

Abstract
The fractional volume occupied by extracellular space in tissues, termed α, is an important parameter of tissue architecture that affects cellular functions and drug delivery. We report a technically simple fluorescent dye partitioning method to measure α in tissue slices based on microfiberoptic detection of dye fluorescence in tissue versus overlying solution. Microfiberoptic tip geometry and dyes were selected for α determination from fluorescence intensity ratios, without the need to correct for illumination profile, light scattering/absorption, or dye binding. The method was validated experimentally using cell-embedded gels of specified α-values and optical properties. In mouse brain slices, α was strongly location-dependent, ranging from 0.16 in thalamus to 0.22 in brainstem, and was sensitive to cell volume changes. Aquaporin-4 water channel gene deletion caused significant extracellular space expansion, with α = 0.181 ± 0.002 in cortex in wild-type mice and 0.211 ± 0.003 in Aquaporin-4 knockout mice. In slices of LLC1 cell tumors grown in mice to ∼5 mm diameter, α decreased remarkably from ∼0.45 in superficial tumor to <0.25 in deeper (>100 μm) tumor. Fluorescent dye partitioning with microfiberoptic detection permits rapid, accurate, and anisotropy-insensitive determination of α-values in tissue slices.
Introduction
The extracelluar space (ECS) is the region between cells in solid tissues, consisting of a jelly-like aqueous medium. The fraction of tissue volume that is extracellular, termed α, is an important determinant of solute diffusion between tissue microvasculature and cells, and therefore of cellular metabolism and drug delivery (1–3). In the brain, α is an important determinant of nonsynaptic cell-cell communication, as well as extracellular K+ and glutamate buffering after physiological and pathological neuroexcitation during seizures and cortical spreading depression (1,4). Information on α has come mainly from tetramethylammonium (TMA+) iontophoresis measurements in brain slices, where α-values in the range of 0.15–0.28 have been reported (5). The TMA+ method involves iontophoretic infusion of TMA+ into brain tissue through a micropipette, and microelectrode detection of [TMA+] using an ion-selective, dual-lumen micropipette near the delivery site (6). As described further in the Discussion, the TMA+ method is technically challenging, requires difficult-to-justify assumptions and complex analysis procedures, and is very sensitive to anisotropy in tissue structure. Limited and inconsistent information about α in tissues outside of the central nervous system is available, largely from earlier studies (3,7,8).
Here we report a simple optical method to measure α accurately in tissue slices, and apply the method to study α in brain and tumor tissue. As diagrammed in Fig. 1, the method relies on fluorescent dye equilibration between the aqueous ECS and the external solution overlying the slice. The ECS volume fraction α is equal to the ratio of dye molecules per unit volume in the slice to that in the external solution. The dye ratio is measured using an etched microfiberoptic with a micron-sized tip that is inserted into the tissue slice through the overlying solution. If the effective detection volume of the microfiberoptic is the same in the tissue and overlying solution, and if the fluorescent dye partitions evenly between the tissue ECS and the solution, then the fluorescence intensity ratio measured in the slice versus the solution is equal to α. This fluorescent dye partitioning with microfiberoptic detection (DPMD) method is validated herein, with consideration given to possible effects of tissue light scattering and absorbance, and fluorescent dye quenching/binding in the ECS. The DPMD method is conceptually and technically simple, and is readily implemented without the need for costly or specialized instrumentation.
Figure 1.
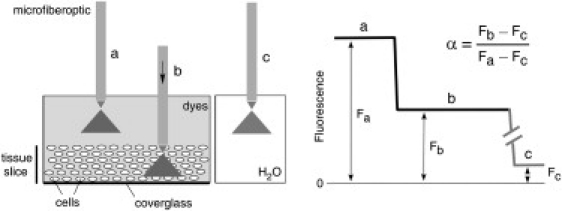
Principle of α determination by DPMD. Left: Microfiberoptic detection of the fluorescence of a noninteracting, aqueous-phase dye in the tissue slice and overlying solution. The illumination/detection volume is approximately conical, with most of the light collected from near the fiberoptic tip. Right: Schematic of DPMD data showing α determination from the ratio of the (background-subtracted) dye fluorescence in the slice to that in the overlying solution.
Materials and Methods
Instrumentation and measurement procedure for DPMD
As diagrammed in Fig. 1, a microfiberoptic was inserted into the dye-equilibrated tissue slice through the overlying solution. The distal end of a multimode fiberoptic (core diameter: 62.5 μm; Thorlabs, Newton, NJ) was stripped and chemically etched as previously described (9) to create a tapered tip down to ∼3 μm diameter. The tip was visualized using a Leica DM 4000B microscope (Leica, Wetzlar, Germany). Different fabricated microfiberoptics had slightly different detection efficiencies and background values, generally within 10%, because of differences in the exact tip geometry. Dye fluorescence was detected using an epifluorescence microscope (Nikon, Melville, NY) in which the excitation light was focused onto the back of the fiberoptic with a ferule connector using a 40× air objective lens (0.55 numerical aperture; Nikon) (Fig. 2).
Figure 2.
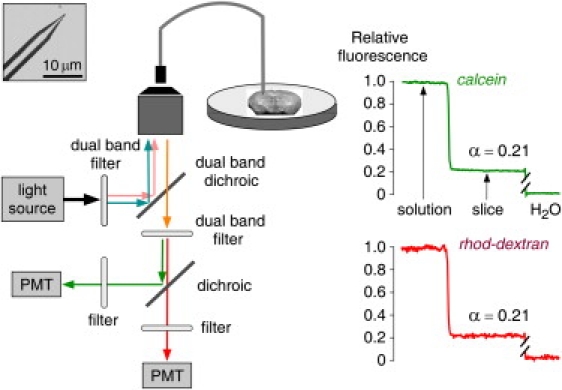
Instrumentation and procedure for α determination by DPMD. Left: A multimode etched microfiberoptic with micron-sized tip is inserted into the overlying solution and tissue slice. Inset shows the microfiberoptic tip geometry. Fluorescence from two probes of different colors is detected simultaneously using two photomultipliers (PMT). Right: Relative fluorescence of calcein (green) and rhod-dextran (red; normalized to unity in the overlying solution and zero in water) during insertion of the microfiberoptic into the overlying solution and into a brain slice (in hippocampus). Fluorescence ratios gave α = 0.21 for both colors.
