

Summary
Developmental abnormalities, cancer and premature aging each have been linked to defects in the DNA damage response (DDR). Mutations in the ATR checkpoint regulator cause developmental defects in mice (pre-gastrulation lethality) and humans (Seckel syndrome). Herein we show that eliminating ATR in adult mice leads to defects in tissue homeostasis and the rapid appearance of age-related phenotypes, such as hair graying, alopecia, kyphosis, osteoporosis, thymic involution, fibrosis and other abnormalities. Histological and genetic analyses indicate that ATR deletion causes acute cellular loss in tissues where continuous cell proliferation is required for maintenance. Importantly, thymic involution and alopecia and hair graying in ATR knockout mice were associated with dramatic reductions in tissue-specific stem and progenitor cells and exhaustion of tissue renewal and homeostatic capacity. In aggregate, these studies suggest that reduced regenerative capacity in adults via deletion of a developmentally essential DDR gene is sufficient to cause characteristics of premature aging.
Keywords: ATR, aging, stem cells, genome integrity, checkpoints, Seckel syndrome
Introduction
DNA damage response (DDR) genes are essential for maintenance of genomic integrity. Mutations in many DDR genes have been linked to disorders that are predominantly associated with failures in development. These syndromes are typically characterized by microcephaly, mental and growth retardation, skeletal abnormalities and dysmorphic facial features. The DDR genes linked to these diseases possess a variety of functions in DNA repair and checkpoint signaling (O'Driscoll and Jeggo, 2006) and include BLM (Bloom's syndrome), FANC genes (Fanconi anaemia), NBS (Nijmegen breakage syndrome), LIG4 (LIG4 syndrome), and ATR (Seckel syndrome). However, mutations in DDR genes are also associated with progeroid syndromes that phenocopy aspects of normal aging, e.g. Werner syndrome, Cockayne syndrome, trichothyodystrophy, dyskeratosis congenita and ataxia-telangiectasia (Kipling et al., 2004). Progeroid syndromes such as these suggest that, in addition to appropriate development, DDR genes are important for preventing age-related diseases.
The function of DDR genes in assuring normal development and preventing age-related diseases has been further elucidated by gene-targeting studies in mice (Hasty et al., 2003; Lombard et al., 2005). Again, these studies have shown that DDR mutations lead to a variety of disorders, including developmental failures, increased cancer incidence and accelerated aging (Lombard et al., 2005; O'Driscoll and Jeggo, 2006). For example, while deletion of BRCA1, XPD or XRCC4 causes embryonic lethality, hypomorphic mutations in these same genes in combination with genetic backgrounds that permit the completion of embryonic development lead to a broad range of age-related phenotypes (Cao et al., 2003; Chao et al., 2006; de Boer et al., 2002). Thus, the presentation of age-related phenotypes and developmental abnormalities appear to vary in a manner that depends on the function of the gene (specific type of DNA repair, checkpoint role, etc.), the cell type in which this function is particularly required and whether the mutation is null or hypomorphic.
In humans, hypomorphic mutations in the ATR (ATM- and Rad3- related) protein kinase have been linked to Seckel syndrome, a disease characterized by severe growth retardation, microcephaly, facial and osteoskeletal abnormalities (O'Driscoll and Jeggo, 2006; O'Driscoll et al., 2003). As a central upstream regulator of cellular responses to replication stress and DNA damage, ATR phosphorylates BRCA1, Chk1, BLM, p53 and other DDR factors to inhibit cell cycle progression and assure genome maintenance (Osborn et al., 2002). While ATR heterozygous mice develop normally and demonstrate a significantly increased tumor risk by 18 months of age (Brown and Baltimore, 2000), homozygous elimination of ATR leads to chromosome breaks, proliferative failure in culture and early embryonic lethality (Brown and Baltimore, 2000; Cortez et al., 2001; de Klein et al., 2000). The effect of ATR loss on cell proliferation is consistent with findings indicating that ATR and ATR orthologs in other organisms play an especially important role in maintaining genome integrity during DNA synthesis (Brown and Baltimore, 2003; Casper et al., 2002; Lopes et al., 2001), putatively by stabilizing stalled DNA replication forks and preventing their collapse into double strand breaks (Osborn et al., 2002). Because chromosome breaks are observed in cultured ATR knockout cells even in the absence of exogenous replication stress (Brown and Baltimore, 2000; Brown and Baltimore, 2003; Casper et al., 2002), stalled DNA replication may be a common event during normal cellular proliferation.
Because ATR is essential for embryonic development, the effect of ATR deletion in adult mice has not been tested previously. Herein, we demonstrate that ATR deletion in adult animals leads to the rapid onset of a broad range of age-related phenotypes that coincides stem and progenitor cell depletion and exhaustion of tissue renewal and homeostatic capacity. Interestingly, reports of skin degeneration, alopecia, premature hair graying and hematopoietic defects have been previously noted in some Seckel patients (Arnold et al., 1999; Boscherini et al., 1981; Butler et al., 1987; Fathizadeh et al., 1979). Our studies suggest that maintenance of tissue homeostasis and regeneration remains important for preventing aging throughout adulthood and may be a common means by which ATR and other developmentally essential DDR genes prevent age-related phenotypes.
Results
Generation of Cre-ERT2 Transgenic Mice to Conditionally Delete ATR
To circumvent embryonic lethality and determine the effect of ATR loss on adult mice, a Cre-ERT2 transgenic mouse line was generated through lentitransgenesis (Lois et al., 2002) using a lentivirus that expresses the Cre-ERT2 from the human ubiquitin C promoter (Figure 1A). Cre-ERT2 is a fusion protein composed of Cre recombinase and a mutant form of the estrogen receptor that is selectively activated only in the presence of tamoxifen, but not estrogen (Feil et al., 1997). In combination with a flox-conditional allele of ATR (Brown and Baltimore, 2003), the Cre-ERT2 line provides a system to efficiently delete ATR both spatially and temporally in the mouse.
Figure 1.
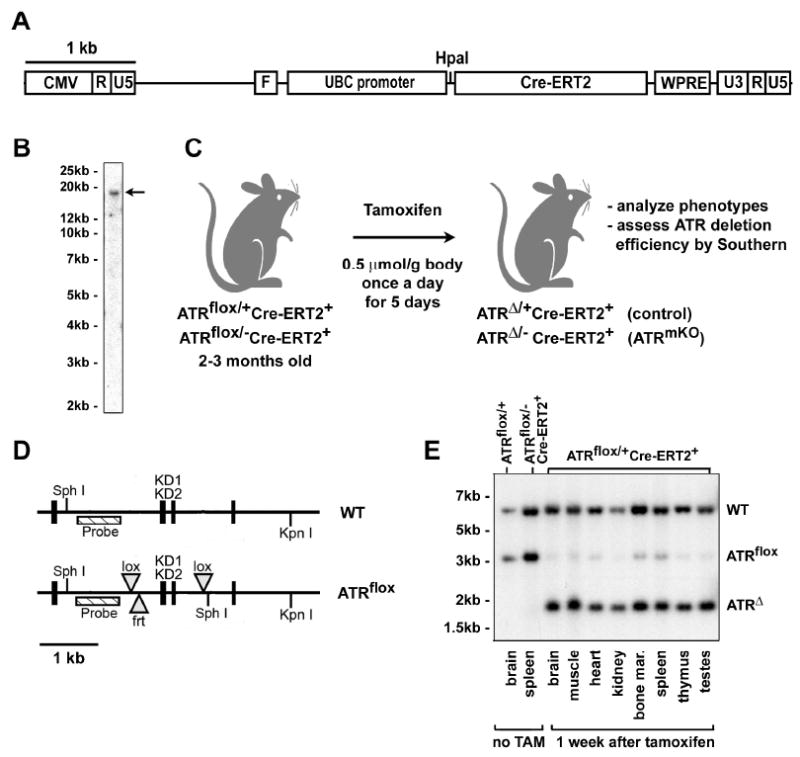
A drug-inducible system to delete ATR in adult mice. (A) Lentiviral construct used to generate Cre-ERT2 fusion protein-expressing lentivirus. (B) A founder with a single copy integrant that expressed high levels of Cre-ERT2 was chosen to establish Cre-ERT2 lentitransgenic mouse line (arrow). (C) TAM treatment regimen used to stimulate recombination of the ATRflox allele. Mice were treated TAM at 2-3 months of age by oral gavage or intraperitoneal injection and analyzed subsequently at various time points. (D) Schematic of the ATRflox region (Brown and Baltimore, 2003). The kinase domain-encoding exons (KD1 & KD2) and probed region are shown. The null allele of ATR (ATR-) is wild-type in this probed region. (E) Southern blot of genomic DNA isolated from TAM-treated ATRflox/+Cre-ERT2+ mice. DNA samples from various tissues were digested with Sph I and Kpn I, Southern blotted and detected for the ATRflox region using the probe indicated in (D). Sph I ATRflox allele fragment = 3.1 kb, Sph I ATRΔ allele fragment = 1.8 kb.
