 | 90 |
| V (Å3) | 6424(2) | 1541.5(5) | 1842.9(3) | 6372(2) | 1801.6(2) |
| Space group | C2/c (# 15) | P-1 (# 2) | P-1 (# 2) | Pnc2 (# 30) | P21 (# 4) |
| Z | 8 | 1 | 2 | 4 | 2 |
| Dcalcd (g/cm3) | 1.186 | 1.232 | 1.336 | 1.215 | 1.157 |
| μ (cm−1) | 7.3 | 6.9 | 13.5 | 8.3 | 5.2 |
| R1, wR2 (I>2σ(I))a | 0.0422, 0.0720 | 0.0356, 0.0674 | 0.0305, 0.0586 | 0.0938, 0.0783 | 0.0328, 0.0570 |
| 6 | 7 | 8 | 9 | 10 | |
|---|---|---|---|---|---|
| chemical formula | C39H71BMnNP3 | C41H63BMnNP3 | C41H63BMnNP3·0.50C4H10O | C31H53BBrMnO4P3Tl | C42H80BMnN3P3 |
| formula weight | 712.63 | 728.58 | 765.64 | 932.67 | 785.75 |
| T (K) | 100 | 100 | 98 | 100 | 100 |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| a (Å) | 10.690(1) | 10.4294(9) | 19.6218(9) | 10.2574(5) | 11.2996(7) |
| b (Å) | 11.249(1) | 16.812(1) | 77.737(4) | 18.6613(8) | 17.578(1) |
| c (Å) | 18.718(2) | 23.393(2) | 11.2629(6) | 19.9243(9) | 23.108(1) |
| α (°) | 98.393(2) | 90 | 90 | 90 | 90 |
| β (°) | 101.614(2) | 97.476(1) | 90 | 100.590(1) | 93.660(2) |
| γ (°) | 107.482(2) | 90 | 90 | 90 | 90 |
| V (Å3) | 2050.8(4) | 4066.7(6) | 17180(2) | 3748.9(3) | 4580.4(5) |
| Space group | P-1 (# 2) | P21/c (# 14) | Fdd2 (# 43) | P21/n (# 14) | P21/n (# 14) |
| Z | 2 | 4 | 16 | 4 | 4 |
| Dcalcd (g/cm3) | 1.154 | 1.190 | 1.184 | 1.652 | 1.139 |
| μ (cm−1) | 4.7 | 4.7 | 4.5 | 58.6 | 4.2 |
| R1, wR2 (I>2σ(I))a | 0.0419, 0.0694 | 0.0409, 0.0724 | 0.0514, 0.0731 | 0.0355, 0.0532 | 0.0437, 0.0707 |
R1 = Σ∥Fo| - |Fc∥ / Σ|Fo|, wR2 = {Σ[w(Fo2 — Fc2)2] / Σ[w(Fo2)2]}1/2
Acknowledgment
We gratefully acknowledge the NSF (CHE-0132216)) and the NIH (GM 070757) for financial support; the Beckman Institute (Caltech) for use of its crystallographic facility and SQUID magnetometer; the Materials and Process Simulation Center (Caltech, Goddard) for use of its computational facility. We thank Dr. Theodore A. Betley and Larry Henling for crystallographic assistance, and Dr. David M. Jenkins for assistance with SQUID and EPR measurements.
