

Abstract
Phosphatidylinositol-4,5-bisphosphate (PIP2) is a phospholipid that has been shown to modulate several ion channels, including some voltage-gated channels like Kv11.1 (hERG). From a biophysical perspective, the mechanisms underlying this regulation are not well characterized. From a physiological perspective, it is critical to establish whether the PIP2 effect is within the physiological concentration range. Using the giant-patch configuration of the patch-clamp technique on COS-7 cells expressing hERG, we confirmed the activating effect of PIP2. PIP2 increased the hERG maximal current and concomitantly slowed deactivation. Regarding the molecular mechanism, these increased amplitude and slowed deactivation suggest that PIP2 stabilizes the channel open state, as it does in KCNE1-KCNQ1. We used kinetic models of hERG to simulate the effects of the phosphoinositide. Simulations strengthened the hypothesis that PIP2 is more likely stabilizing the channel open state than affecting the voltage sensors. From the physiological aspect, we established that the sensitivity of hERG to PIP2 comes close to that of KCNE1-KCNQ1 channels, which lies in the range of physiological PIP2 variations.
Introduction
Phosphatidylinositol-4,5-bisphosphate (PIP2) is a phospholipid present in the inner leaflet of the cell plasma membrane that is implicated in many physiological functions, such as production of the second messengers inositol trisphosphate and diacylglycerol, membrane trafficking, cytoskeleton attachment, and regulation of ion channels and transporters (1). PIP2 notably regulates several cardiac potassium channels, such as Kir6.2, Kir2.1, Kv11.1 (hERG), and Kv7.1 (KCNQ1), implicated in the IKATP, IK1, IKr, and IKs currents, respectively (2–6). McDonald and co-workers were the first to describe the PIP2 sensitivity of a voltage-gated potassium (Kv) channel, hERG (6). This channel, encoded by the human ether-à-go-go related gene (hERG, or KCNH2), is responsible for the rapid delayed rectifier K+ current, IKr, in the heart and several other cell types. In Chinese hamster ovary (CHO) cells expressing hERG channels, PIP2 application in the pipette led to an increase in current and a change in activation and inactivation gating (6).
A key question is the molecular mechanism of this PIP2 regulation. Previous work suggested that this mechanism was similar in ion channels as various as Kir (inwardly rectifying potassium) and Kv channels: PIP2 stabilizes the channel open state of Kir6.2 (7) and the Kv channel KCNQ1 (8). For KCNQ1 recorded in the inside-out configuration, this stabilization of the open state resulted in a slowed deactivation with no effect on activation. It was surprising to find, in the work by MacDonald and colleagues, that PIP2 addition (from the pipette on CHO cells in a whole-cell configuration) led to completely different effects on hERG biophysical parameters: an accelerated activation with no effect on deactivation (6). Two main hypotheses can be proposed to explain these differences: 1), the molecular mechanism underlying Kir6.2 and KCNQ1 modulation by PIP2 does not apply to hERG channels; or 2), the molecular mechanism underlying Kir6.2 and KCNQ1 modulation applies to hERG, but the difference in the biophysical effects of PIP2 is due to the configuration of the experimental setup (whole-cell in hERG versus inside-out in KCNQ1). To address this question, we studied the effects of PIP2 on hERG under experimental conditions used previously for KCNQ1, mainly the giant-patch configuration. This configuration is characterized by rapid PIP2 depletion after patch excision and allows direct addition of exogenous PIP2 after this depletion. In this configuration, hERG and KCNQ1 biophysical changes induced by PIP2 insertion are quite similar (slowed deactivation with no effect on activation). We also show that, similar to the case for KCNQ1, the effects of PIP2 on hERG are much more convincingly modeled by an influence on a voltage-independent transition step late in the activation pathway, suggesting a stabilization of the open state, as in KCNQ1.
Another key question is whether this PIP2 regulation is physiologically relevant. A method to gain insight about this is to study the effects of physiological variations of PIP2. Indeed, in isolated rabbit cardiac myocytes, α1-adrenergic stimulation activates phospholipase C and results in a slight decrease in IKr (6). One limitation to this study was that activation of a receptor may trigger several regulatory pathways in parallel (9), sometimes leading to an apparent PIP2 insensitivity (10). We used several approaches to quantify the channel PIP2 sensitivity, and compared it with the well-characterized KCNQ1 PIP2 sensitivity (2,5,9). The results led us to the conclusion that hERG would be sensitive to moderate PIP2 changes in the range of physiological concentrations, but that at low PIP2 levels, a fraction of hERG current persists, which may lead to an apparent lower PIP2 sensitivity compared to that of KCNQ1.
Methods
Cell culture and transfection
The cell line COS-7, derived from the African green monkey kidney, was obtained from the American Type Culture Collection (CRL-1651, Rockville, MD) and cultured in DMEM supplemented with 10% serum and antibiotics (100 IU/mL penicillin and 100 μg/mL streptomycin), all from GIBCO, (Paisley, Scotland). Cells were transiently transfected with the plasmids using Fugene-6 (Roche Molecular Biochemical, Indianapolis, IN) according to the standard protocol recommended by the manufacturer.
hERG cDNA (a kind gift from D. Roden, Vanderbilt University School of Medicine, Nashville, TN) was subcloned into the mammalian expression vector pSI (Promega, Madison, WI). The human pCDNA3.1-KCNE1-KCNQ1 concatemer, human KCNE1 linked to the N-terminus of the human KCNQ1 (11), is a kind gift from R. S. Kass (The Center for Molecular Therapeutics, Columbia Presbyterian Medical Center, New York, NY). The plasmid coding for the green fluorescent protein (pEGFP) used to identify transfected cells was purchased from Clontech (Palo Alto, CA).