A Cre-ERT2 mouse line with a single integrated copy of the virus was chosen for further analysis (Figure 1B). To test the efficiency of Cre recombinase activation, adult ATRflox/+Cre-ERT2+ mice (8-12 weeks of age) were treated once per day for 5 days with tamoxifen (TAM) by oral gavage (Figure 1C). Three days after the final treatment, mice were sacrificed and DNA from a variety of tissues was analyzed by Southern hybridization to quantify the ATR+, ATRflox and ATRΔ alleles (Figure 1D,E). Efficient recombination (>70%) was observed in all tissues examined (Figure 1E) including the intestine, liver, pancreas and lung (Figure 4C and data not shown). Thus, the Cre-ERT2 mouse line provides a system to conditionally activate Cre recombinase in a wide range of tissues upon TAM treatment. In addition, because the flox allele remained unrecombined in a small percentage of cells in all tissues examined (Figures 1E and 4C), this system allowed production of mice that are mosaic for ATRflox recombination.
Figure 4.
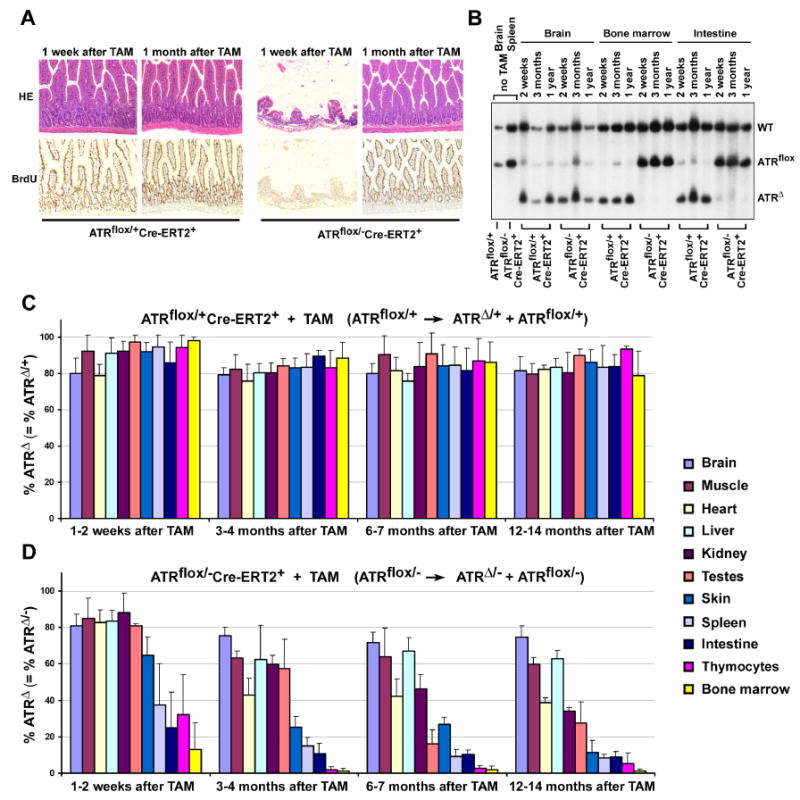
ATR deletion leads to loss of proliferating cells in mice. (A) Rapid loss and reconstitution of proliferating intestinal epithelial cells after ATR deletion. Mice were treated with BrdU in drinking water for up to 1 month following TAM treatment. Loss of proliferating intestinal epithelial cells was observed 1 week after ATR deletion in ATRmKO mice (n = 4), however, a full recovery was observed 3 weeks later (n = 5). Pictures were taken at 100× magnification. (B) ATRΔ/- cells are rapidly lost in proliferating tissues (bone marrow, intestine), but not in the brain of ATR knockout mice. Southern blot of genomic DNA isolated from ATRflox/+Cre-ERT2+ (control) and ATRflox/-Cre-ERT2+ (ATRmKO) mice treated or left untreated with TAM. Appearance of the ATRΔ allele represents ATRΔ/+ cells in control mice and ATRΔ/- cells in test mice. (C, D) Percentage of ATRΔ/+ cells remains constant (C), while ATRΔ/- cells are lost (D) in various tissues following lox recombination. Southern blot band intensities of lox recombined (ATRΔ) and unrecombined (ATRflox) were used to quantify the total percentage of cells that maintained a recombined copy of ATR (ATRΔ). Band intensities were quantified by phosphoimager, and mean percentages were calculated from the ratio ATRΔ over ATRΔ + ATRflox. For each tissue and time point, 2-8 mice were evaluated; each error bar indicates one standard deviation.
Age-related Phenotypes in ATRmKO Mice: Hair Loss and Graying, Kyphosis, Osteoporosis, Fibrosis of the Heart and Kidney, Reduced Thymopoiesis and Spermatogensis
ATR mosaic knockout (ATRmKO) mice were generated by treatment of ATRflox/-Cre-ERT2+ at 8-12 weeks of age (Fig. 1C) and were compared to TAM-treated ATRflox/+Cre-ERT2+ (control) mice. Within 3-4 months after TAM treatment, several age-related phenotypes were observed in ATRmKO mice including pervasive hair graying and alopecia (n = 43), which continued to increase in expressivity for more than one year after treatment (Figure 2A). The extent of hair graying far exceeded that observed in normally aged mice. In contrast, no obvious phenotypes were observed in control mice (n = 50), which remained similar in appearance to untreated age-matched littermates. In comparison to control skin, thinning of the subcutaneous adipose layer (up to 70% reduced), thickening of the epidermis and loss of hair follicles were observable within 3-6 months of TAM treatment in ATRmKO mice; these abnormalities consistently progressed to greater severity subsequently (Figure 2B). Similar phenotypes have been documented in aging humans and in previously described mouse models of aging (Hasty et al., 2003; Lombard et al., 2005). The follicles remaining in ATRmKO mice often yielded gray hair shafts and displayed abnormal architecture, such as sebaceous gland cell hypertrophy and degeneration of dermal and epidermal structures (Figure 2B and data not shown). Single topical application of 4-hydroxytamoxifen (4-OHT) to a 1-2 cm2 patch of skin caused localized alopecia and gray hair re-growth in ATRflox/-Cre-ERT2+ mice, but not in ATRflox/+Cre-ERT2+ control mice (Supplemental Figure S1). Together, these results indicate that ATR is required to prevent age-related phenotypes in the hair and skin.
Figure 2.