Footnotes
Supporting Information Available: Crystallographic data and CIFs for complexes 1a, 1b, 2-4, 6−10, and {(Hdbabh)MnI(μ-N-dbabh)}2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nugent WA, Mayer JM. Metal-Ligand Multiple Bonds. Wiley; New York: 1988. [Google Scholar]
- 2.Holm RH. Chem. Rev. 1987;87:1401. [Google Scholar]
- 3.Jorgensen KA. Chem. Rev. 1989;89:431. [Google Scholar]
- 4.Gao Y, Hanson RM, Klunder JM, Ko SY, Masamune H, Sharpless KB. J. Am. Chem. Soc. 1987;109:5765. [Google Scholar]
- 5.Collman JP, Zhang XM, Lee VJ, Uffelman ES, Brauman JI. Science. 1993;261:1404. doi: 10.1126/science.8367724. [DOI] [PubMed] [Google Scholar]
- 6.Collman JP, Brauman JI, Meunier B, Raybuck SA, Kodadek T. Proc. Natl. Acad. Sci. U. S. A. 1984;81:3245. doi: 10.1073/pnas.81.10.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Z, Quan RW, Jacobsen EN. J. Am. Chem. Soc. 1995;117:5889. [Google Scholar]
- 8.Evans DA, Woerpel KA, Hinman MM, Faul MM. J. Am. Chem. Soc. 1991;113:726. [Google Scholar]
- 9.Jacobsen EN. Comprehensive Asymmetric Catalysis. Springer; Hamburg: 1999. [Google Scholar]
- 10.Müller P, Fruit C. Chem. Rev. 2003;103:2905. doi: 10.1021/cr020043t. [DOI] [PubMed] [Google Scholar]
- 11.Brookhart M, Studabaker WB. Chem. Rev. 1987;87:411. [Google Scholar]
- 12.Doyle MP. Chem. Rev. 1986;86:919. [Google Scholar]
- 13.Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds. Wiley; New York: 1997. [Google Scholar]
- 14.Nguyen ST, Grubbs RH, Ziller JW. J. Am. Chem. Soc. 1993;115:9858. [Google Scholar]
- 15.Grubbs RH, Chang S. Tetrahedron. 1998;54:4413. [Google Scholar]
- 16.Hoveyda AH, Schrock RR. Chem. Eur. J. 2001;7:945. doi: 10.1002/1521-3765(20010302)7:5<945::aid-chem945>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 17.Feldman J, Schrock RR. Prog. Inorg. Chem. 1991;39:1. [Google Scholar]
- 18.Davies HML, Hansen T, Churchill MR. J. Am. Chem. Soc. 2000;122:3063. [Google Scholar]
- 19.Groves JT, Han Y-Z. Cytochrome P450: Structure, Mechanism, and Biochemistry. Plenum Press; New York: 1995. p. 3. [Google Scholar]
- 20.Abu-Omar MM, Loaiza A, Hontzeas N. Chem. Rev. 2005;105:2227. doi: 10.1021/cr040653o. [DOI] [PubMed] [Google Scholar]
- 21.Nakamoto K. Coord. Chem. Rev. 2002;226:153. [Google Scholar]
- 22.Costas M, Mehn MP, Jensen MP, Que L. Chem. Rev. 2004;104:939. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 23.Glueck DS, Wu JX, Hollander FJ, Bergman RG. J. Am. Chem. Soc. 1991;113:2041. [Google Scholar]
- 24.Glueck DS, Hollander FJ, Bergman RG. J. Am. Chem. Soc. 1989;111:2719. [Google Scholar]
- 25.Hay-Motherwell RS, Wilkinson G, Hussain-Bates B, Hursthouse MB. Polyhedron. 1993;12:2009. [Google Scholar]
- 26.Verma AK, Nazif TN, Achim C, Lee SC. J. Am. Chem. Soc. 2000;122:11013. [Google Scholar]