For giant-patch experiments on hERG, relative DNA composition was 60% pSI-hERG and 40% pEGFP (of a total of 2 μg of DNA). For giant-patch experiments on KCNE1-KCNQ1, relative DNA composition was 60% pCDNA3-KCNE1-KCNQ1 and 40% pEGFP (of a total of 2 μg of DNA). For giant patch experiments on cotransfected hERG and KCNE1-KCNQ1, relative DNA composition was 60% pCDNA3-KCNE1-KCNQ1, 30% pSI-hERG, and 10% pEGFP (of a total of 2 μg of DNA).
Electrophysiology
The effect of PIP2 on hERG was studied using the inside-out configuration of the patch-clamp technique, which allows direct access to the inner leaflet of the plasma membrane. hERG channels were expressed in COS-7 cells. To obtain current amplitudes compatible with an accurate determination of the biophysical parameters, giant patches of membrane (diameter ∼10 μm) were excised.
From 24 to 72 h after transfection, COS-7 cells were mounted on the stage of an inverted microscope and constantly superfused at a rate of ∼2 mL/min. Experiments were performed at room temperature (23 ± 2°C). Acquisition and analysis were performed using Acquis1 software (Bio-Logic Science Instruments, Claix, France). Electrodes were connected to a patch-clamp amplifier (RK-400, Bio-Logic). For giant-patch experiments, the procedure described in Hilgemann (12) was adapted to excise giant patches from COS-7 cells. Pipettes were pulled from borosilicate glass capillaries (glass type 8250, King Precision Glass, Claremont, CA) on a vertical puller (P30, Sutter Instruments, Novato, CA) and fire-polished using a microforge (MF-83, Narishige, Tokyo, Japan) to obtain tip diameters of ∼10 μm for patch pipettes and 20 μm for excision pipettes. The excision pipette, filled with the standard solution, was connected to a 20-mL syringe to apply suction for excision. A microperfusion system allowed local application and rapid change of the different experimental solutions (13).
Four protocols for hERG currents
Activation protocol. From a holding potential of −100 mV, five voltage steps (to −85, −70, −55, −40, and +30 mV) of 2 s each were followed by a 1-s step at the holding potential, every 16 s.
This protocol facilitated several measurements:
Maximal current, measured as the peak tail current after the +30 mV step.
Half-activation potential, where peak tail currents at each potential were fitted by a Boltzmann function:
Deactivation time constant at −100 mV, where deactivation time constants for a single protocol were measured twice, after voltage steps at −55 mV and −40 mV, and averaged. These values were obtained from a single exponential fit of the tail current after complete recovery from inactivation (after 1.5 times the time to peak).
Activation time constant at −55 mV, at which potential hERG was not completely inactivated, allowing accurate measurement of activation kinetics. The activating current was fitted by a single exponential fit after inactivation reached a steady state (t > 30 ms after the beginning of the −55-mV step).
Recovery from inactivation time constant at −100 mV, obtained by a single exponential fit of the tail current after the +30-mV step. The tail current was fitted from the start of repolarization to the peak.
Inactivation protocol. From a holding potential of +30 mV, 13 hyperpolarized voltage steps (from −130 mV to −10 mV in 10-mV increments) of 70 ms each were followed by a 230-ms step at the holding potential. This protocol enabled characterization of the steady-state inactivation of hERG from a fully activated/inactivated holding state. The half-inactivation potential was obtained by a Boltzmann fit of the peak current at each step divided by the electromotive force:
Envelope protocol. From a holding potential of −100 mV, 13 depolarized steps (to −10, 0, 10, or 20 mV) of increasing duration (from 10 ms to 500 ms). These envelope protocols made it possible to discover the activation kinetics of hERG otherwise hidden by inactivation at potentials of −10 mV to 20 mV.
PIP2-Mg2+ and polylysine protocol. From a holding potential of −100 mV, every 3 s the potential was stepped to 30 mV for 1 s. In this protocol, the maximal hERG current was measured as the peak tail current at −100 mV.
Protocol for all KCNE1-KCNQ1 currents
From a holding potential of −60 mV, every 3.5 s the potential was stepped to +40 mV for 1.5 s. This protocol was used for the PIP2-Mg2+ and polylysine experiments; the KCNE1-KCNQ1 current was measured as the peak tail current at −60 mV.
Protocol for hERG and KCNE1-KCNQ1 together during cotransfection experiments
From a holding potential of −100 mV, every 3.5 s the potential was stepped to +60 mV for 1.5 s. KCNE1-KCNQ1 current was measured as the current increase during the step (because of inactivation, hERG current was stable and very low at this potential, so that any current increase after a few milliseconds could be reliably attributed to the activation of KCNE1-KCNQ1 channels). hERG current was measured from the recovery from inactivation measured during the tail potential (difference between peak tail current and current at the beginning of the tail). KCNE1-KCNQ1 alone did not show any recovery from inactivation (since it does not inactivate) when submitted to this protocol (data not shown), so that the observed recovery in cotransfection experiments could be reliably attributed to hERG (Fig. S1 in the Supporting Material).
Statistical significance of the observed effects was assessed by Student's t-test (N > 30), Wilcoxon's test, or two-way ANOVA.
Solutions and drugs
For giant-patch experiments, cells were superfused with a standard solution containing (in mmol/L) 145 KCl, 10 HEPES, and 1 EGTA, pH 7.3 with KOH. A solution of (in mmol/L) 145 K-gluconate, 10 HEPES, and 1 EGTA, pH 7.3 with KOH, was used to superfuse the cell during K+ current measurements and to fill the tip of the patch pipette. PIP2 (Roche Molecular Biochemicals, Meylan, France) was diluted to 5 μmol/L in the K+ current measurement solution and sonicated on ice for 30 min before application to inside-out patches. 1,2-dioctanoyl-4,5-bisphosphate (diC8-PIP2) was purchased from Cayman Chemicals (Ann Arbor, MI). Polylysine 10 kDa was purchased from Sigma-Aldrich (St. Louis, MO) and dissolved in a 3-mg/mL stock in water.