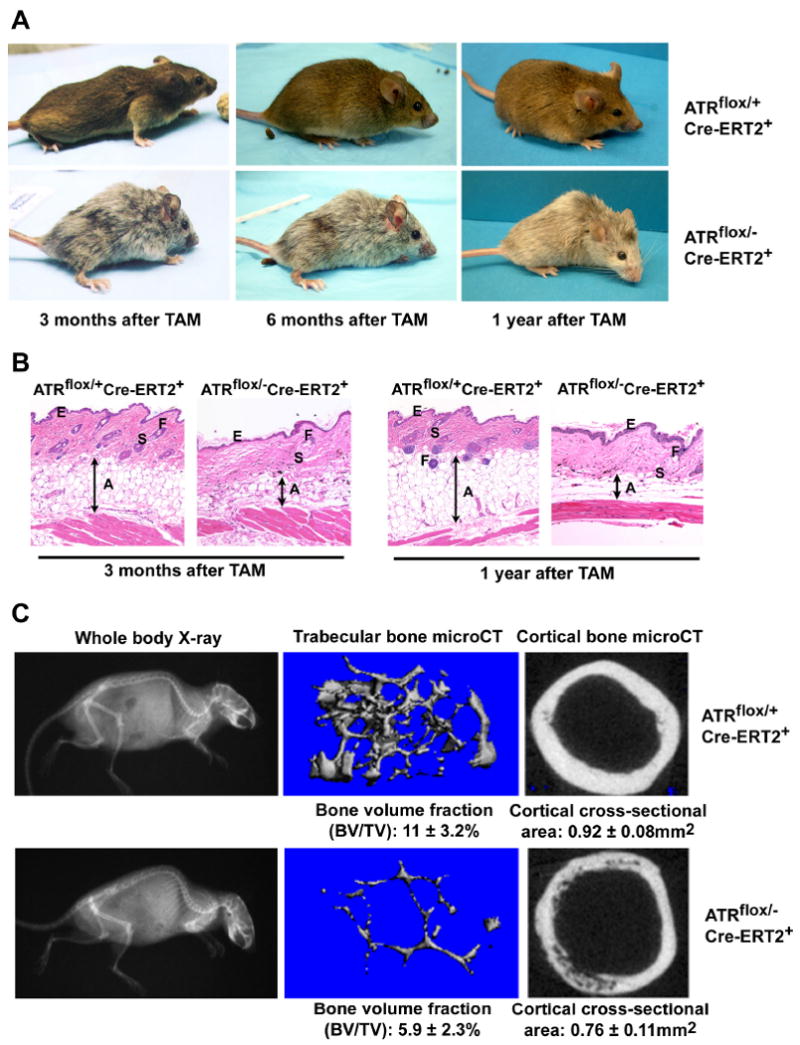
ATR deletion leads to hair graying, alopecia, kyphosis and osteoporosis. (A) ATR deletion following TAM treatment leads to pervasive hair graying and patchy hair loss and kyphosis in ATRmKO mice. (B) Age-related abnormalities in the skin of ATRmKO mice. Thinning of the subcutaneous adipose layer (A), thickening of the epidermis (E), loss of hair follicles (F) and sebaceous gland cell hypertrophy (S) were observed in ATRmKO mice (n = 19) but not in control mice (n = 22). Sections from sex-matched mice are shown. (C) Increased kyphosis and osteoporosis in ATRmKO mice. Control and ATRmKO mice were X-rayed 1 year after TAM treatment (left panel). Increased kyphosis over controls was observed in all ATRmKO mice analyzed (n = 6). To analyze bone volume and cross-sectional area, femurs from ATRmKO and control mice were subjected to microCT analysis (4-7 mice analyzed/group). Trabecular bone in the distal metaphysic (middle panel) and cortical bone cross-sectional area (right panel) was imaged and analyzed. Bone volume/total volume (BV/TV) and cortical area are shown as mean ± SD. P ≤ 0.04 as calculated by Student's T-test.
Other common characteristics of aging in humans and mice are increased kyphosis (an abnormal rearward curvature of the spine), weight loss and osteoporosis (Hasty et al., 2003; Lombard et al., 2005). By simple visual inspection, ATRmKO mice 3 months to 1 year following TAM treatment displayed significantly increased kyphosis in comparison to control mice (Figure 2A). This phenotype was accompanied by a decrease in the total body weight. Body weight averages for ATRmKO were 20% lower than control mice by 1 year after treatment (21.3 ± 3.2 g for ATRmKO mice versus 26.6 ± 3.1 g for control mice, p = 0.0006). To confirm that the apparent kyphosis in ATRmKO mice was not a visual artifact produced by reduced body fat, ATRmKO and control mice were analyzed by X-ray 1 year after TAM treatment. In all mice analyzed, ATRmKO mice displayed a higher degree of kyphosis compared to that observed in control mice (Fig. 2C). These data indicate that ATR loss leads to kyphosis in adult mice.
In humans, kyphosis is associated with bone degeneration. To test if ATR deletion leads to osteoporosis, femurs from ATRmKO mice and control mice one year after TAM treatment were analyzed for cortical and trabecular bone volume and architecture. In comparison to control mice, femurs of ATRmKO mice displayed an 18% decrease in cortical bone cross-sectional area (p = 0.0001) and 46%-76% reduction in trabecular bone volume (p = 0.04) as determined by micro-CT (Figure 2C) and histomorphometric analyses (data not shown). In aggregate, these data indicate that ATR deletion leads to kyphosis and osteoporosis in adult mice.
Thymic involution is a hallmark of hematopoietic aging and is associated with decreased thymocyte abundance and deterioration of the thymic stroma (Zediak and Bhandoola, 2005). Consistent with the appearance of other age-related phenotypes (above), premature suppression of thymocyte abundance was observed in ATRmKO mice in comparison to control mice (Figure 3A). Thymocyte numbers in ATRmKO mice declined precipitously 1-2 weeks after TAM treatment. Although thymocyte numbers largely recovered within 3 months, they were again observed to be significantly suppressed 6 months to 1 year after TAM treatment (Figure 3A). Of note, thymocyte abundance in ATRmKO mice 6 months after ATR deletion was comparable to that observed in control mice 12-14 months after treatment. Thus, the naturally occurring age-associated decrease in thymocyte numbers occurred significantly earlier in ATRmKO mice.
Figure 3.

Reduced thymopoiesis, accelerated thymic involution and failed spermatogenesis in ATRmKO mice. (A) Quantification of thymocytes at differing time points after ATR deletion. Thymi from ATRmKO and control animals (6-12 mice per group) were processed through mesh and thymocytes were counted by hemacytometer. Standard error bars are shown; P values were calculated by Student's T test. NS – not significant. (B) Premature thymic involution in ATRmKO mice. A significant reduction in the volume of stromal cortical layer, where early T cell precursors reside, was observed in all ATRmKO mice analyzed 1 year after ATR deletion (n = 3, double arrow). K5 – medullary epithelium, K8 – cortical epithelium. (C) Testicular degeneration in ATRmKO mice. Dramatic testicular atrophy was observed in all ATRmKO mice examined (n = 7), but not in control mice (n = 5). Pictures in (B) and (C) were taken at 100× magnification.
Age-associated architectural deterioration of the thymic stroma was also observed prematurely in ATRmKO mice. The thymus is comprised of the cortex, in which early T cell precursors undergo differentiation, and the medulla, which harbors more mature T cell progenitors. Aged thymi exhibit cellular damage and architectural disruption predominantly in the cortical layer (Nabarra and Andrianarison, 1996). A significant reduction in the total thymus volume, particularly in the thickness of the cortical layer (1.5 – 2 fold), was observed in the thymi of ATRmKO mice in comparison to control mice (Figure 3B). Together, these data indicate that ATR loss affects cellularity and architecture of the thymus in manners that emulate normal aging.
Several other age-associated disorders were observed in ATRmKO mice. These disorders included architectural deterioration and fibrosis in the kidney and heart (Supplemental Figure S2) and an early and permanent loss of spermatogenesis (Figure 3C). These phenotypes were not observed in control mice (Supplemental Figure S2 and Figure 3C). In addition, as previously reported in other mouse models of aging (Hasty et al., 2003; Lombard et al., 2005), we observed no significant increase in tumor risk in ATRmKO mice compared to ATR heterozygous control mice. No tumors were observed in any ATRmKO mice in the course of these studies, even in older cohorts between the ages of 12 to 18 months (n = 17). In contrast, tumors were observed in 3 out of 28 control mice (Supplemental Table 1), which approximately represents the frequency expected in ATR heterozygotes in this age group (Brown and Baltimore, 2000). These data indicate that complete loss of ATR does not promote tumorigenesis in vivo.