Fluorescent dyes of two colors were detected simultaneously using a dual FITC/TRITC filter set (Chroma Technology, Rockingham, VT). The dual-excitation interference filter passes bands of 475–490 nm and 540–565 nm. The dual emission dichroic and emission filters enable detection of green and orange-red emission from the two dyes simultaneously with minimal cross talk. The filtered emission light was further split at 90° by a second dichroic mirror (580 nm; Chroma) and detected by two photomultipliers through interference filters (green, 540 ± 25 nm band-pass; red, 595 nm long-pass; Chroma). The amplified signals were digitized at 5 Hz.
The microfiberoptic was held vertically and positioned using a micromanipulator (World Precision Instruments, Sarasota, FL), allowing insertion to specific depths in tissue with an accuracy of ∼2 μm. Brain and tumor tissue slices were immobilized and incubated with dyes in an open bath slice chamber (RC-26; Warner Instruments, Hamden, CT) that was maintained at 37°C and surrounded by an enclosure saturated with humidified air or 95% O2/5% CO2.
Fluorescent dyes
Calcein, sulforhodamine 101, lucifer yellow, fluorescein-dextran (10 kDa), rodamine B dextran (10 kDa), and tetramethylrhodamine dextran (3 kDa) were purchased from Sigma (St. Louis, MO). Bodipy-FL-dextran (10 kDa) and bodipy-TX-dextran (10 kDa) were purchased from Invitrogen (Carlsbad, CA). Fluorophore selection was based on the following criteria: 1), cell impermeability; 2), noninteraction with the fiber tip; 3), nonbinding to the surface of cells; and 4), pH insensitivity. In some experiments, solution light scattering was increased by adding 0–16% (wt/vol) nonfat milk to phosphate-buffered saline (PBS), or solution light absorption was increased by adding 0–0.5% (vol/vol) India ink.
Cell-embedded gels
Cell-embedded agarose gels were prepared as previously described (10). SP 2/0 Ag14 cells (ATCC No. CRL-1581) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 2 mM L-glutamine, penicillin (100 U/mL), streptomycin (0.1 mg/mL), and 10% horse serum. These cells were chosen for their rapid growth in suspension medium and uniform spherical shape and size. After centrifugation, the cell pellet was suspended in Hank's balanced salt solution containing 0.3% low-melting-point agarose, which was allowed to solidify. Cell-embedded gels were kept in eight-well culture slides (BD, Franklin Lakes, NJ) in a humidified atmosphere to prevent drying, and used within 45 min after preparation. A sample taken before gelation was used for determination of cell density using a hemocytometer. The ECS volume fraction, α, was computed from the cell density and average cell volume (diameter 10.4 ± 0.4 μm, as determined by imaging). In some experiments, 15% wt/wt Ficoll-70 (Sigma) was added to the gel to increase the ECS viscosity.
Brain slices
Wild-type (WT) mice in a CD1 genetic background (age 8–12 weeks) were used to prepare brain slices. In comparative studies using age-matched WT and Aquaporin-4 (AQP4) null mice (11), the investigators were blinded to genotype information. All procedures were approved by the Committee on Animal Research, University of California, San Francisco. For preparation of brain slices, mice were anesthetized with 2,2,2-tribromoethanol (125 mg/kg intraperitoneal; Sigma) and decapitated. Coronal slices of 400 μm thickness were cut using a vibratome (Leica VT-1000S; Leica) in ice-cold sucrose-CerebroSpinal Fluid (CSF) containing (in mM) 206 sucrose, 2.8 KCl, 1 CaCl2, 1 MgCl2, 2 MgSO4, 1.25 NaH2PO4, 10 D-glucose, 10 Na pyruvate, and 26 NaHCO3 (320 mOsm osmolarity, pH 7.4, saturated with 5%CO2/95%O2). Slices were incubated in artificial CerebroSpinal Fluid (aCSF) consisting of (in mM) 124 NaCl, 2 CaCl2, 5 KCl, 1 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, 10 Na pyruvate, and 10 glucose, in 5% CO2/95% O2, for 2 h before measurements were obtained. In some experiments, N-methyl-D-aspartic acid (NMDA), ouabain, or mannitol (all from Sigma) was added to the aCSF. High-K+ CSF was made by replacing 50 mM NaCl in aCSF with 50 mM KCl.
Tumor generation in mice and preparation of tumor slices
Tumors were generated in female WT mice in a C57BL/6 genetic background. The mice were maintained in air-filtered cages and fed normal mouse chow in the University of California-San Francisco Animal Care facility. Tumors were implanted as described previously (9). Briefly, mouse Lewis lung carcinoma cells (LLC1 and ATCC CRL-1642) were cultured in DMEM supplemented with 4 mM L-glutamine, penicillin (100 U/mL), streptomycin (0.1 mg/mL), and 10% fetal bovine serum. LLC1 cells (106 cells in 200 μL PBS) were injected subcutaneously between the shoulder blades. Experiments were done 10–12 days postimplantation, at which time the tumor volume was 0.5–2 cm3. After the mice were anesthetized, the tumors were carefully isolated and embedded in 3% argarose gels to cut 400-μm-thick slices with the vibratome. The slices were incubated in Hank's balanced salt solution until measurements were obtained. After experiments were conducted, the tumor slices were fixed in formalin for preparation 7-μm-thick paraffin sections, which were stained with hematoxylin and eosin.