Kinetic models
Three Markov kinetic models were used (see Fig. 3, Fig. S3, and Fig. S4). They were adapted from previously described hERG models (14–16). For the model shown in Fig. 3, activation and inactivation are uncoupled and the hERG current is I = N × g × PO × PNI × (Vm − Vr), where N × g (N is the number of channels present and g the single-channel conductance) represents the maximal conductance of the membrane, PO the probability that the activation gate is in the open state (O), and PNI the probability that the inactivation gate is in the noninactivated state (NI). Vm is the membrane potential and Vr the reversal potential of the hERG current. The same concentration of potassium is applied on both sides of the patch for all experiments and Vr is set to 0 mV.
Figure 3.
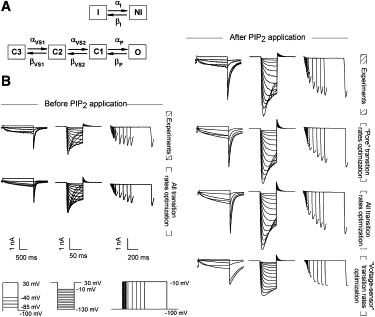
(A) Kinetic model of hERG currents before and after PIP2 application. Activation and inactivation are modeled as uncoupled processes. The channel is open when it is in the O and NI states at the same time. To focus on activation properties, transition rates regarding inactivation (αI, βI) are determined before and after PIP2 addition from a concomitant fit of voltage-dependent inactivation rates and recovery-from-inactivation kinetics (Fig. S2). (B) Simulated and recorded hERG currents for the three protocols before and after PIP2 application. The transition rates and biophysical parameters determined from these simulations are shown in Table S1. Optimization of the late voltage-independent transitions (αP, βP), supposed to be related to the pore domain, leads to a reasonable fit of the PIP2 effect that is as good as the fit obtained by optimizing all transition rates. Conversely, optimization of early voltage-dependent transition rates (αVS1, βVS1, αVS2, βVS2), supposed to be mainly related to the voltage sensing of hERG, leads to a poor fit of the PIP2 effect.
Optimization was performed with ModelMaker v4.0 (AP Benson, Wallingford, United Kingdom) using the Marquardt method. Optimization was aimed at minimizing the mean square distance between currents measured for three protocols (see Results) and corresponding simulated currents. All transition rates had the form. α2 was set to 0 mV−1 in the case of voltage-independent transitions.
The inactivation transition rates of the model with uncoupled activation/inactivation (see Fig. 3) and the weakly coupled model (Fig. S3 A) were determined by two fits (see Fig. S2). The four free parameters (αI1, αI2, βI1, and βI2) were adjusted to fit the inactivation curve and recovery from inactivation kinetics:
Results
PIP2 modulation of hERG channels
The effect of PIP2 on hERG was studied using the inside-out configuration of the patch-clamp technique. For several channels it has been demonstrated that patch excision results in a current decrease (rundown) attributable at least partially to a decrease in membrane PIP2 levels, and that PIP2 at least partially reverses this rundown (8). Consequently, we first examined the effect of excision and PIP2 application on the activity of hERG channels. Currents were measured during three distinct step protocols detailed in the Methods section. 1), An activation protocol (Fig. 1 A, left) was used to construct the activation curve (Fig. 1 B, left). Applied every 16 s, this protocol indicated the evolution of half-activation potential and deactivation, activation, and recovery from inactivation kinetics (Fig. 1 C). 2), An inactivation protocol (Fig. 1 A, middle) was used to construct the inactivation curve (Fig. 1 B, middle) at specific time points: after excision (control), after rundown, and after PIP2 application. 3), An envelope protocol (Fig. 1 A, right) was used to measure the activation kinetics (Fig. 1 B, right) at potentials where inactivation masks activation (−10, 0, +10, and +20 mV).
Figure 1.
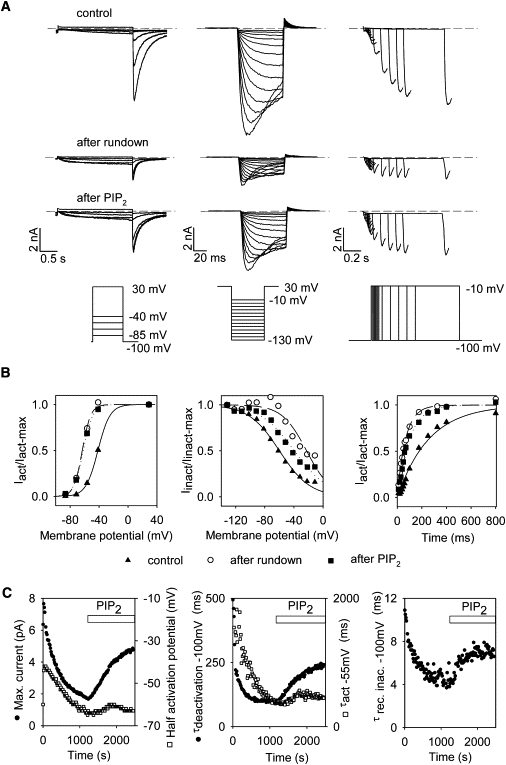
Rundown of hERG and reversal by PIP2 application. Data from a giant patch of a representative COS-7 cell transfected with hERG and studied in the inside-out configuration. (A) hERG current in response to an activation protocol (left), an inactivation protocol (middle), and an envelope protocol (right). Tail currents are proportional to the activation of hERG in the first protocol. Peak currents divided by the electromotive force are proportional to the extent of inactivation in the second protocol. Peak currents represent the time course of channel activation in the envelope protocol. Shown are the control current after excision (t = 60 s), the current after rundown (t = 1076 s), and the current after addition of PIP2 (t = 1757 s, 5 μmol/L PIP2 is added at t = 1200 s). (B) Activation curve (left), inactivation curve (middle), and time course of the peak inward current amplitude (right) at −10 mV, obtained from the recordings shown in A. Activation and inactivation curves are fitted by Boltzmann equations. The measurement points (symbols) and fits (lines) are indicated for control (triangles, solid line), after rundown (circles, dashed line); and after PIP2 addition (squares, dotted line). (C) Kinetics of hERG biophysical parameters during rundown and after 5 μmol/L PIP2 addition. These parameters are measured from the activation protocol repeated every 16 s. Solid symbols indicate maximal current, −100 mV deactivation, and recovery-from-inactivation time constants; open symbols indicate half-activation potential and −55 mV activation time constant.