In summary, deletion of ATR in adult mice leads to a number of disorders including hair loss and graying, kyphosis, osteoporosis, premature involution of the thymus, fibrosis of the heart and kidney and decreased spermatogenesis. Although it is currently not clear whether these age-related phenotypes ultimately affect longevity, these disorders present in a manner that is reminiscent of aging in humans and mice. Taken together, these data indicate that ATR loss in adult mice causes the premature appearance of age-related phenotypes.
ATR Deletion Leads to Loss of Proliferating Cells in vivo
Besides the age-associated phenotypes described above, we observed a dramatic acute effect of ATR deletion on the intestines of ATRmKO mice. Approximately 80% of the villus epithelium was lost one week after ATR deletion; however, full recovery was observed 1 month later (Figure 4A). This effect was not observed in control mice (Figure 4A). Initial acute loss of intestinal structures was strongly associated with loss of proliferating cells (BrdU, Figure 4A) and a similar loss of proliferating cells after ATR deletion was observed in other organs, such as skin, kidney, and liver (data not shown). Increased abundance of apoptotic cells was not detected following ATR deletion (data not shown), possibly due to rapid clearance mechanisms. Our findings suggest that, while the abundance of proliferating cells is compromised in most tissues following ATR loss, tissue homeostasis affords some degree of recovery over time.
We hypothesized that, while ATR knockout cells appear to be lost in the course proliferation, the ATRflox/- cells that escape lox recombination may permit some tissue reconstitution in ATRmKO mice. To examine this possibility, the ratio of ATRflox and ATRΔ alleles was assessed by Southern hybridization at various time points following ATR deletion. The average deletion rate of the ATRflox allele in ATRflox/+Cre-ERT2+ mice was high in a variety of tissues 1-2 weeks after TAM treatment, and the representation of these ATRΔ/+ cells remained similarly high for more than one year (Figures 1E, 4B,C). Thus, ATRΔ/+ cells displayed no observable competitive disadvantage in comparison to ATRflox/+ cells in TAM-treated ATRflox/+Cre-ERT2+ mice. In contrast, the representation of ATRΔ/- cells in ATRmKO mice rapidly declined over time in tissues in which cellular proliferation occurs constitutively and rapidly, such as in the bone marrow, intestines, spleen and thymus (Figure 4B,D). The observed reconstitution of these tissues (Figure 3A, 4A and data not shown) must be attributable to cells that escaped lox recombination (ATRflox/-) and continue to express ATR. Importantly, representation of ATRΔ/- cells did not decrease appreciably in the brain, a tissue in which cellular proliferation is generally slow (Figure 4D), suggesting that ATR is less important for the continued viability of non-dividing cells. Consistent with this interpretation, deletion of ATR specifically in post-mitotic neurons led to no observable effect on behavior, activity, coordination or circadian rhythm (Supplemental Figure S3). In addition, although a decrease in ATRΔ/- cells was observed in the heart and kidney over time, these tissues also displayed increased fibrosis (Supplemental Figure S2), which was likely attributable to fibroblasts that escaped lox recombination. Together, these data indicate that ATR loss in ATRmKO mice leads to the loss of proliferating cells in vivo and, in some tissues, a subsequent reconstitution with the cells that escaped lox recombination and remain ATR positive.
It has been previously observed that ATR deletion in cultured murine embryonic fibroblasts leads to cell cycle exit in a manner reminiscent of cellular senescence (Brown and Baltimore, 2003). Therefore, it is within reason that part of the decreased representation of ATRΔ/- cells in vivo may be due to cell cycle exit via senescence. To investigate this possibility, we examined several tissues for senescence-associated β-galactosidase (SA-β-gal) activity and p21 expression, which are established markers of senescent cells in vivo. In most tissues examined, including the thymus, intestine, skin, heart and brain, no difference in SA-β-gal staining was observed between ATRmKO and control mice either soon after TAM treatment or up to 1 year later. High levels of staining in the red pulp of the spleen and within cortical regions of the kidney were observed equally in control and ATRmKO mice, as was staining of specific cell types with high lysosomal content, such as paneth cells in the intestine and lysing cells of the sebaceous gland in the skin (Supplemental Figure S4 and data not shown). In comparison to control mice, a significant increase in SA-β-gal stained cells was found only in the liver of ATRmKO mice 1 year after ATR deletion. Although the widespread appearance of cells expressing increased levels of p21 were not observed in this tissue or others (Supplemental Figure S4 and data not shown), it is formally possible that the increase in expression of p21 that accompanies senesecence was insufficient to be detectable by immunohistochemistry. We do not rule out the possibilities that senescent cells in ATRmKO mice may have been generated and subsequently cleared (Xue et al., 2007) or were present in small subpopulations that were not readily identifiable; nevertheless, a widespread accumulation of senescent cells in ATRmKO mice using the methods described herein was not observed.
Together, our results indicate that ATR deletion leads to the loss of ATRΔ/- cells and a subsequent deterioration of tissue homeostasis (Figures 2-4). Given these results, we reasoned that the aging phenotypes observed in ATRmKO mice do not appear to be the consequence of persistent ATR knockout cells but, instead, may be caused by impaired regenerative capacity and depletion of stem cell potential. To address these possibilities, we made use of two systems in which stem cells have been well characterized: hematopoiesis/thymopoiesis and the hair follicle cycle as described below.
Premature Thymic Involution in ATRmKO Mice Correlates with Thymic Progenitor Cell Attrition
As shown in Figure 3A, ATRmKO mice exhibited a significant reduction in thymocyte numbers, indicative of an accelerated involution process. In normal aged mice, thymic involution is thought to result from deterioration of the thymic stroma and declining numbers of bone marrow-derived hematopoietic progenitors and early intrathymic T cell progenitors (Zediak and Bhandoola, 2005). To determine whether ATRmKO exhibited reductions in T cell progenitor populations, the frequencies of T cell precursors in the bone marrow and thymus were assessed in ATRmKO and control mice.
Lin-Sca-1+c-Kithi (LSK) stem and progenitor cells in the bone marrow are early precursors in T cell development (Zediak and Bhandoola, 2005). In comparison to control mice, a significant decline in the LSK fraction was observed in the bone marrow of ATRmKO mice 12-13 months after TAM treatment (Figure 5A). In addition, consistent with premature thymic involution, the number and frequency of early T cell progenitors (ETPs, Lin-CD25-c-Kithi) and downstream double negative 2 progenitors (DN2, Lin- CD25+c-Kithi) were dramatically decreased in the thymi of ATRmKO mice (Figure 5B,C). This observation is consistent with the previously reported reduction of ETP frequencies in aged mice (Min et al., 2004) and suggests that the premature thymic involution observed in ATR knockout mice is caused, at least in part, by progenitor cell attrition.
Figure 5.
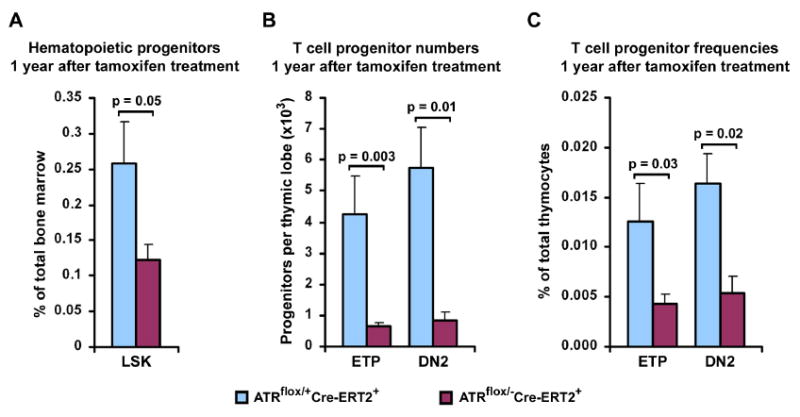
Decreased levels of hematopoietic and thymic progenitors in ATRmKO mice. (A) Quantification of hematopoietic LSK (Lin-Sca+c-Kithi) progenitors in the bone marrow of ATRflox/+Cre-ERT2+ (control) and ATRflox/-Cre-ERT2+ (ATRmKO) mice 1 year after TAM treatment. 5-9 mice per group were analyzed. (B, C) Quantification of T cell progenitors in the thymus of control and test mice. A decline in numbers (B) and frequencies (C) of ETPs (early T cell progenitors, Lin-CD25-c-Kithi) and DN2 (downstream double negative 2 progenitors, Lin-CD25+c-Kithi) was observed in the thymi of ATRmKO mice 1 year after TAM treatment (5-7 mice per group). Standard error bars are shown; P values were calculated by Mann-Whitney U test.