- 27.Brown SD, Peters JC. J. Am. Chem. Soc. 2005;127:1913. doi: 10.1021/ja0453073. [DOI] [PubMed] [Google Scholar]
- 28.Betley TA, Peters JC. J. Am. Chem. Soc. 2004;126:6252. doi: 10.1021/ja048713v. [DOI] [PubMed] [Google Scholar]
- 29.Brown SD, Betley TA, Peters JC. J. Am. Chem. Soc. 2003;125:322. doi: 10.1021/ja028448i. [DOI] [PubMed] [Google Scholar]
- 30.Klinker EJ, Kaizer J, Brennessel WW, Woodrum NL, Cramer CJ, Que L. Angew. Chem. Int. Ed. 2005;44:3690. doi: 10.1002/anie.200500485. [DOI] [PubMed] [Google Scholar]
- 31.Rohde JU, In JH, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Münck E, Nam W, Que L. Science. 2003;299:1037. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- 32.Kaizer J, Klinker EJ, Oh NY, Rohde JU, Song WJ, Stubna A, Kim J, Münck E, Nam W, Que L. J. Am. Chem. Soc. 2004;126:472. doi: 10.1021/ja037288n. [DOI] [PubMed] [Google Scholar]
- 33.Bart SC, Lobkovsky E, Bill E, Chirik PJ. J. Am. Chem. Soc. 2006;128:5302. doi: 10.1021/ja057165y. [DOI] [PubMed] [Google Scholar]
- 34.Shay DT, Yap GPA, Zakharov LN, Rheingold AL, Theopold KH. Angew. Chem. Int. Ed. 2005;44:1508. doi: 10.1002/anie.200462529. [DOI] [PubMed] [Google Scholar]
- 35.Dai XL, Kapoor P, Warren TH. J. Am. Chem. Soc. 2004;126:4798. doi: 10.1021/ja036308i. [DOI] [PubMed] [Google Scholar]
- 36.Jenkins DM, Betley TA, Peters JC. J. Am. Chem. Soc. 2002;124:11238. doi: 10.1021/ja026852b. [DOI] [PubMed] [Google Scholar]
- 37.Termaten AT, Aktas H, Schakel M, Ehlers AW, Lutz M, Spek AL, Lammertsma K. Organometallics. 2003;22:1827. [Google Scholar]
- 38.Sanchez-Nieves J, Sterenberg BT, Udachin KA, Carty AJ. J. Am. Chem. Soc. 2003;125:2404. doi: 10.1021/ja028303b. [DOI] [PubMed] [Google Scholar]
- 39.Mindiola DJ, Hillhouse GL. Chem. Commun. 2002:1840. doi: 10.1039/b204846a. [DOI] [PubMed] [Google Scholar]
- 40.Mindiola DJ, Hillhouse GL. J. Am. Chem. Soc. 2002;124:9976. doi: 10.1021/ja0269183. [DOI] [PubMed] [Google Scholar]
- 41.Melenkivitz R, Mindiola DJ, Hillhouse GL. J. Am. Chem. Soc. 2002;124:3846. doi: 10.1021/ja017787t. [DOI] [PubMed] [Google Scholar]
- 42.Mindiola DJ, Hillhouse GL. J. Am. Chem. Soc. 2001;123:4623. doi: 10.1021/ja010358a. [DOI] [PubMed] [Google Scholar]
- 43.Kogut E, Wiencko HL, Zhang LB, Cordeau DE, Warren TH. J. Am. Chem. Soc. 2005;127:11248. doi: 10.1021/ja0533186. [DOI] [PubMed] [Google Scholar]
- 44.Dai XL, Warren TH. J. Am. Chem. Soc. 2004;126:10085. doi: 10.1021/ja047935q. [DOI] [PubMed] [Google Scholar]
- 45.Straub BF, Hofmann P. Angew. Chem. Int. Ed. 2001;40:1288. doi: 10.1002/1521-3773(20010401)40:7<1288::aid-anie1288>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 46.Eikey RA, Abu-Omar MM. Coord. Chem. Rev. 2003;243:83. [Google Scholar]
- 47.Groves JT, Takahashi T. J. Am. Chem. Soc. 1983;105:2073. [Google Scholar]