Data from a representative patch, on which all protocols were run, are shown in Fig. 1. The averaged biophysical parameters measured in the three conditions (control, after rundown, and after PIP2 application) are shown in Fig. 2. The currents measured with the three protocols illustrate the decrease in hERG current ∼15 min after excision (Fig. 1 A). The maximal current decreased below one-quarter of its initial level within ∼10 min (Fig. 1 C), but in most of the cells, a current persisted after rundown. The remaining current may correspond to a PIP2-independent component, or it may be due to residual membrane PIP2 levels. Concomitantly with its rundown, hERG current showed an acceleration of both the activation and deactivation kinetics (Figs. 1 C and 2). hERG voltage dependence of activation shifted by about −18 mV toward negative potentials. Channel inactivation was also affected: the half-inactivation potential was shifted by about +15 mV toward positive potentials, and recovery from inactivation was accelerated (Figs. 1, B and C, and 2).
Figure 2.
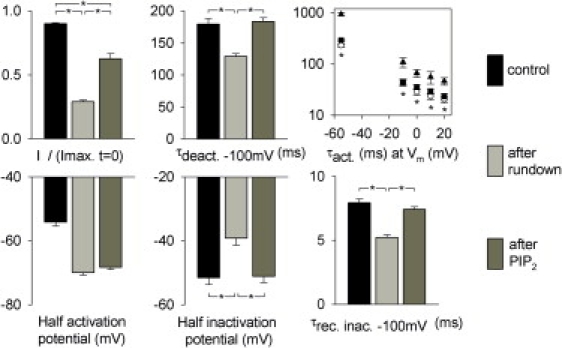
Biophysical parameters variation for hERG during rundown and after PIP2 addition. Biophysical parameters are averaged. Maximal current, half-activation potential, and time constants for deactivation at −100 mV and activation at −55 mV were obtained from 81–90 patches. Activation time constants at −10 to 20 mV were obtained from 14−23 patches. Half-inactivation potential and recovery from inactivation time constant at −100 mV were obtained from 27 and 45 patches, respectively. Black bars represent control, measured after excision; light gray bars represent the current after 5–10 min rundown and before PIP2 addition; and dark gray bars indicate the current >2 min after PIP2 addition (∗p < 0.05). A Bonferroni correction was applied for multiple comparison between time constants of activation. There was a significant difference between time constants of activation measured in control and after rundown, but not between the after-rundown and after-PIP2 conditions.
Patch excision may lead to additional changes in the channel environment than beside PIP2 depletion. To confirm the PIP2 modulation of hERG, we added 5 μmol/L PIP2 when the biophysical parameters were stable. Within a few minutes of PIP2 superfusion, the current amplitude increased but did not reach the initial current level observed right after excision (Figs. 1 C and 2). Regarding the activation gate, deactivation slowed down to initial levels. However, addition of PIP2 did not restore channel activation kinetics, indicating that the changes in this parameter observed upon patch rupture are unrelated to the depletion of PIP2. All these observations (incomplete current recovery, complete deactivation kinetics recovery, and no change in activation kinetics) were made previously for the PIP2 effect on KCNQ1 (8). In that work, the incomplete current recovery was attributed to a PIP2-independent rundown that could be prevented by addition of MgATP. This was not the case for hERG (data not shown). Regarding inactivation, PIP2 slowed the recovery from inactivation at −100 mV and shifted the half-inactivation potential toward negative potentials back toward initial control values (Figs. 1 C and 2).
PIP2 favors the open state of the hERG channel
We observed that PIP2 effects on hERG were very close to those observed on KCNQ1 in the same experimental conditions (mainly the giant-patch configuration): increased current, slowed deactivation, and no effect on activation kinetics. However, the effects of PIP2 on hERG were quite different if PIP2 was applied in the patch pipette (i.e., accelerated activation and no effect on deactivation) (6). Altogether, these observations stress the importance of experimental conditions in studying the effect of PIP2 on a current. Of most importance, the similarity between the effect of PIP2 on hERG and that on KCNQ1 suggests a similar molecular mechanism for this regulation.
Using kinetic models, we showed previously that PIP2 affects the late conformational changes of the KCNQ1 pore domain leading to channel opening rather than the early conformational changes implicating S4 movement (8). We used the same strategy in this study, to explore whether PIP2 affects the late conformational changes also in hERG. In the model used, we hypothesize that the voltage sensor movements are predominantly described by the early and voltage-dependent transitions in the activation pathway, whereas the pore opening is predominantly described by the late and voltage-independent transition, as suggested for other channels (8,17). Modification of several biophysical parameters of hERG implies that the effect of PIP2 cannot be simply a modification of the number of active channels (since in that case only the maximal current would be altered). This observation justifies the need for kinetic models to determine the transition rates between hERG conformations that are sensitive to PIP2. We already mentioned that after 10 min of rundown, hERG biophysical parameters were almost stable. As a consequence, the modification of hERG current after PIP2 addition could be mainly attributed to the PIP2 effect. We thus used current recordings after ∼15 min of rundown (before PIP2 addition) and after PIP2 addition (∼10 min later) as constraints for our kinetic models. To limit the number of free parameters, we first used the simplest model that fitted our data correctly (Fig. 3). In this model, hERG activation and inactivation processes are uncoupled. This is consistent with a recent study (18) and makes it possible to address independently the effect of PIP2 on channel activation and inactivation. However, the question of the activation/inactivation coupling being still open, we also used coupled models (cf. below and the Supporting Material). To take into account the PIP2 effect on the inactivation process, the transition rates of this process (αI and βI) were determined before and after PIP2 addition from a concomitant fit of the inactivation curve and the voltage dependency of the kinetics of recovery from inactivation (Fig. S2). Subsequently, these inactivation kinetics were fixed, and only the transition rates of the activation process were optimized to fit our data.