Graying Hair and Alopecia in ATRmKO Mice are Associated with Loss of Follicle Bulge Stem Cells
The hair follicle cycle in mice and humans consists of three major phases: anagen (follicle growth), catagen (follicle regression by apoptosis) and telogen (quiescent resting phase). Anagen is induced naturally following catagen or initiated prematurely in telogen by hair depilation, i.e. plucking (Ito et al., 2004). During normal telogen to anagen transition or following depilation, follicle bulge stem cells become activated and proliferate to initiate growth of a new hair shaft (Alonso and Fuchs, 2006). In this light, depilation may be viewed as an inducible system to assess hair regenerative capacity.
As described above, hair follicle loss and hair graying were observed in ATRmKO mice (Figure 2). To test the effect of ATR loss on the hair follicle cycle, 50-55 day old (telogen phase) ATRflox/+Cre-ERT2+ control and ATRflox/-Cre-ERT2+ test mice were treated on 2-3 cm2 patches of skin with 4-OHT. Telogen hair follicles in control and ATRmKO skin exhibited normal morphology after 4-OHT treatment, indicating that ATR is not required during the quiescent phase (Supplemental Figure S5A). To induce anagen, mice were depilated on the central 1 cm2 patch of 4-OHT-treated skin. Following depilation, anagen in control ATRflox/+Cre-ERT2+ mice was initiated and proceeded normally and indicated by the enlargement of the dermal papillae (DP) and elongation of the epithelial strands above DP (Figure 6A, 1st depilation). In contrast, early anagen phase in ATRmKO skin was accompanied by the widespread appearance of degenerative hair follicles 4 days after depilation and a delay in anagen progression 8 days after depilation (Figure 6A, 1st depilation). Hair shaft generation in ATRmKO skin was also delayed and compromised both in abundance and quality (Figure 6B and data not shown). Although sufficient regenerative capacity exists for a majority of hair follicles in ATRmKO skin, follicle development was significantly delayed.
Figure 6.
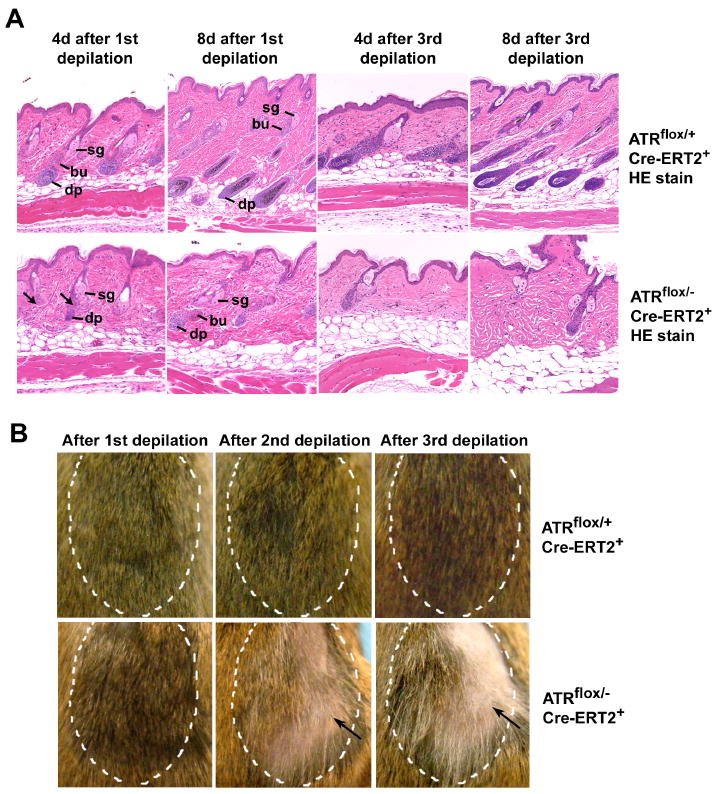
ATR deletion causes dysfunctional hair follicle regenerative cycling. (A) Formation of the new lower hair follicle was delayed after the 1st depilation (n = 5 mice for each time point) and frequently defective after the 3rd depilation in ATRmKO skin (n = 3 mice for each time point) (arrows). ATR was deleted in 50-55 day old mice by topical treatment of 4-OH-tamoxifen on 2-3 cm2 of dorsal skin. Four days later, hair shafts (telogen phase) were plucked in the treated areas. Second and third round of depilation were performed in subsequent telogen phases. H&E-stained histological sections of control and ATRmKO skins 4 and 8 days after depilation are shown, 4-10 sections were analyzed per mouse. Sg – sebaceous gland, bu – bulge, dp – dermal pappila. (B) Alopecia and graying of hair in ATRmKO skin 3-4 weeks after the 1st, 2nd and 3rd rounds of depilation (arrows). Depilated areas are marked by hatched lines. Defective hair regrowth was observed in all ATRmKO mice (n = 3-7 for each time point), but not in control mice (n = 3-8).
After a second round of depilation in the subsequent telogen phase, hair regeneration in ATRmKO skin was more dramatically affected, both in regards to hair paucity and the increased representation of gray hair shafts (Figure 6B). A final third round of depilation once again caused a profound delay in hair regeneration in ATRmKO regions in which hair either did not grow back or grew back predominantly gray (Figure 6B; Supplemental Figure S5B), implying the depletion of hair bulb melanocytes. Histological examination revealed that hair shaft developmental defects in ATRmKO regions were preceded by delays in follicle formation in early anagen (Figure 6A, 3rd depilation). In contrast, the hair in 4-OHT-treated regions of ATRflox/+Cre-ERT2+ control mice grew back normally following the second and third depilation. These data demonstrate that ATR deletion leads to significant defects in hair follicle regeneration.
Hair follicle regeneration is governed by stem cells that reside in the follicle bulge (Morris et al., 2004). Normally, anagen induction by depilation causes apoptotic cell death of a fraction of follicle bulge stem cells and the proliferation of the remaining stem and progenitor cells in the bulge and secondary germ. These cells begin to divide within 24 hours and continue to proliferate for at least 4 additional days to self-renew and form the new lower anagen follicle (Ito et al., 2004). CD34-staining (Blanpain et al., 2004) was then used to assess bulge stem cell abundance in ATRmKO and control skin following depilation. Prior to depilation, the representation of telogen phase follicle bulge stem cells was similar between ATRmKO and control skin (Figure 7A,B). However, four days after depilation, while the follicle bulge stem cell compartment in the control skin was maintained in the expected manner, a significant depletion of stem cells in the follicles of ATRmKO skin was observed (Figure 7A,B). Subsequently, bulge stem cells in many follicles were restored with delayed kinetics in ATRmKO skin (data not shown). This restoration presumably allowed for the delayed regeneration of most hair follicles following the first round of depilation (Figure 7C). However, after three rounds of depilation, the continued inability of stem cell compartments to self-renew appropriately in ATRmKO skin (Figure 7A,B) correlated with the degeneration of hair follicles (Figures 6A,B and 7C; Supplemental Figure S5C) as well as significant delays in hair shaft development in the surviving hair follicles (data not shown). In contrast, stem cell and hair regeneration was normal in the 4-OHT-treated ATRflox/+Cre-ERT2+ control skin after three rounds of depilation (Figure 7). Together, these results demonstrate that alopecia and hair graying observed in ATRmKO mice correlates with a progressive loss of regenerative capacity and stem cells in hair follicles.