- 48.Buchler JW, Dreher C, Lay KL, Lee YJA, Scheidt WR. Inorg. Chem. 1983;22:888. [Google Scholar]
- 49.Hill CL, Hollander FJ. J. Am. Chem. Soc. 1982;104:7318. [Google Scholar]
- 50.Buchler JW, Dreher C, Lay KL. Z. Naturforsch., B: Chem. Sci. 1982;37:1155. [Google Scholar]
- 51.Dehnicke K, Strahle J. Angew. Chem., Int. Ed. Eng. 1981;20:413. [Google Scholar]
- 52.Meyer K, Bendix J, Metzler-Nolte N, Weyhermüller T, Wieghardt K. J. Am. Chem. Soc. 1998;120:7260. [Google Scholar]
- 53.Niemann A, Bossek U, Haselhorst G, Wieghardt K, Nuber B. Inorg. Chem. 1996;35:906. doi: 10.1021/ic9508031. [DOI] [PubMed] [Google Scholar]
- 54.Bendix J, Meyer K, Weyhermüller T, Bill E, Metzler-Nolte N, Wieghardt K. Inorg. Chem. 1998;37:1767. doi: 10.1021/ic970850o. [DOI] [PubMed] [Google Scholar]
- 55.Du Bois J, Tomooka CS, Hong J, Carreira EM. Acc. Chem. Res. 1997;30:364. [Google Scholar]
- 56.Du Bois J, Tomooka CS, Hong J, Carreira EM, Day MW. Angew. Chem., Int. Ed. Eng. 1997;36:1645. [Google Scholar]
- 57.Chang CJ, Connick WB, Low DW, Day MW, Gray HB. Inorg. Chem. 1998;37:3107. [Google Scholar]
- 58.Chang CJ, Low DW, Gray HB. Inorg. Chem. 1997;36:270. doi: 10.1021/ic970824q. [DOI] [PubMed] [Google Scholar]
- 59.Danopoulos AA, Wilkinson G, Sweet TKN, Hursthouse MB. J. Chem. Soc., Dalton Trans. 1995:205. [Google Scholar]
- 60.Danopoulos AA, Green JC, Hursthouse MB. J. Organomet. Chem. 1999;591:36. [Google Scholar]
- 61.Danopoulos AA, Wilkinson G, Sweet TKN, Hursthouse MB. J. Chem. Soc., Dalton Trans. 1995:937. [Google Scholar]
- 62.Danopoulos AA, Wilkinson G, Sweet TKN, Hursthouse MB. J. Chem. Soc., Dalton Trans. 1994:1037. [Google Scholar]
- 63.Abu-Omar MM, Shields CE, Edwards NY, Eikey RA. Angew. Chem. Int. Ed. 2005;44:6203. doi: 10.1002/anie.200462647. [DOI] [PubMed] [Google Scholar]
- 64.Edwards NY, Eikey RA, Loring MI, Abu-Omar MM. Inorg. Chem. 2005;44:3700. doi: 10.1021/ic0484506. [DOI] [PubMed] [Google Scholar]
- 65.Eikey RA, Khan SI, Abu-Omar MM. Angew. Chem. Int. Ed. 2002;41:3592. doi: 10.1002/1521-3773(20021004)41:19<3591::AID-ANIE3591>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 66.Du Bois J, Hong J, Carreira EM, Day MW. J. Am. Chem. Soc. 1996;118:915. [Google Scholar]
- 67.Thomas CM, Mankad NP, Peters JC. J. Am. Chem. Soc. 2006;128:4956. doi: 10.1021/ja0604358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.MacBeth CE, Thomas JC, Betley TA, Peters JC. Inorg. Chem. 2004;43:4645. doi: 10.1021/ic049936p. [DOI] [PubMed] [Google Scholar]
- 69.Shapiro IR, Jenkins DM, Thomas JC, Day MW, Peters JC. Chem. Commun. 2001:2152. doi: 10.1039/b104447h. [DOI] [PubMed] [Google Scholar]
- 70.Betley TA, Peters JC. Inorg. Chem. 2003;42:5074. doi: 10.1021/ic0343096. [DOI] [PubMed] [Google Scholar]