We fitted all the current recordings obtained with the three different pulse protocols shown in Fig. 1 A. First, transition rates regarding activation before PIP2 addition were optimized (Table S1) and the current recordings were well fitted for each protocol (Fig. 3). The biophysical parameters obtained from the simulations were also close to those determined from the experiments (Table S1). Second, to determine which transition rates varied after PIP2 addition, we optimized separately the values of the transition rates related to the voltage sensing (early voltage-dependent transitions: αS1, βS1, αS2, and βS2), the transition rates related to the pore (late voltage-independent transitions: αP, βP), and all the transition rates regarding activation (αS1, βS1, αS2, βS2, αP, and βP). The fit obtained by optimizing only the pore transition rates was reasonably good, almost as good as the fit obtained by optimizing all the transition rates (Fig. 3). Biophysical parameters were also determined (Table S1). Modifying the late transition rates was sufficient to simulate the two main effects of PIP2: increase of maximal current and slowing of deactivation (Table S1, bold print). On the contrary, optimization of the voltage sensor transition rates was unable to fit the experimental current recordings despite having more free parameters. These simulations indicated that the effect of PIP2 on hERG current could be fully explained by a change of a late and voltage-independent transition rate that is probably related to the pore, leading to a stabilization of the open state. We also demonstrated that this conclusion was valid for other kinetic models of hERG that show weak or strong coupling between activation and inactivation (see Discussion and Fig. S3 and Fig. S4); hence, this conclusion certainly did not result from the choice of an oversimplified description of hERG. A kinetic model is not an absolute proof for one or another mechanism, and it is still possible that PIP2 exerts a dual effect both on the pore and the voltage-sensor. However, these models suggest that a stabilization of the pore is sufficient to explain the PIP2 effect.
hERG presents a PIP2 affinity close to that of KCNE1-KCNQ1
When studying the PIP2 dependency of a channel, it is important to make sure that the affinity is compatible with a physiological regulation by PIP2. A common method is to add the PIP2 analog diC8-PIP2 on the cytosolic side of the membrane. This facilitates dose-response measurements and sorting of the affinities of different channels. We observed that hERG was only weakly activated by addition of up to 100 μmol/L diC8-PIP2 on the cytosolic face of the membrane (Fig. S5). We also noticed that the application of diC8-PIP2 led to an acceleration of channel deactivation, as opposed to slowing of deactivation by PIP2, showing that the effect of diC8-PIP2 on hERG does not reproduce the effect of PIP2.
To quantify the affinity of hERG for PIP2, we used another approach based on the fact that 1), the PIP2 sensitivity of KCNQ1 is well characterized; and 2), this sensitivity is proven to be in a physiological range (2,5,9). We cotransfected hERG and KCNE1-KCNQ1 in the same cells and measured, in the same patch, current rundown upon patch excision and recovery after PIP2 addition for both hERG and KCNE1-KCNQ1. We first compared channel rundown, which for both currents started immediately after excision and was concomitant (Fig. 4 B). A difference, however, was the presence of a PIP2-independent fraction of hERG current, as described above. An even more reliable test was to look at the effect of PIP2 addition, since PIP2 insertion may be quick, and this avoids the bias of the KCNE1-KCNQ1 PIP2- independent rundown (8). Both channels responded at the same time (Fig. 4 B) and thus appeared to be sensitive to the same range of PIP2 concentration. This was further supported by the correlation in magnitude of hERG and KCNE1-KCNQ1 current amplitudes in response to PIP2 (Fig. 4 C).
Figure 4.
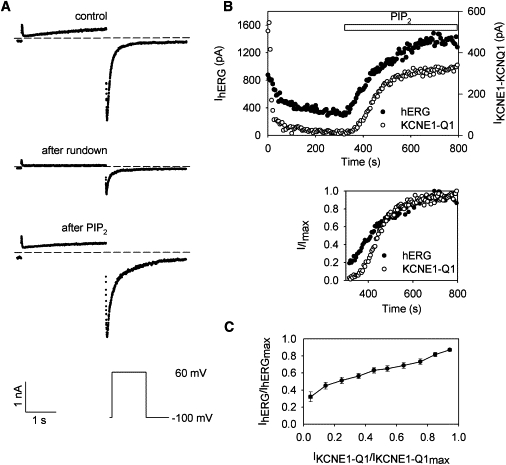
Concomitant responses to PIP2 of cotransfected hERG and KCNE1-KCNQ1. (A) Measured currents of a representative cell (protocol shown in the inset). hERG current was computed from the recovery from inactivation (KCNE1-KCNQ1 does not inactivate); KCNE1-KCNQ1 current was computed from the current increase during the prepulse (hERG current is stable after a few milliseconds at high potential). Currents are shown after excision (control, t = 5 s), after rundown (t = 290 s), and after PIP2 addition (t = 780 s; 5 μmol/L PIP2 is added at t = 315 s). (B) Kinetics of hERG (solid circles) and KCNE1-KCNQ1 (open circles) currents of a representative cell (the same as in A). (Upper graph) Current amplitudes. (Lower graph) Current amplitudes after PIP2 addition normalized to their steady-state values after complete PIP2 effect. (C) hERG versus KCNE1-KCNQ1 normalized current amplitudes during PIP2 application (N = 11 cells). hERG and KCNE1-KCNQ1 currents are normalized to their steady state after PIP2 addition.