Analysis of DPMD data
As depicted in the Fig. 1 (right), α was computed from the ratio of dye fluorescence in slices (Fb) to that in the overlying solution (Fa) after subtraction of background fluorescence (Fc): α = (Fb − Fc) / (Fa − Fc). In the same slice, in the same general area of brain, measurement variability arises from the detailed ultrastructural difference in the illuminated areas. Therefore, at each location, two to four measurements were made, with the tip of the microfiberoptic moved 5–10 μm axially to avoid repeat measurements at the same location. In consecutive slices or slices from different mice, additional variability in measurements could result from differences in the quality of the brain slice preparation; therefore, at least four slices were studied at the same location. The α-values were first averaged from the same brain slice at the same region, and then averaged over different slices. Data from the green and red channels were averaged as well, after agreement was confirmed. Measurements were made at three to nine locations in each slice.
Results
Method development
Measurement of α in tissue slices by DPMD requires an appropriately shaped microfiberoptic and noninteracting fluorescent probes, such that the measured fluorescence signal is proportional to the aqueous-phase ECS volume fraction at the location of the microfiberoptic tip. As diagrammed in Fig. 1, a microfiberoptic with a micron-sized tip is inserted up to 200–300 μm beneath the surface of a tissue slice bathed under a thicker (>500 μm) solution layer. Various tip geometries, as produced by hydrofluoric acid etching of a 62.5-μm-core-diameter glass multimode fiberoptic, were tested for their ability to detect fluorescence at the tip and not along the shaft, and to have sufficient mechanical rigidity for tissue penetration. A smoothly tapered microfiberoptic tip shape worked well for this purpose (photo of tip shown in Fig. 2 A, inset). Previous in vivo microfiberoptic photobleaching studies utilized a microfiberoptic with a metal-coated shaft to ensure detection of signal from the tip only (9,12); however, a metal coating was not required for the modified tip geometry and tissue slice application used here.
The requirements for fluorescent probes in DPMD include high polarity and water solubility, lack of interaction with tissue components, and bright fluorescence. In addition, to ensure the robustness and accuracy of α measurements, suitable fluorescent probes with distinct fluorescence spectra were chosen for simultaneous determinations of α with two separate fluorescent probes. A series of small fluorophores (calcein, sulforhodamine 101, and lucifer yellow) and dextran-bound fluorophores (rhodamine B, Bodipy-fl, Bodipy-TX, fluorescein, and tetramethylrhodamine) were tested in cell-embedded gels as well as in brain and tumor tissue slices for accurate α measurement, signal stability, and complete washout. Whereas some of the dyes, such as sulforhodamine 101, were taken up by cells, other dyes, including Bodipy-fl, Bodipy-TX, and tetramethylrhodamine dextrans, slowly interacted with the microfiber tip, causing a baseline shift. Calcein and rhodamine B-dextran (rhod-dextran, 10 kDa) fulfilled the requirements and yielded consistent and stable α-values under the multiple conditions tested (see below). Fig. 2 shows a DPMD measurement of α in a brain slice from a WT mouse (in the hippocampus) bathed under physiological conditions in aCSF containing calcein (0.1 mM) and rhod-dextran (2 μg/μL). The green fluorescence and red fluorescence of calcein and rhod-dextran, respectively, were stable during insertion into the fluorescent solution overlying the brain slice, and then were reduced within the slice to ∼21% of that in the overlying solution. The fluorescence background, which was subtracted for the α computation, was <10% for both colors as measured in brain slices bathed in non-fluorophore-containing aCSF (or after washout of the fluorescently stained slices).
Experimental validation
Cell-embedded gels were used to model solid tissues having cellular and extracellular compartments, as described previously (10). Within the 0.3% agarose gels were suspended live spherical cells with α specified from cell density and size. Fig. 3 A (left) shows representative DPMD data for α = 1.0 (agarose gel not containing cells), 0.5, and 0.3. In each case, the measured α-values from fluorescence intensity ratios in gel versus solution, for both fluorescent probes, were in agreement with α-values deduced from cell density. Fig. 3 A (right) shows agreement between the experimentally measured α-values and those deduced from cell size and density. The measured α-values did not depend on the ECS viscosity, as verified when viscosity was increased fivefold by addition of 15% Ficoll-70 to the cell-embedded agarose gels (data not shown).
Figure 3.
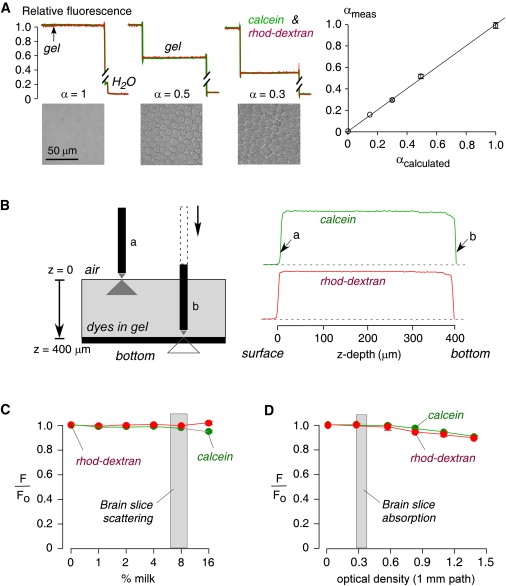
Experimental validation using cell-embedded gels and solutions simulating tissue optical properties. (A) Agarose gels containing SP2/0 cells giving specified α were bathed in PBS containing calcein and rhod-dextran. Top: Dye fluorescence shown during microfiberoptic insertion into the overlying solutions and gels. Bottom: Brightfield photographs show different cell densities, giving α = 1 (gel not containing cells), 0.5, and 0.3. Bar: 50 μm. Right: Experimentally measured (from fluorescence ratios) versus calculated (from cell density and size) α (SE, n = 4, except n = 2 for α = 0.15). (B) Effects of illumination/detection geometry. Left: A coverglass supports a 400-μm-thick agarose gel equilibrated with calcein and rhod-dextran in PBS. Right: Fluorescence shown during insertion of the microfiberoptic through the gel down to the bottom surface. The example is representative of six separate experiments. (C) Effect of light scattering. PBS/dye solutions were supplemented with nonfat milk (up to 16%) to produce light scattering. Calcein and rhod-dextran fluorescence changed by <1% for <12% milk (equivalent scattering from brain slices 7–10% milk, SE, n = 6 for each point). (D) Effect of light absorbance. PBS/dye solutions were supplemented with India ink (0–0.5%) to produce light absorbance (optical density at 1 mm pathlength from 0 to 1.5 at 595 nm). Calcein and rhod-dextran fluorescence changed by <1% for 0.2% India ink solution (equivalent optical density from brain slices: 0.3; SE, n = 6 for each point).