Figure 7.
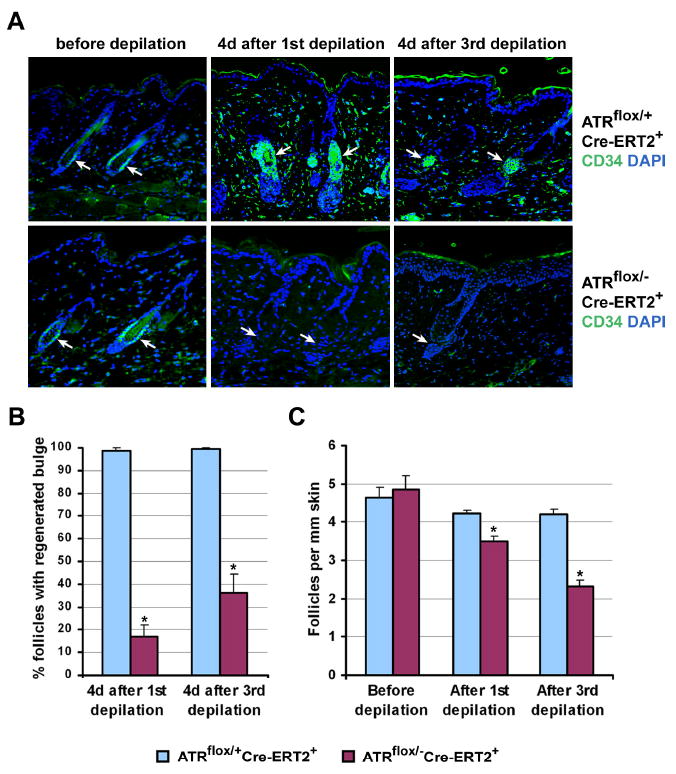
ATR deletion leads to bulge stem cell and follicle loss after depilation. (A) Bulge stem cells were detected by CD34 immunohistochemistry (green) in telogen phase (before depilation) and anagen phase (4 days after the 1st and 3rd depilations) follicles; nuclei were detected with DAPI (blue). Significant loss and delayed regeneration of follicle bulge stem cells were observed in ATRmKO skin following depilation, while the bulge regenerated normally in the control mice (arrows). All pictures were taken at 100× magnification. (B) Quantification of the data presented in (A) Two mice per group and 8 skin sections per mouse were used, and at least 12 follicles per section were analyzed. (C) Quantification of the average number of follicles per 1 mm of skin in control and ATRmKO mice before depilation and 4-6 weeks after each depilation. H&E stained skin sections were used. A significant decrease in follicle density in ATRmKO skin (n = 2-4 for each group) was observed. Error bars represent SEM. (*) p < 0.001 as calculated by Student's T-test.
These studies indicate that loss of tissue homeostasis and regenerative capacity in the thymus and hair follicles of ATR knockout mice correlates with a loss of stem and progenitor cells. Although the mosaic nature of ATR deletion affords some reconstitution of these tissues, the inability of ATRΔ/- cells to contribute to regeneration appeared to effectively shorten the time in which homeostasis and regeneration can be maintained in the lives of these animals. The implications of these findings on the cause of premature aging phenotypes in ATRmKO mice are discussed below.
Discussion
We have shown that deletion of the developmentally essential ATR checkpoint kinase and the subsequent loss of ATR knockout cells in adult mice leads to the exhaustion of tissue renewal and homeostatic capacity and the premature onset of age-related phenotypes. Distinct from other mouse models of aging, the age-related phenotypes observed in our studies are not influenced by or the direct result of developmental abnormalities. Therefore, our studies imply that reduced regenerative capacity in adults may be sufficient to cause characteristics of premature aging. The implications of these observations on the role of developmentally essential DDR genes in preserving regenerative capacity and preventing age-related diseases are discussed below.
Effect of ATR Loss on Tissue Homeostasis and Regeneration
Previously, ATR has been shown to be required for embryonic development prior to gastrulation and indispensable for cellular proliferation in cultured cells (Brown and Baltimore, 2000; Brown and Baltimore, 2003; Cortez et al., 2001). Herein, we have shown that ATR is essential for continued cellular proliferation in vivo in the mouse (Figure 4). Importantly, the inability of ATRΔ/- cells to proliferate correlated with the deterioration of several tissues in a manner that resembled premature aging. These data imply that aging phenotypes in ATRmKO mice result from the proliferative failure of ATRΔ/- cells and the consequential deficiencies in tissue regeneration.
Despite the proliferative failure of ATRΔ/- cells, ATRmKO mice are not completely devoid of regenerative potential. While ATRΔ/- cells were lost particularly from tissues with constitutive cell proliferation, many of these same tissues were later reconstituted with cells that escaped lox recombination (Figures 3-7). Thus, aging phenotypes were observed in ATRmKO despite ample tissue reconstitution with ATR-expressing cells. For example, after an initial drop in cellularity following ATR deletion, the ATRmKO thymi were reconstituted within 3-4 months with cells that failed to undergo lox recombination and remained ATRflox/- (Figure 4D). Subsequently, however, the cellularity of ATRmKO thymi dropped below control levels (Figure 3A). Therefore, in the end, the inability of ATRmKO mice to maintain thymopoiesis at normal levels resulted from the failure of cells that continued to express ATR. Insufficient tissue homeostasis as governed by residual ATR-expressing cells is also the likely cause of deterioration in other tissues (Figures 2, 3, 6) and, in some cases, was accompanied by replacement via fibrosis (Supplemental Figure S2). As discussed below, loss of regenerative capacity in ATRmKO mice correlated with stem cell attrition, providing a potential mechanism for loss of regenerative capacity and the emergence of age-related phenotypes.
Stem Cell Depletion and Aging
Loss of tissue homeostasis through stem and progenitor cell attrition has previously been proposed as a model to explain the general organismal decline associated with aging (Chen, 2004; Pelicci, 2004; Rando, 2006; Sharpless and DePinho, 2004; Van Zant and Liang, 2003). Because ATR deletion led to the depletion of stem and progenitor cells, our results indicate that stem and progenitor cell attrition may be the primary cause of reduced regenerative capacity in ATRmKO mice. Although alternative explanations are certainly possible, accelerated stem cell and progenitor cell attrition provides a simple model for the homeostatic and regenerative failure in ATRmKO mice. In general support of this model, apoptosis (Kujoth et al., 2005) and p16- and p21-mediated senescence (Choudhury et al., 2007; Janzen et al., 2006; Krishnamurthy et al., 2006; Molofsky et al., 2006) of stem and progenitor cells have been shown recently to be strongly associated with several age-related phenotypes in mice. The causes of stem and progenitor cell reduction in ATRmKO mice are not clear at the present time, but may result from the replicative incompetence of ATR–deleted cells or indirectly from niche deterioration. Evidence in support of each of these models is described below.
ATR deletion may render a majority of stem cells replication incompetent, leading to an over-reliance on the remaining ATR-expressing ATRflox/- stem cells that escaped lox recombination. These surviving ATR-expressing stem and progenitor cells may subsequently be lost either by natural causes or by additional replicative stress acquired during pool regeneration; thus, these cells may be insufficient to maintain tissue homeostasis over the long term. In support of this model, we observed a dramatic loss of CD34+ follicle bulge stem cells soon after depilation-induced anagen. While these bulge stem cells regenerated to a sufficient degree to drive a delayed anagen phase, they recovered poorly in subsequent regenerative cycles and their depletion was ultimately associated with follicle degeneration and hair loss (Figures 6 and 7). Stem and progenitor cell attrition by such means may also contribute to reduced LSK and ETP abundance (Figure 5), the downstream progeny of which were almost entirely composed of ATR-expressing ATRflox/- cells (Figure 4D).