To further compare hERG and KCNE1-KCNQ1 PIP2 affinities, we applied polylysine (3 μg/mL) to shield PIP2 negative charges and to accelerate current rundown (5). This should minimize the bias of the KCNE1-KCNQ1 PIP2-independent rundown. Fig. 5 A shows that polylysine-triggered rundowns were similar in hERG and KCNE1-KCNQ1 within the first 20 s, but that hERG and KCNE1-KCNQ1 then diverged. KCNE1-KCNQ1 reached zero within 60 s, a time at which a fraction of hERG current persisted. This suggests a similar PIP2 sensitivity at high open probability, but a divergent PIP2 sensitivity at low open probability. To control that polylysine was specifically targeting PIP2, we checked whether deactivation was accelerated. Indeed, we observed an acceleration of deactivation similar to that observed during the spontaneous rundown (compare Figs. 2 and 5 B).
Figure 5.
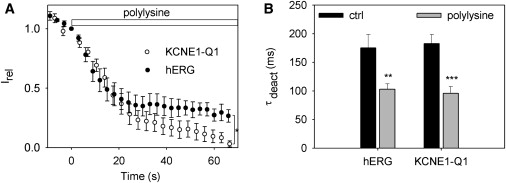
Comparison of the polylysine-induced rundown of hERG and KCNE1-KCNQ1 currents. (A) Time course of hERG and KCNE1-KCNQ1 current rundown after 3 μg/mL polylysine application at time 0. hERG current was evaluated from the peak tail current of a 1-s step at +30 mV followed by repolarization at –100 mV repeated every 3 s (N = 8 cells). KCNE1-KCNQ1 current was evaluated from the current at the end of a 1.5-s step at +40 mV followed by repolarization at –60 mV repeated every 3.5 s (N = 11 cells). ∗p < 0.05; two-way ANOVA. (B) Deactivation time constants obtained from a monoexponential fit of the deactivating tail current before (ctrl) and after the polylysine-induced rundown. For KCNQ1, deactivation was measured when a minimal current persisted (hERG, N = 8 cells; KCNQ1, N = 11 cells). ∗∗p < 0.01; ∗∗∗p < 0.001.
A final approach in comparing hERG and KCNE1-KCNQ1 PIP2 sensitivity was to apply different concentrations of Mg2+ to control the concentration of available PIP2 in the membrane patch. It has been shown in Kir channels that intracellular Mg2+ inhibition is correlated with the strength of channel-PIP2 interaction (19). Mg2+ inhibits the channel by screening the PIP2 negative charge in a manner similar to that of polylysine, but reversibly and in a concentration-dependent manner (20). We thus applied a range of Mg2+ concentrations to a patch—after addition of PIP2—to obtain a range of available PIP2 concentrations for hERG and KCNE1-KCNQ1 (Fig. 6, A and B). After stabilization of hERG maximal current, increasing doses of Mg2+ were applied to chelate PIP2, and this resulted in an increasing (and reversible) inhibition of hERG and KCNE1-KCNQ1 currents. Mg2+ dose-response curves are shown in Fig. 6 C. The KCNE1-KCNQ1 dose-response curve was close to that of hERG: the half-maximal inhibitory concentrations (IC50) for Mg2+ were 4.7 mmol/L and 4.8 mmol/L, with Hill coefficients of 0.8 and 1.0, for hERG and KCNE1-KCNQ1, respectively. To control that Mg2+ was specifically targeting PIP2, we checked whether deactivation was accelerated. Indeed, we observed an acceleration of deactivation similar to that observed during the spontaneous rundown (compare Fig. 2 and Fig. 6 D). These results indicate that the affinity of hERG for PIP2 is close to that of KCNE1-KCNQ1 and that hERG is likely to be sensitive to PIP2 variations in the same concentration range.
Figure 6.
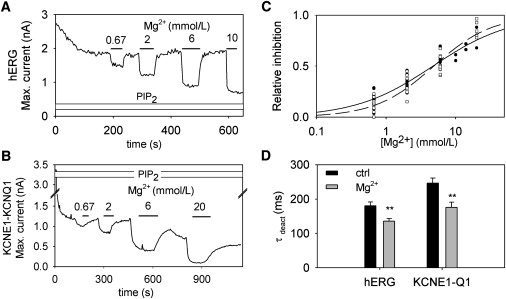
Mg2+ dose response of hERG and KCNE1-KCNQ1 in the presence of PIP2 (A) hERG maximal current of a representative cell. hERG maximal current was evaluated from the peak tail current of a 1-s step at 30 mV followed by repolarization at –100 mV repeated every 3 s. PIP2 was continuously applied. Various concentrations of Mg2+ were added as indicated. (B) KCNE1-KCNQ1 maximal current of a representative cell. KCNE1-KCNQ1 maximal current was evaluated from the current at the end of a 1.5-s step at +40 mV followed by repolarization at –60 mV repeated every 3.5 s. Various concentrations of Mg2+ were added as indicated. (C) Mg2+ dose response data from hERG (N = 10 cells) and KCNE1-KCNQ1 (N = 8 cells). Hill fits are presented for hERG (solid line) and KCNE1-KCNQ1 (dashed line). (D) Deactivation time constants obtained from a monoexponential fit of the deactivating tail current before (control) and after 6 mM Mg2+ application in hERG (N = 10 cells) and KCNE1-KCNQ1 (N = 8 cells). ∗∗p < 0.01.
Based on the convergent results of 1), cotransfection experiments, 2), polylysine effects, and 3), [PIP2] modulation by Mg2+, we concluded that hERG has a PIP2 sensitivity close to that of KCNE1-KCNQ1 and is thus likely to respond to physiological changes in PIP2 concentration. However, experiments with the spontaneous rundown and with polylysine clearly showed that a fraction of hERG current is poorly sensitive or insensitive to PIP2, which may lead to some difference in the channel physiological regulation.