To investigate the z-distance dependence of our system (i e., the effective z-resolution), we measured fluorescence as a function of distance between the tip of the microfiberoptic and the coverglass supporting a 400-μm thick fluorescently stained agarose gel. Fig. 3 B shows constant fluorescence throughout the gel depth until the microfiberoptic tip is very close to the coverglass, where fluorescence is decreased because the illuminated volume includes the fluorescent gel and the nonfluorescent coverglass. The z-dependence of the measurement is determined by the cone-like illumination profile, the numerical aperture, and the ∼1/r2 detection efficiency (where r is the distance between the fluorescent probe and the microfiberoptic tip). We found from measurements such as that shown in Fig. 3 B that >90% of the collected fluorescence was within 3–5 μm of the microfiberoptic tip.
Light scattering and absorbance by tissue could affect the illumination and detection volumes, secondarily affecting measured fluorescence intensity. To investigate light scattering effects, nonfat milk (0–16%) was added to the PBS. Little effect (<1%) on the red and green fluorescence signals was seen for <12% milk, with small changes in signal at higher milk concentrations (Fig. 3 C). Brain slices had scattering comparable to 7% and 10% milk in gray and white matter, respectively, at 495 nm, as deduced from microfiberoptic scattering measurements done with monochromatic light and a 50/50 beamsplitter replacing the filter/dichroic-containing cube. To investigate absorbance effects, India ink (0–0.5%) was added to the PBS. Fig. 3 D shows little effect (<1%) on the red and green fluorescence signals for 0.3% India ink, which has an optical density of 0.8 (1 mm pathlength) at 595 nm. Brain and tumor tissue had an optical density of 0.38 (1 mm pathlength) at 595 nm, equivalent to ∼0.1% India ink solution, as deduced from absorbance measurements done on membrane-free supernatants of brain and tumor tissue homogenates. The results indicate that tissue light scattering or absorbance had little effect on α determination under the experimental conditions of our DPMD measurements.
Determination of α-values in brain slices
Brain slices of 400 μm thickness were generated from mice and processed using standard procedures to maintain their viability, as done for α measurements using the TMA+ method. The slices were immobilized in an open perfusion chamber, as diagrammed in Fig. 2 A. To determine the incubation time with fluorescent dye and ensure equilibration throughout the ECS, the time course of fluorescence was measured with the microfiberoptic tip inserted 200 μm into the brain slices. After the fluorescent probes were added to the aCSF bathing solution, the fluorescence of calcein and rhod-dextran was stable by 10 min, with 50% fluorescence equilibration in ∼1 min (Fig. 4 A). Fig. 4 B shows the time course for fluorescence dye washout, with complete washout by 20 min, indicating that the dyes were not significantly taken up by the cells under the conditions of the experiment.
Figure 4.
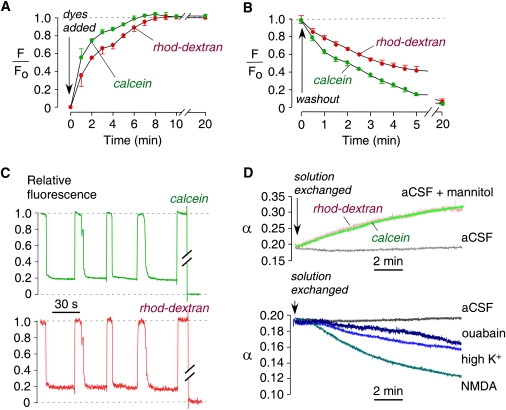
ECS volume measurements in brain slices from mice. (A) Equilibration time course of fluorescence after dye addition to the bathing solution. Microfiberoptic tip positioned at a depth of 200 μm in 400-μm-thick brain slices (SE, n = 5). (B) Washout time course of fluorescence (SE, n = 4). (C) Fluorescence of calcein and rhod-dextran during repeated microfiberoptic insertion into a brain slice (in cortex at 400 μm from brain surface). (D) Kinetics of changes in α in response to brain cell volume-altering maneuvers, including addition of 50 mM mannitol (top), 100 μM NMDA (bottom), 50 mM high K+ (bottom), and 100 μM ouabain (bottom). Control slices with stable α shown for comparison (labeled aCSF). Examples are representative of four or more separate experiments.