In addition to stem cell exhaustion, decreased regenerative capacity in ATRmKO mice may result from deterioration of the stem cell niche, which maintains stem cells and regulates the production of downstream progenitor cells. Such a mechanism may contribute to decreased LSK abundance in ATRmKO mice (Figure 5A) since the volume of trabecular bone, which is proposed to be a hematopoietic stem cell niche (Wilson and Trumpp, 2006), is reduced in these mice (Figure 2C). In addition, deterioration of the thymic epithelial stroma could contribute to the observed decreases in ETPs; thus, the dramatic effect of ATR deletion on ETP numbers may result from the combined effects of LSK reduction and thymic stromal deterioration. These mechanisms, stem cell exhaustion and niche deterioration, may each be at work and may synergistically decrease stem and progenitor cell potential and tissue regeneration capacity in ATRmKO mice.
ATRmKO Mice and Seckel Syndrome
In humans, hypomorphic mutations in the ATR gene have been linked to Seckel syndrome, a disease characterized by intrauterine and postnatal growth retardation, microcephaly, mental retardation and dysmorphic facial and skeletal features (O'Driscoll and Jeggo, 2006; O'Driscoll et al., 2003). The nature of these characteristics suggests that abnormalities in development are the predominant cause of these Seckel-associated disorders (O'Driscoll and Jeggo, 2006). Our results are consistent with this hypothesis as post-development elimination of ATR resulted in few of the above characteristics. For example, ATR deletion in post-mitotic neurons had little or no effect on neurological function (Supplemental Figure S3), arguing against a role for ATR in maintaining post-mitotic arrest as previously suggested (O'Driscoll and Jeggo, 2003). Our results support a model in which the major disorders associated with Seckel syndrome are predominantly caused by irregularities during development, possibly as a consequence of defects in cellular proliferation.
Nevertheless, other characteristics of Seckel syndrome that are normally overshadowed by the above developmental abnormalities have been reported. For example, a senile, or progeroid, appearance has frequently been noted in Seckel patients (Arnold et al., 1999; Boscherini et al., 1981; Butler et al., 1987; Fathizadeh et al., 1979); however, this aspect has been difficult to distinguish from the effects of growth retardation. Hematopoietic abnormalities (pancytopenia) have been reported in approximately 20% of patients (Arnold et al., 1999; Butler et al., 1987), and hair graying and alopecia have been observed in a few cases (Boscherini et al., 1981; Fathizadeh et al., 1979). The variance in appearance of these characteristics is partly due to the use of broad diagnostic parameters, but may also be influenced by genetic heterogeneity, since mutations in genes other than ATR are associated with Seckel (Ch. 14q21-q22 and Ch. 18p11.31-q11.2, OMIM). It is important to note that the penetrance and expressivity of these minor Seckel phenotypes would not be expected to reach the level observed in ATRmKO mice given the hypomorphic nature of the ATR mutation in Seckel patients. Should any of the phenotypes observed in ATRmKO mice be mirrored in the subset of Seckel patients that harbor mutations in the ATR gene, then it is possible that these abnormalities may be rooted in a post-developmental effect of ATR mutations on tissue homeostasis and regeneration in these individuals.
ATRmKO and Other DDR Mutant Mouse Models
As described above, germline mutations in a number of DDR genes lead to premature aging phenotypes (Hasty et al., 2003; Lombard et al., 2005). Interestingly, the penetrance, expressivity and speed of onset of aging phenotypes were frequently greater in ATRmKO mice than in other DDR mutant mouse models. While most phenotypes in ATRmKO mice were observed within 6 months of ATR deletion, previously reported DDR mutants often displayed age-related phenotypes only after 12-18 months (Hasty et al., 2003; Lombard et al., 2005). In light of our studies, it is possible that premature aging phenotypes in DDR mutants are generally determined by the importance of the gene in maintaining cell viability and tissue homeostasis in adulthood. The rapid onset of age-related phenotypes observed in XPDR722W/R722W (3 months to 1 year after birth) are consistent with this theory as this model harbors a potent hypomorphic mutation in an otherwise essential gene (de Boer et al., 2002). Thus, the essential functions of XPD and ATR under normal physiologic conditions prevent aging by maintaining cell viability and proliferation. Deletion of ATR using the mouse model system described herein brings about an extreme effect on cell proliferation in adulthood previously not possible with germline mutants. As described above, a simple explanation for the ultimate failure of tissue homeostasis in ATRmKO mice is the depletion of stem and progenitor cell pools. If true, the importance of a given DDR gene in preventing aging phenotypes will be directly related to its importance in maintaining genome integrity and the viability of stem cells. Because the inducible system described herein may be adapted to delete DDR genes in a tissue-specific or cell type-specific manner, such systems in combination with lox recombination reporter alleles may be used to examine cellular turnover rates during tissue homeostasis and the importance of specific stem and progenitor compartments in preventing age-related disease in adult animals.
Experimental Procedures
Generation of Cre-ERT2 Lentitransgenic Mice
The Cre-ERT2 mouse line was generated through lentitransgenesis (Lois et al., 2002) by microinjection of Cre-ERT2-expressing lentivirus (100,000 TU/μl) into the perivitelline space of one cell zygotes (129SvEv/C57BL/6 mixed). After three days in culture, morula and blastocyst stage embryos (12 - 20) were implanted into each pseudopregnant recipient mouse. Integrated copies of the recombinant lentivirus in the resulting 22 founder mice were detected by Southern blotting of Hpa I digested tail DNA and hybridization to radiolabeled DNA encoding Cre recombinase. Expression of Cre-ERT2 in founders with single copy integrants was confirmed by western blot detection using 1:2000 dilution anti-Cre polyclonal antisera (Novagen).
Deletion of ATR in Adult Mice
To recombine the ATRflox allele using the Cre-ERT2 transgenic system, TAM (MP Biomedicals) was solubilized at 20 mg/ml in a mixture of 98% corn oil and 2% ethanol and delivered into mice by intraperitoneal injection or oral gavage treatment (0.5 μM/gram body weight, once/day for 5 days). Lactated Ringer's solution (hydration) was delivered by subcutaneous injection when necessary. Southern blot detection and quantification of ATR+, ATRflox and ATRΔ alleles was performed as described in Supplemental Experimental Procedures.
Histological Analysis and Immunofluorescence
Tissues were fixed in 4% paraformaldehyde at 4°C overnight, dehydrated and embedded in paraffin. For general morphology, tissues were sectioned and stained with hematoxylin and eosin (HE). For proliferation analysis, mice were treated with BrdU (1mg/ml in drinking water) for one month prior to sacrifice. For stromal staining, thymuses were collected and frozen fresh in OCT compound (VWR). Antibodies used for detection are described in Supplemental Experimental Procedures.
Bone X-ray, MicroCT and Histomorphometry
Mice were imaged by X-ray (MP500 High Frequency, Universal). For histological analysis, bones were fixed in 4% paraformaldehyde at 4°C overnight, dehydrated and stored in 70% EtOH. Bones were scanned and histomorphometry was performed at the MicroCT Facility and the Center of Bone Histology and Histomorphometry of the University of Connecticut, respectively.
Analysis of Hematopoietic Tissues
Harvested thymi were dissociated into single cell suspensions and cells were counted by hemacytometer. Bone marrow was flushed from the hindlimbs and red blood cells were lysed using ACK lysing buffer (Cambrex). Bone marrow cells and thymocytes were stained as described in Supplemental Experimental Procedures. Dead cells were excluded with 4,6-diamidino-2-phenylindole (DAPI). Cells were analyzed on a four laser LSR II (Becton Dickinson) and cytometric data were analyzed using FlowJo (Tree Star).