Discussion
This study suggests that the molecular effect of PIP2, that is, a stabilization of the open state, is applicable to various, if not all, PIP2-sensitive potassium channels: the pore-forming subunit of the KATP channel Kir6.2, the voltage-dependent channels KCNQ1 (Kv7.1), and, as shown in this study, hERG (Kv11.1). In addition, this study suggests that both KCNQ1 and hERG are regulated by physiological PIP2 levels.
Comparison of the PIP2 effects observed in the whole-cell and giant-patch configurations
The activating effect of PIP2 on hERG has been demonstrated in CHO cells using the whole-cell configuration with PIP2 in the pipette (6) and in rabbit atrial myocytes submitted to α1A-adrenergic stimulation (21). The effect was clear but moderate. In this study, the giant-patch experiments on COS cells confirmed the activating effect of PIP2 on hERG and showed that PIP2 could strongly increase hERG current, notably by increasing its maximal amplitude by up to three times (Fig. 4 B). The changes in the recovery-from-inactivation time constant were also qualitatively the same as those measured in CHO cells using the whole-cell configuration, with PIP2 dialyzed from the pipette. However, the observed effect of PIP2 on half-activation potential and on deactivation and activation time constants differed largely between the two configurations. Two major differences between the whole-cell and giant-patch configurations may explain the divergent observations. 1), In the whole-cell configuration PIP2 is added in excess, causing an increase in membrane PIP2, whereas PIP2 levels are decreased in the giant-patch configuration. This may explain why deactivation is not modified in whole-cell experiments but is accelerated during rundown and slowed by PIP2 application in giant-patch experiments. Deactivation may be at its maximal level at physiological PIP2 concentrations, explaining the absence of effect when PIP2 is added in the pipette. 2), In the whole-cell configuration, it was not possible to distinguish between diffusion of PIP2 from the pipette and the dilution of many cytosolic molecules. In the giant-patch experiments, on the other hand, we were able to observe that the prominent acceleration of activation kinetics and the leftward shift of the activation curve caused by patch excision could not be recovered by PIP2 application, indicating that these processes are unrelated to PIP2 depletion. In a similar way, it is possible that in the whole-cell experiments the changes in these two parameters are not due to PIP2 depletion but rather to the dilution of cytosolic factors.
The biophysical effects of PIP2 on hERG current are consistent with a stabilization of the open state
To explore which state(s) is affected by PIP2, kinetic models of hERG were evaluated. The ongoing debate on whether or not channel activation and inactivation are coupled complicates the choice of the best model for testing the PIP2 effect (18). Therefore, several scenarios (no, weak, or strong coupling) were explored. Data from the uncoupled model are presented above.
The model shown in Fig. S3 is a weak inactivation/activation coupling model. The coupling is said to be weak because transition rates of the last activation step (αP and βP) do not depend on the inactivation state. The inactivation rates αI and βI were still set by the fit of the voltage-dependent inactivation rates and recovery from inactivation kinetics to limit the number of free parameters. The early voltage-dependent transition rates were also supposed to be linked to the voltage sensor and the late voltage-independent step to the pore. Based on the simulated currents and biophysical parameters (Fig. S3 and Table S2), it still seems likely that the late voltage-independent step controlled the PIP2 effect.
The model of Fig. S4 is a classical model of coupled inactivation. Late transitions are voltage-dependent and activation and inactivation processes cannot be clearly distinguished for these transitions. However, the early transitions may still be linked to the voltage sensor and the late transitions to the pore. There were no imposed inactivation parameters for this model. The late, but not the early, transition changes accounted well for the PIP2 effect. Altogether, all these models support the idea that PIP2 acts on late transitions and stabilizes the open pore conformation.
In all models (no, weak, and strong coupling), the best fit to the electrophysiological data was obtained when late transition rates probably corresponding to pore opening were optimized. Optimization of the voltage sensor transition rates resulted in an inadequate fit of the experimental data. This observation led us to conclude that PIP2 exerts its effect by acting on a late voltage-independent transition, favoring the open conformation of the pore domain. However, since optimizing all transition rates gave an equally satisfying result we cannot fully rule out that PIP2 also modulates other transition rates in addition to the late voltage-independent rate.
Physiological relevance of the PIP2 effect on hERG
To address the difficult question of the physiological relevance of the PIP2 effect on hERG, it is critical to evaluate the channel PIP2 sensitivity. To obtain a quantification of the sensitivity of a channel to PIP2, addition of soluble diC8-PIP2 is commonly used (5). To our surprise, hERG was shown to be weakly sensitive to diC8-PIP2 in our experiments, most probably because of the channel's lower affinity for this short-chain PIP2. Some experimental data from inward rectifiers show that the maximal effect of short-chain PIP2 is lower than that of PIP2, suggesting a lower affinity (22). To explore hERG sensitivity to native PIP2, we designed three experiments to compare the PIP2 sensitivity of hERG with that of KCNE1-KCNQ1, which is well characterized: 1), cytosolic PIP2 application on giant patches presenting both KCNE1-KCNQ1 and hERG channels showed a concomitant increase in both currents; 2), polylysine-induced rundown presented similar kinetics for both channels; and 3), concomitant addition of the phospholipid and various concentrations of Mg2+ controlled PIP2 levels, indicating a similar sensitivity of hERG and KCNE1-KCNQ1 channels to Mg2+. All these data support the idea that hERG and KCNE1-KCNQ1 channels have a similar affinity to PIP2. However, the experiments performed also shed light on the persistence of a fraction of hERG current at low PIP2 levels, which may give rise to a different response to physiological events, leading to a profound decrease in membrane PIP2 levels.
Acknowledgments
We thank Béatrice Leray, Marie-Joseph Louerat, Sylvie Leroux, and Agnes Carcouët for expert technical assistance.