- 71.Jenkins DM, Di Bilio AJ, Allen MJ, Betley TA, Peters JC. J. Am. Chem. Soc. 2002;124:15336. doi: 10.1021/ja026433e. [DOI] [PubMed] [Google Scholar]
- 72.Hebendanz N, Köhler FH, Müller G. Inorg. Chem. 1984;23:3043. [Google Scholar]
- 73.Sur SK. J. Magn. Reson. 1989;82:169. [Google Scholar]
- 74.Evans DF. J. Chem. Soc. 1959:2003. [Google Scholar]
- 75.Kahn O. Molecular Magnetism. VCH; New York: 1993. [Google Scholar]
- 76.Armentano D, de Munno G, Guerra F, Faus J, Lloret F, Julve M. Dalton Trans. 2003:4626. [Google Scholar]
- 77.Girolami GS, Wilkinson G, Galas AMR, Thornton-Pett M, Hursthouse MB. J. Chem. Soc., Dalton Trans. 1985:1339. [Google Scholar]
- 78.Brown SD, Mehn MP, Peters JC. J. Am. Chem. Soc. 2005;127:13146. doi: 10.1021/ja0544509. [DOI] [PubMed] [Google Scholar]
- 79.Godfrey SM, McAuliffe CA, Ndifon PT, Pritchard RG. J. Chem. Soc., Dalton Trans. 1993:3373. [Google Scholar]
- 80.Godfrey SM, McAuliffe CA, Pritchard RG. J. Chem. Soc., Dalton Trans. 1993:371. [Google Scholar]
- 81.Cecconi F, Ghilardi CA, Midollini S, Orlandini A. J. Chem. Soc., Dalton Trans. 1992:33. [Google Scholar]
- 82.Beagley B, McAuliffe CA, Minten K, Pritchard RG. J. Chem. Soc., Dalton Trans. 1987:1999. [Google Scholar]
- 83.Beagley B, McAuliffe CA, Minten K, Pritchard RG. J. Chem. Soc., Chem. Commun. 1984:658. [Google Scholar]
- 84.Komatsuzaki H, Sakamoto N, Satoh M, Hikichi S, Akita M, Moro-oka Y. J. Am. Chem. Soc. 1998;37:6554. doi: 10.1021/ic980509j. [DOI] [PubMed] [Google Scholar]
- 85.Matsunaga Y, Fujisawa K, Ibi N, Miyashita Y, Okamoto K. Inorg. Chem. 2005;44:325. doi: 10.1021/ic049814x. [DOI] [PubMed] [Google Scholar]
- 86.Nabika M, Seki Y, Miyatake T, Ishikawa Y, Okamoto K, Fujisawa K. Organometallics. 2004;23:4335. [Google Scholar]
- 87.Brunker TJ, Hascall T, Cowley AR, Rees LH, O'Hare D. Inorg. Chem. 2001;40:3170. doi: 10.1021/ic010125k. [DOI] [PubMed] [Google Scholar]
- 88.Komatsuzaki H, Nagasu Y, Suzuki K, Shibasaki T, Satoh M, Ebina F, Hikichi S, Akita M, Moro-oka Y. J. Chem. Soc., Dalton Trans. 1998:511. [Google Scholar]
- 89.Wieghardt K, Bossek U, Nuber B, Weiss J. Inorg. Chim. Acta. 1987;126:39. [Google Scholar]
- 90.Hubin TJ, McCormick JM, Alcock NW, Busch DH. Inorg. Chem. 2001;40:435. doi: 10.1021/ic9912225. [DOI] [PubMed] [Google Scholar]
- 91.Mantel C, Hassan AK, Pecaut J, Deronzier A, Collomb MN, Duboc-Toia C. J. Am. Chem. Soc. 2003;125:12337. doi: 10.1021/ja034652+. [DOI] [PubMed] [Google Scholar]
- 92.Saha S, Mal D, Koner S, Bhattacherjee A, Gutlich P, Mondal S, Mukherjee M, Okamoto KI. Polyhedron. 2004;23:1811. [Google Scholar]
- 93.Chai JF, Zhu HP, Fan HJ, Roesky HW, Magull J. Organometallics. 2004;23:1177. [Google Scholar]