We first measured α in brain cortex, since α was previously measured in mouse brain cortex to be ∼0.18 using the TMA+ method (6) and ∼0.20 using the two-color pulsed-infusion microfiberoptic photodetection (PIMP) method (10). Fig. 4 C shows a representative measurement in which the microfiberoptic was repeatedly moved between the overlying solution (relative fluorescence: 1.0) and the brain slice at sites ∼400 μm from the brain surface. Data from both fluorescent dyes gave α = 0.19 ± 0.01 (standard error (SE), n = 4). Several maneuvers that should affect α by changing the brain cell volume were then tested. As expected, α increased in response to addition of mannitol to the aCSF, which osmotically reduced brain cell volume (Fig. 4 D, top), resulting in an increase in α from 0.19 to 0.32 over 10 min. In control slices (labeled aCSF), α remained constant. As expected, α decreased in response to maneuvers that cause brain cell swelling. High-dose NMDA, which activates glutamate receptors and causes neuronal death (13), decreased α from 0.20 to 0.12 over 10 min (Fig. 4 D, bottom). High-K+, acting by neuronal excitation, produced a slow decrease in α from 0.19 to 0.15, and ouabain, which inhibits Na+/K+ pumps in all cell types, caused a slowed decrease in α from 0.19 to 0.16.
The technical simplicity of DPMD allowed us to rapidly determine α in different regions of brain as identified in brain slices from mapped coordinates (and verified by visual identification of brain structures; Fig. 5 A, top). Since α determination by DPMD is not confounded by anisotropic tissue structure, we were able to measure α in regions of brain that have not previously been investigated. Representative DPMD data are shown for different regions of brain, with α-values summarized in Fig. 5 A (bottom). Data from red and green dyes produced consistent α-values in all cases. We found a quite remarkable variation of α-values in different regions of the brain. In gray matter, α was significantly higher in the hippocampus (0.207 ± 0.004) than in the cortex (0.194 ± 0.005), slightly higher in the striatum but not significantly so (0.205± 0.006), and considerably lower in the thalamus (0.170 ± 0.003). In the olfactory bulb, α was greater in the central layer (0.209 ± 0.010) than in the superficial glomerular layer (0.161 ± 0.005). In the white matter, such as brain stem, α was higher (0.216±0.006) than in gray matter.
Figure 5.
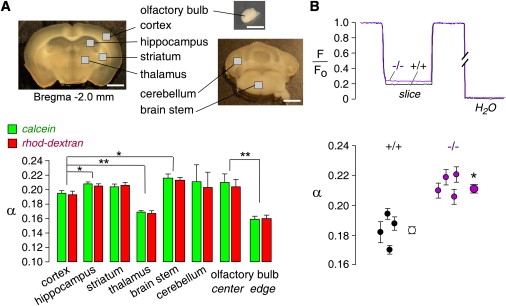
Regional variation of α in brain, and effects of AQP4 gene deletion. (A) Top: Photograph of brain slices with indicated measurement regions (bar: 1 mm). Bottom: Regional differences in α. Summary of α (SE, 6–10 slices per location, ∗p < 0.05, ∗∗p < 0.001) in indicated regions. (B) Effects of AQP4 gene deletion. Top: DPMD data comparing brain slices from WT and AQP4 null mice. Bottom: Summary of α in individual mice (SE, four slices per mouse, six measurements in cortical gray matter per slice), and averaged α for all slices (SE, four mice, ∗p < 0.001).
We compared α-values in the brain cortex of WT mice versus mice lacking AQP4, a glial water channel that has been implicated in brain edema, neural signal transduction, and glial cell migration (14). Previous studies showed increased solute diffusion in the ECS of brain cortex in AQP4 null mice (12,15) and an enlarged ECS (16). Representative data and deduced α-values in brain slices from individual WT and AQP4 null mice are summarized in Fig. 5 B. The averaged α-values were remarkably greater in brain cortex from AQP4 null mice (0.211 ± 0.003) compared to WT mice (0.181 ± 0.002; p < 0.001).
Determination of α-values in tumor tissue slices
We also used DPMD to measure α-values in slices from tumor tissue, using a model in which we previously showed greatly slowed dye diffusion in superficial versus deep tumor (9). Fig. 6 A shows a hematoxylin and eosin-stained section of the original LLC1 cell tumor (diameter: ∼5 mm) that was used to prepare slices in a direction transverse to the skin surface. The squares indicate the depths of microfiberoptic insertion. Insets a–f show the fine structure of each measurement region. In superficial tumor (Fig. 6 A, a, ∼50 μm from the tumor surface), cells were relatively less dense, with much matrix surrounding the cells. In deeper tumor, from 200 to 500 μm (Fig. 6 A, b and c), cells were more dense with less apparent ECS. At the center of the tumor (Fig. 6 A, d–f, 600 μm to 2 mm), the tumor showed necrosis with lower cellular density. Fig. 6 B summarizes α as a function of depth from the tumor surface, showing remarkably reduced α from 0.44 ± 0.04 in superficial tumor (50 μm) to 0.25 ± 0.03 in viable, highly cellular deeper tumor (200 μm). As expected, we found considerable heterogeneity in tumor sizes in different mice, with corresponding differences in the detailed α versus depth dependence; however, in each case the depth at which α changed correlated with histology.
Figure 6.
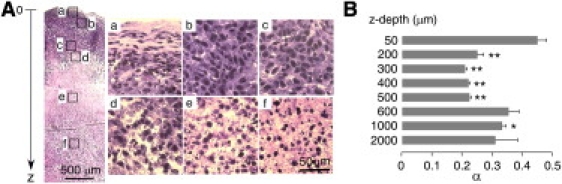
ECS volume measurement in tumor slices. Slices cut from a 5-mm-diameter subcutaneous tumor in a mouse at 12 days after injection of LLC1 cells. (A) Histology showing hematoxylin and eosin staining of a tumor slice in a direction transverse to the skin surface. Squares indicate sites of microfiberoptic insertion. Bar: 500 μm. Panels a–f show high-magnification views of microfiberoptic insertion areas. Bar: 50 μm. (B) Summary of α-values at different depths from the tumor surface (SE, 8–10 slices per location, ∗p < 0.05, ∗∗p < 0.001 compared with α at 50 μm). Representative of studies in tumors from four mice.