This work was supported by grants from the Institut National de la Santé et de la Recherche Médicale (INSERM) and from the Agence Nationale de la Recherche to G.L. and N.R. (ANR-05-JCJC-0160-01) and to I.B. (ANR COD/A05045GS). G.L. and I.B. are recipients of a tenured position supported by the Centre National de la Recherche Scientifique (CNRS). J.P. was supported by the Association Française contre les Myopathies (AFM).
Supporting Material
References
- 1.McLaughlin S., Wang J., Murray D. PIP2 and proteins: interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 2002;31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 2.Park K.H., Piron J., Loussouarn G. Impaired KCNQ1-KCNE1 and phosphatidylinositol-4,5-bisphosphate interaction underlies the long QT syndrome. Circ. Res. 2005;96:730–739. doi: 10.1161/01.RES.0000161451.04649.a8. [DOI] [PubMed] [Google Scholar]
- 3.Huang C.L., Feng S., Hilgemann D.W. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- 4.Hilgemann D.W., Ball R. Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science. 1996;273:956–959. doi: 10.1126/science.273.5277.956. [DOI] [PubMed] [Google Scholar]
- 5.Zhang H., Craciun L.C., Logothetis D.E. PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron. 2003;37:963–975. doi: 10.1016/s0896-6273(03)00125-9. [DOI] [PubMed] [Google Scholar]
- 6.Bian J., Cui J., McDonald T.V. HERG K+ channel activity is regulated by changes in phosphatidyl inositol 4,5-bisphosphate. Circ. Res. 2001;89:1168–1176. doi: 10.1161/hh2401.101375. [DOI] [PubMed] [Google Scholar]
- 7.Enkvetchakul D., Loussouarn G., Nichols C.G. The kinetic and physical basis of KATP channel gating: toward a unified molecular understanding. Biophys. J. 2000;78:2334–2348. doi: 10.1016/S0006-3495(00)76779-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loussouarn G., Park K.H., Escande D. Phosphatidylinositol-4,5-bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage-gated potassium channels: a functional homology between voltage-gated and inward rectifier K+ channels. EMBO J. 2003;22:5412–5421. doi: 10.1093/emboj/cdg526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matavel A., Lopes C.M. PKC activation and PIP2 depletion underlie biphasic regulation of IKs by Gq-coupled receptors. J. Mol. Cell. Cardiol. 2009;46:704–712. doi: 10.1016/j.yjmcc.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gamper N., Li Y., Shapiro M.S. Structural requirements for differential sensitivity of KCNQ K+ channels to modulation by Ca2+/calmodulin. Mol. Biol. Cell. 2005;16:3538–3551. doi: 10.1091/mbc.E04-09-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang W., Xia J., Kass R.S. MinK-KvLQT1 fusion proteins, evidence for multiple stoichiometries of the assembled IsK channel. J. Biol. Chem. 1998;273:34069–34074. doi: 10.1074/jbc.273.51.34069. [DOI] [PubMed] [Google Scholar]
- 12.Hilgemann D.W. Giant excised cardiac sarcolemmal membrane patches: sodium and sodium-calcium exchange currents. Pflugers Arch. 1989;415:247–249. doi: 10.1007/BF00370601. [DOI] [PubMed] [Google Scholar]
- 13.Loussouarn G., Baró I., Escande D. KCNQ1 K+ channel-mediated cardiac channelopathies. Methods Mol. Biol. 2006;337:167–183. doi: 10.1385/1-59745-095-2:167. [DOI] [PubMed] [Google Scholar]
- 14.Kiehn J., Lacerda A.E., Brown A.M. Pathways of HERG inactivation. Am. J. Physiol. 1999;277:H199–H210. doi: 10.1152/ajpheart.1999.277.1.H199. [DOI] [PubMed] [Google Scholar]
- 15.Clancy C.E., Rudy Y. Cellular consequences of HERG mutations in the long QT syndrome: precursors to sudden cardiac death. Cardiovasc. Res. 2001;50:301–313. doi: 10.1016/s0008-6363(00)00293-5. [DOI] [PubMed] [Google Scholar]
- 16.Wang S., Liu S., Rasmusson R.L. A quantitative analysis of the activation and inactivation kinetics of HERG expressed in Xenopus oocytes. J. Physiol. 1997;502:45–60. doi: 10.1111/j.1469-7793.1997.045bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gagnon D.G., Bezanilla F. A single charged voltage sensor is capable of gating the Shaker K+ channel. J. Gen. Physiol. 2009;133:467–483. doi: 10.1085/jgp.200810082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choveau F.S., El Harchi A., Loussouarn G. Transfer of rolf S3-S4 linker to HERG eliminates activation gating but spares inactivation. Biophys. J. 2009;97:1323–1334. doi: 10.1016/j.bpj.2009.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du X., Zhang H., Logothetis D.E. Characteristic interactions with phosphatidylinositol 4,5-bisphosphate determine regulation of kir channels by diverse modulators. J. Biol. Chem. 2004;279:37271–37281. doi: 10.1074/jbc.M403413200. [DOI] [PubMed] [Google Scholar]
- 20.Suh B.C., Hille B. Electrostatic interaction of internal Mg2+ with membrane PIP2 seen with KCNQ K+ channels. J. Gen. Physiol. 2007;130:241–256. doi: 10.1085/jgp.200709821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bian J.S., McDonald T.V. Phosphatidylinositol 4,5-bisphosphate interactions with the HERG K+ channel. Pflugers Arch. 2007;455:105–113. doi: 10.1007/s00424-007-0292-5. [DOI] [PubMed] [Google Scholar]
- 22.Rohács T., Lopes C., Logothetis D.E. Assaying phosphatidylinositol bisphosphate regulation of potassium channels. Methods Enzymol. 2002;345:71–92. doi: 10.1016/s0076-6879(02)45008-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.