Depilation
50-55 days old (telogen phase) ATRflox/+Cre-ERT2+ control and ATRflox/-Cre-ERT2+ test mice were topically treated on 2-3 cm2 of dorsal skin with 4-OH-tamoxifen dissolved in 100% EtOH (4 μmol once/day for 2 days; Alexis biochemicals). Four days later, hair in the treated areas was manually plucked. The same skin areas were then depilated for the second and third time when in telogen (45-50 days after depilation). Treated and depilated skins were collected at several time points after the first and the third depilations. Skins were fixed in 4% paraformaldehyde at 4°C overnight, processed for paraffin embedding, then sectioned and stained for HE and CD34 (Supplemental Experimental Procedures).
Supplementary Material
Acknowledgments
Due to space limitations and the broad scope of this paper, we were often unable to cite primary research literature; we thank the authors for their understanding of the use of reviews. We also thank the following individuals for their expert advice and assistance in the indicated areas and for reagents: Douglas Adams and Gloria Gronowicz (bone analysis at UConn MicroCT Facility and Center for Histology and Histomorphometry), David Baltimore, Carlos Lois and Shirley Pease (lentitransgenesis), Mayumi Ito (hair regeneration experiments), Maja Bucan (behavioral analysis of ATRflox/-Syn-Cre+ mice), Qian-Chun Yu, the AFCRI histology core facility and Susan Bender (tissue processing and analysis), Martin Carroll (PBL analysis), Lawrence Donehower, Manuel Serrano, Manuel Collado, Pier Paolo Pandolfi and Zhenbang Chen (detection of senescence markers p21 and SA-β-gal), Igor Kuznetsov (statistical analysis), Pierre Chambon (Cre-ERT2 cDNA), Brad Johnson and Ben Stanger (critically reading the manuscript). Financial support of these studies was provided by the National Institute on Aging (1R01AG027376-01), the Abramson Family Cancer Research Institute and the General Motors Cancer Research Scholars Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alonso L, Fuchs E. The hair cycle. J Cell Sci. 2006;119:391–393. doi: 10.1242/jcs.02793. [DOI] [PubMed] [Google Scholar]
- Arnold SR, Spicer D, Kouseff B, Lacson A, Gilbert-Barness E. Seckel-like syndrome in three siblings. Pediatr Dev Pathol. 1999;2:180–187. doi: 10.1007/s100249900107. [DOI] [PubMed] [Google Scholar]
- Blanpain C, Lowry WE, Geoghegan A, Polak L, Fuchs E. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell. 2004;118:635–648. doi: 10.1016/j.cell.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Boscherini B, Iannaccone G, La Cauza C, Mancuso G, Girotti F, Finocchi G, Pasquino AM. Intrauterine growth retardation. A report of two cases with bird-headed appearance, skeletal changes and peripheral GH resistance. Eur J Pediatr. 1981;137:237–242. doi: 10.1007/BF00441325. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003;17:615–628. doi: 10.1101/gad.1067403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Hall BD, Maclean RN, Lozzio CB. Do some patients with Seckel syndrome have hematological problems and/or chromosome breakage? Am J Med Genet. 1987;27:645–649. doi: 10.1002/ajmg.1320270318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Li W, Kim S, Brodie SG, Deng CX. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003;17:201–213. doi: 10.1101/gad.1050003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell. 2002;111:779–789. doi: 10.1016/s0092-8674(02)01113-3. [DOI] [PubMed] [Google Scholar]
- Chao C, Herr D, Chun J, Xu Y. Ser18 and 23 phosphorylation is required for p53-dependent apoptosis and tumor suppression. Embo J. 2006;25:2615–2622. doi: 10.1038/sj.emboj.7601167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. Senescence and functional failure in hematopoietic stem cells. Exp Hematol. 2004;32:1025–1032. doi: 10.1016/j.exphem.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Choudhury AR, Ju Z, Djojosubroto MW, Schienke A, Lechel A, Schaetzlein S, Jiang H, Stepczynska A, Wang C, Buer J, et al. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat Genet. 2007;39:99–105. doi: 10.1038/ng1937. [DOI] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–1716. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- de Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, van Steeg H, Weeda G, van der Horst GT, van Leeuwen W, Themmen AP, et al. Premature aging in mice deficient in DNA repair and transcription. Science. 2002;296:1276–1279. doi: 10.1126/science.1070174. [DOI] [PubMed] [Google Scholar]
- de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, Lehmann AR, Hoeijmakers JH. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol. 2000;10:479–482. doi: 10.1016/s0960-9822(00)00447-4. [DOI] [PubMed] [Google Scholar]
- Fathizadeh A, Soltani K, Medenica M, Lorincz AL. Pigmentary changes in Seckel's syndrome. J Am Acad Dermatol. 1979;1:52–54. doi: 10.1016/s0190-9622(79)70004-1. [DOI] [PubMed] [Google Scholar]
- Feil R, Wagner J, Metzger D, Chambon P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun. 1997;237:752–757. doi: 10.1006/bbrc.1997.7124. [DOI] [PubMed] [Google Scholar]
- Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J. Aging and genome maintenance: lessons from the mouse? Science. 2003;299:1355–1359. doi: 10.1126/science.1079161. [DOI] [PubMed] [Google Scholar]
- Ito M, Cotsarelis G, Kizawa K, Hamada K. Hair follicle stem cells in the lower bulge form the secondary germ, a biochemically distinct but functionally equivalent progenitor cell population, at the termination of catagen. Differentiation. 2004;72:548–557. doi: 10.1111/j.1432-0436.2004.07209008.x. [DOI] [PubMed] [Google Scholar]
- Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- Kipling D, Davis T, Ostler EL, Faragher RG. What can progeroid syndromes tell us about human aging? Science. 2004;305:1426–1431. doi: 10.1126/science.1102587. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell. 2005;120:497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M. The DNA replication checkpoint response stabilizes stalled replication forks. Nature. 2001;412:557–561. doi: 10.1038/35087613. [DOI] [PubMed] [Google Scholar]
- Min H, Montecino-Rodriguez E, Dorshkind K. Reduction in the developmental potential of intrathymic T cell progenitors with age. J Immunol. 2004;173:245–250. doi: 10.4049/jimmunol.173.1.245. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–452. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RJ, Liu Y, Marles L, Yang Z, Trempus C, Li S, Lin JS, Sawicki JA, Cotsarelis G. Capturing and profiling adult hair follicle stem cells. Nat Biotechnol. 2004;22:411–417. doi: 10.1038/nbt950. [DOI] [PubMed] [Google Scholar]
- Nabarra B, Andrianarison I. Ultrastructural study of thymic microenvironment involution in aging mice. Exp Gerontol. 1996;31:489–506. doi: 10.1016/0531-5565(95)02038-1. [DOI] [PubMed] [Google Scholar]
- O'Driscoll M, Jeggo PA. Clinical impact of ATR checkpoint signalling failure in humans. Cell Cycle. 2003;2:194–195. [PubMed] [Google Scholar]
- O'Driscoll M, Jeggo PA. The role of double-strand break repair - insights from human genetics. Nat Rev Genet. 2006;7:45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- Osborn AJ, Elledge SJ, Zou L. Checking on the fork: the DNA-replication stress-response pathway. Trends Cell Biol. 2002;12:509–516. doi: 10.1016/s0962-8924(02)02380-2. [DOI] [PubMed] [Google Scholar]
- Pelicci PG. Do tumor-suppressive mechanisms contribute to organism aging by inducing stem cell senescence? J Clin Invest. 2004;113:4–7. doi: 10.1172/JCI200420750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando TA. Stem cells, ageing and the quest for immortality. Nature. 2006;441:1080–1086. doi: 10.1038/nature04958. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA. Telomeres, stem cells, senescence, and cancer. J Clin Invest. 2004;113:160–168. doi: 10.1172/JCI20761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Zant G, Liang Y. The role of stem cells in aging. Exp Hematol. 2003;31:659–672. doi: 10.1016/s0301-472x(03)00088-2. [DOI] [PubMed] [Google Scholar]
- Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol. 2006;6:93–106. doi: 10.1038/nri1779. [DOI] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zediak VP, Bhandoola A. Aging and T cell development: interplay between progenitors and their environment. Semin Immunol. 2005;17:337–346. doi: 10.1016/j.smim.2005.05.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.