Discussion
For measuring α-values in brain slices, DPMD has significant advantages over the TMA+ method (6), which because of its technical complexity has been used by only two laboratories. The DPMD method is technically and conceptually simple, relying on the measurement of a steady-state fluorescence signal ratio to quantify dye partitioning. The dye-partitioning concept for experimental determination of α was introduced more than 35 years ago, and originally was applied in studies using various radioactive solutes to investigate the ECS in brain tissue (17,18). However, those earlier studies involved processing of fixed tissues and hence were subject to various artifacts and could not provide dynamic information about the ECS. In contrast to the TMA+ method, measurement of α by DPMD is not affected by tissue anisotropy, nor does it require mathematical deconvolution of complex TMA+ kinetic data to retrieve information on α, TMA+ diffusion, and TMA+ partitioning between cells and the ECS. Also, the TMA+ method is poorly suited for measuring α in anisotropic tissue where diffusion is direction-dependent, as found in many regions of the central nervous system. TMA+ measurements in anisotropic tissue require the acquisition of TMA+ kinetic measurements in multiple orientations of the triple-barrel microelectrode, and the use of simplifying assumptions about diffusion tensor symmetry. Determination of α by dye partitioning is not affected by tissue structure, ECS diffusion properties, or dye penetration into cells.
In developing the DPMD method, our main challenges were the fabrication of a suitable microfiberopic and the selection of noninteracting, membrane-impermeant fluorescent probes. A tapered microfiberoptic with a micron-sized tip, which was fabricated by hydrofluoric acid etching of a commercial multimode fiberoptic, was found to be suitable for α measurements in tissue slices without the need for metallic coating, resulting in a small measurement volume equivalent to a 3–5 μm apparent z-resolution. The fluorescent probes we selected for measuring α are well-established, membrane-impermeant dyes. They were fully washed out of the tissue slices and gave internally consistent α-values that agreed with previous measurements obtained in mouse brain cortex using the TMA+ and PIMP methods. The two-color epifluorescence detection system used here allows for simultaneous measurements of α with different probes to confidently determine α in tissue slices.
The application of DPMD for α measurements in different types of tissue requires validation of the fluorescent probes, as it cannot be assumed with certainty that the probes we found to be suitable for brain and tumor slices will not interact in the ECS in other tissues, such as kidney, lens, or certain tumors. DPMD further assumes free probe access throughout the ECS, which may not be the case in dense tissues with low α-values, such as the nuclear region of lens. Another limitation of the DPMD method is its time resolution, which is determined by fluorescent dye diffusion between the extracellular solution and the site of the microfiberoptic tip. The time resolution in brain slices was no better than ∼1 min. The DPMD method is not suitable for α determination in vivo, since convective effects and vascular transport preclude the assumption of uniform, passive dye partitioning. Finally, we note that DPMD provides no information about dye diffusion; however, DPMD can be readily combined with microfiberoptic photobleaching (9) to obtain information about fluorescent dye diffusion.
The brain slice measurements indicated a considerable heterogeneity in α-values. We found significantly greater α-values in white than in gray matter. Within gray matter, α was slightly greater in hippocampus, striatum, and cerebellum than in cerebral cortex. The lowest α-value was found in thalamus, which has a very high cell density. Previous TMA+ measurements showed α-values of 0.18–0.22 in rat and mouse brain cortex (1,5), 0.21 in rat brain striatum (19), and 0.18 in rat cerebellum (6), in general agreement with the data reported here. However, conflicting values were reported in rat hippocampus, with reported α-values of 0.20–0.22 (20), 0.13–0.14 (21), and 0.24 (22). Conflicting values were also found in highly anisotropic rat spinal cord white matter, with reported α-values ranging from 0.18 (23) to 0.27 (24). Our data show an average α-value of ∼0.21 in mouse hippocampus and ∼0.22 in brainstem white matter. The size and dimensions of the ECS are major determinants of tortuosity (25). Using microfiberoptic fluorescence photobleaching to study diffusion of macromolecules in the ECS in deep brain structures (12), we found relatively higher diffusion in hippocampus and lower diffusion in thalamus compared to cortex. These differences in diffusion are explained by the differences in α-values in these structures as found here.
The ECS in olfactory bulb has not been studied previously. We found that α was greater in the central layers of olfactory bulb (0.209 ± 0.010) than in the superficial glomerular layer (0.161 ± 0.005). The glomerular layer contains hundreds of glomeruli, and within each glomerulus thousands of receptor axons synapse with dendrites of neurons in a fine, dense meshwork (26). K+ accumulation and diffusion in the ECS of the glomerulus is important for odorant signal transduction. A small ECS, as found here, is predicted to accelerate changes in ECS K+ concentration in response to K+ transport between the intracellular space and the ECS.
Significant ECS expansion was found in the brain cortex of gray matter in AQP4-deficient mice. ECS expansion in AQP4 deficiency may account, in part, for several neuroexcitation-related phenotypes in AQP4-deficient mice, including altered seizure dynamics (15), and impaired auditory (27), olfactory (28), and visual (29) signal transduction. AQP4 is not expressed in electrically excitable cells but in adjacent supportive cells, such as astrocytes but not neurons in brain, and Müller cells but not bipolar cells in retina. We first observed brain ECS expansion in AQP4-deficient mice by cortical surface photobleaching measurements of FITC-dextran diffusion in live mice (30), which was further characterized by microfiberoptic photobleaching measurements and mathematical modeling (12,31). A study using the TMA+ method also reported mild ECS expansion in AQP4-deficient mice with α = 0.18 in brain cortex of WT mice and 0.23 in AQP4 knockout mice (16); however, we question the accuracy of those data because the expected corresponding changes in TMA+ diffusion were not found, and only a small percentage of analyzed tissues were deemed suitable for inclusion in the averaged data. Our findings here confirm and extend these prior studies.