- 94.Howard CG, Girolami GS, Wilkinson G, Thornton-Pett M, Hursthouse MB. J. Chem. Soc., Dalton Trans. 1983:2631. [Google Scholar]
- 95.Chai JF, Zhu HP, Roesky HW, He C, Schmidt HG, Noltemeyer M. Organometallics. 2004;23:3284. [Google Scholar]
- 96.Riollet V, Copéret C, Basset JM, Rousset L, Bouchu D, Grosvalet L, Perrin M. Angew. Chem. Int. Ed. 2002;41:3025. doi: 10.1002/1521-3773(20020816)41:16<3025::AID-ANIE3025>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 97.Daida EJ, Peters JC. Inorg. Chem. 2004;43:7474. doi: 10.1021/ic0488583. [DOI] [PubMed] [Google Scholar]
- 98.Mindiola DJ, Cummins CC. Angew. Chem. Int. Ed. 1998;37:945. doi: 10.1002/(SICI)1521-3773(19980420)37:7<945::AID-ANIE945>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 99.Betley TA, Peters JC. J. Am. Chem. Soc. 2003;125:10782. doi: 10.1021/ja036687f. [DOI] [PubMed] [Google Scholar]
- 100.Hu X, Meyer K. J. Am. Chem. Soc. 2004;126:16322. doi: 10.1021/ja044271b. [DOI] [PubMed] [Google Scholar]
- 101.Baker RJ, Edwards PG, Gracia-Mora J, Ingold F, Malik KMA. J. Chem. Soc., Dalton Trans. 2002:3985. [Google Scholar]
- 102.Bond AM, Colton R, van den Bergen A, Walter JN. Inorg. Chem. 2000;39:4696. doi: 10.1021/ic0000294. [DOI] [PubMed] [Google Scholar]
- 103.Connolly J, Genge ARJ, Levason W, Orchard SD, Pope SJA, Reid G. J. Chem. Soc., Dalton Trans. 1999:2343. [Google Scholar]
- 104.Connor JA, Hudson GA. J. Organomet. Chem. 1974;73:351. [Google Scholar]
- 105.Reimann RH, Singleton E. J. Chem. Soc., Dalton Trans. 1973:841. [Google Scholar]
- 106.Sorace L, Golze C, Gatteschi D, Bencini A, Roesky HW, Chai J, Stückl AC. Inorg. Chem. 2006;45:395. doi: 10.1021/ic051496u. [DOI] [PubMed] [Google Scholar]
- 107.Chai JF, Zhu HP, Stückl AC, Roesky HW, Magull J, Bencini A, Caneschi A, Gatteschi D. J. Am. Chem. Soc. 2005;127:9201. doi: 10.1021/ja042269e. [DOI] [PubMed] [Google Scholar]
- 108.King WA, Scott BL, Eckert J, Kubas GJ. Inorg. Chem. 1999;38:1069. doi: 10.1021/ic981263l. [DOI] [PubMed] [Google Scholar]
- 109.King WA, Luo XL, Scott BL, Kubas GJ, Zilm KW. J. Am. Chem. Soc. 1996;118:6782. [Google Scholar]
- 110.Schollenberger M, Nuber B, Ziegler ML, Hey-Hawkins E. J. Organomet. Chem. 1993;460:55. [Google Scholar]
- 111.Guilard R, Zrineh A, Ferhat M, Tabard A, Mitaine P, Swistak C, Richard P, Lecomte C, Kadish KM. Inorg. Chem. 1988;27:697. [Google Scholar]
- 112.Attempts to minimize formation of [PhBPiPr3]Na by adding CNtBu to 2 prior to the addition of sodium napthalenide or by adding sodium napthalenide to 2 at low temperatures gave rise to more complex reaction mixtures.
- 113.Bailey PL, Coxall RA, Dick CM, Fabre S, Henderson LC, Herber C, Liddle ST, Lorono-Gonzalez D, Parkin A, Parsons S. Chem. Eur. J. 2003;9:4820. doi: 10.1002/chem.200305053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.