Up to now, little information has been available about α in tumor tissue, and data in earlier studies were derived from partitioning of radioactive solutes in fixed tissues (32,33). In fibrosarcomas, the ECS volume fraction was estimated to vary between 0.33 and 0.6, whereas it varies between 0.2 and 0.4 in gliomas, and between 0.36 and 0.5 in carcinomas (34). Within the same fibrosacromas, the ECS volume fraction was reported to vary greatly from 0.4 to 0.8 in different tumor subregions (35), and an intratumoral variation of 0.05–0.7 was observed in melanomas (36). In two studies using the TMA+ method, ECS volume fractions of 0.22–0.48 were reported across a spectrum of human brain tumors, including astrocytomas, ependymomas, oligodendrogliomas, and medulloblastomas (37,38). Using microfiberoptic photobleaching in previous studies, we discovered a remarkable slowing of diffusion beyond a few millimeters for the tumor surface, where the majority of tumor matrix is located (9), and examined by enzymatic digestion the biochemical determinants of extracellular matrix diffusion (39). Here, using live tumor slices, we found remarkably different α-values in different regions of LLC1 tumor, with much lower values in superficial compared to deeper tumor. The lower α-values are correlated with a high density of cell bodies. As previously reported (9,39), extracellular matrix collagen-1 and decorin are enriched in deep LLC1 tumor, where much slower diffusion is found.
In conclusion, the technically and conceptually simple DPMD method reported here should be applicable to measurements of α in slices of various tissues and organs, enabling further investigation of the relatively understudied biology of the ECS.
Acknowledgments
This work was supported by grants EB00415, DK35124, EY13574, DK86125, DK72517, and HL73856 from the National Institutes of Health, and a grant from the Guthy-Jackson Charitable Foundation.
References
- 1.Syková E., Nicholson C. Diffusion in brain extracellular space. Physiol. Rev. 2008;88:1277–1340. doi: 10.1152/physrev.00027.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jain R.K. Delivery of molecular and cellular medicine to solid tumors. Adv. Drug Deliv. Rev. 1997;26:71–90. doi: 10.1016/s0169-409x(97)00027-6. [DOI] [PubMed] [Google Scholar]
- 3.Law R.O. Techniques and applications of extracellular space determination in mammalian tissues. Experientia. 1982;38:411–421. doi: 10.1007/BF01952615. [DOI] [PubMed] [Google Scholar]
- 4.Syková E. Diffusion properties of the brain in health and disease. Neurochem. Int. 2004;45:453–466. doi: 10.1016/j.neuint.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Nicholson C., Syková E. Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 1998;21:207–215. doi: 10.1016/s0166-2236(98)01261-2. [DOI] [PubMed] [Google Scholar]
- 6.Nicholson C., Phillips J.M. Ion diffusion modified by tortuosity and volume fraction in the extracellular microenvironment of the rat cerebellum. J. Physiol. 1981;321:225–257. doi: 10.1113/jphysiol.1981.sp013981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenberg L.E., Downing S.J., Segal S. Extracellular space estimation in rat kidney slices using C saccharides and phlorizin. Am. J. Physiol. 1962;202:800–804. doi: 10.1152/ajplegacy.1962.202.4.800. [DOI] [PubMed] [Google Scholar]
- 8.Jackson M.J., Cassidy M.M., Weller R.S. Studies on intestinal fluid transport. I. Estimation of the extracellular space of everted sacs of rat small intestine. Biochim. Biophys. Acta. 1970;211:425–435. doi: 10.1016/0005-2736(70)90248-8. [DOI] [PubMed] [Google Scholar]
- 9.Thiagarajah J.R., Kim J.K., Verkman A.S. Slowed diffusion in tumors revealed by microfiberoptic epifluorescence photobleaching. Nat. Methods. 2006;3:275–280. doi: 10.1038/nmeth863. [DOI] [PubMed] [Google Scholar]
- 10.Magzoub M., Zhang H., Verkman A.S. Extracellular space volume measured by two-color pulsed dye infusion with microfiberoptic fluorescence photodetection. Biophys. J. 2009;96:2382–2390. doi: 10.1016/j.bpj.2008.12.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma T., Yang B., Verkman A.S. Generation and phenotype of a transgenic knockout mouse lacking the mercurial-insensitive water channel aquaporin-4. J. Clin. Invest. 1997;100:957–962. doi: 10.1172/JCI231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zador Z., Magzoub M., Verkman A.S. Microfiberoptic fluorescence photobleaching reveals size-dependent macromolecule diffusion in extracellular space deep in brain. FASEB J. 2008;22:870–879. doi: 10.1096/fj.07-9468com. [DOI] [PubMed] [Google Scholar]
- 13.Choi D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 14.Verkman A.S., Binder D.K., Papadopoulos M.C. Three distinct roles of aquaporin-4 in brain function revealed by knockout mice. Biochim. Biophys. Acta. 2006;1758:1085–1093. doi: 10.1016/j.bbamem.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 15.Binder D.K., Yao X., Manley G.T. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia. 2006;53:631–636. doi: 10.1002/glia.20318. [DOI] [PubMed] [Google Scholar]
- 16.Yao X., Hrabetová S., Manley G.T. Aquaporin-4-deficient mice have increased extracellular space without tortuosity change. J. Neurosci. 2008;28:5460–5464. doi: 10.1523/JNEUROSCI.0257-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blasberg R.G., Patlak C., Fenstermacher J.D. Intrathecal chemotherapy: brain tissue profiles after ventriculocisternal perfusion. J. Pharmacol. Exp. Ther. 1975;195:73–83. [PubMed] [Google Scholar]
- 18.Levin V.A., Fenstermacher J.D., Patlak C.S. Sucrose and inulin space measurements of cerebral cortex in four mammalian species. Am. J. Physiol. 1970;219:1528–1533. doi: 10.1152/ajplegacy.1970.219.5.1528. [DOI] [PubMed] [Google Scholar]
- 19.Rice M.E., Nicholson C. Diffusion characteristics and extracellular volume fraction during normoxia and hypoxia in slices of rat neostriatum. J. Neurophysiol. 1991;65:264–272. doi: 10.1152/jn.1991.65.2.264. [DOI] [PubMed] [Google Scholar]
- 20.Mazel T., Simonová Z., Syková E. Diffusion heterogeneity and anisotropy in rat hippocampus. Neuroreport. 1998;9:1299–1304. doi: 10.1097/00001756-199805110-00008. [DOI] [PubMed] [Google Scholar]
- 21.McBain C.J., Traynelis S.F., Dingledine R. Regional variation of extracellular space in the hippocampus. Science. 1990;249:674–677. doi: 10.1126/science.2382142. [DOI] [PubMed] [Google Scholar]
- 22.Hrabetová S. Extracellular diffusion is fast and isotropic in the stratum radiatum of hippocampal CA1 region in rat brain slices. Hippocampus. 2005;15:441–450. doi: 10.1002/hipo.20068. [DOI] [PubMed] [Google Scholar]
- 23.Simonová Z., Svoboda J., Syková E. Changes of extracellular space volume and tortuosity in the spinal cord of Lewis rats with experimental autoimmune encephalomyelitis. Physiol. Res. 1996;45:11–22. [PubMed] [Google Scholar]
- 24.Prokopová S., Vargová L., Syková E. Heterogeneous and anisotropic diffusion in the developing rat spinal cord. Neuroreport. 1997;8:3527–3532. doi: 10.1097/00001756-199711100-00022. [DOI] [PubMed] [Google Scholar]
- 25.Hrabe J., Hrabetová S., Segeth K. A model of effective diffusion and tortuosity in the extracellular space of the brain. Biophys. J. 2004;87:1606–1617. doi: 10.1529/biophysj.103.039495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mori K., Nagao H., Yoshihara Y. The olfactory bulb: coding and processing of odor molecule information. Science. 1999;286:711–715. doi: 10.1126/science.286.5440.711. [DOI] [PubMed] [Google Scholar]
- 27.Li J., Verkman A.S. Impaired hearing in mice lacking aquaporin-4 water channels. J. Biol. Chem. 2001;276:31233–31237. doi: 10.1074/jbc.M104368200. [DOI] [PubMed] [Google Scholar]
- 28.Lu D.C., Zhang H., Verkman A.S. Impaired olfaction in mice lacking aquaporin-4 water channels. FASEB J. 2008;22:3216–3223. doi: 10.1096/fj.07-104836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J., Patil R.V., Verkman A.S. Mildly abnormal retinal function in transgenic mice without Müller cell aquaporin-4 water channels. Invest. Ophthalmol. Vis. Sci. 2002;43:573–579. [PubMed] [Google Scholar]
- 30.Binder D.K., Papadopoulos M.C., Verkman A.S. In vivo measurement of brain extracellular space diffusion by cortical surface photobleaching. J. Neurosci. 2004;24:8049–8056. doi: 10.1523/JNEUROSCI.2294-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin S., Zador Z., Verkman A.S. Random-walk model of diffusion in three dimensions in brain extracellular space: comparison with microfiberoptic photobleaching measurements. Biophys. J. 2008;95:1785–1794. doi: 10.1529/biophysj.108.131466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karlsson L., Alpsten M., Peterson H.I. Intratumor distribution of vascular and extravascular spaces. Microvasc. Res. 1980;19:71–79. doi: 10.1016/0026-2862(80)90084-9. [DOI] [PubMed] [Google Scholar]
- 33.Appelgren L., Peterson H.I., Rosengren B. Vascular and extravascular spaces in two transplantable tumors of the rat. Bibl. Anat. 1973;12:504–510. [PubMed] [Google Scholar]
- 34.Jain R.K. Transport of molecules in the tumor interstitium: a review. Cancer Res. 1987;47:3039–3051. [PubMed] [Google Scholar]
- 35.Krol A., Maresca J., Yuan F. Available volume fraction of macromolecules in the extravascular space of a fibrosarcoma: implications for drug delivery. Cancer Res. 1999;59:4136–4141. [PubMed] [Google Scholar]
- 36.Jakobsen I., Lyng H., Rofstad E.K. MRI of human tumor xenografts in vivo: proton relaxation times and extracellular tumor volume. Magn. Reson. Imaging. 1995;13:693–700. doi: 10.1016/0730-725x(95)00019-d. [DOI] [PubMed] [Google Scholar]
- 37.Zámecník J., Vargová L., Syková E. Extracellular matrix glycoproteins and diffusion barriers in human astrocytic tumours. Neuropathol. Appl. Neurobiol. 2004;30:338–350. doi: 10.1046/j.0305-1846.2003.00541.x. [DOI] [PubMed] [Google Scholar]
- 38.Vargová L., Homola A., Syková E. Diffusion parameters of the extracellular space in human gliomas. Glia. 2003;42:77–88. doi: 10.1002/glia.10204. [DOI] [PubMed] [Google Scholar]
- 39.Magzoub M., Jin S., Verkman A.S. Enhanced macromolecule diffusion deep in tumors after enzymatic digestion of extracellular matrix collagen and its associated proteoglycan decorin. FASEB J. 2008;22:276–284. doi: 10.1096/fj.07-9150com. [DOI] [PubMed] [Google Scholar]