

Abstract
Myelin basic protein (MBP) maintains the tight multilamellar compaction of the myelin sheath in the central nervous system through peripheral binding of adjacent lipid bilayers of oligodendrocytes. Myelin instability in multiple sclerosis (MS) is associated with the loss of positive charge in MBP as a result of posttranslational enzymatic deimination. A highly-conserved central membrane-binding fragment (murine N81-PVVHFFKNIVTPRTPPP-S99, identical to human N83-S101) represents a primary immunodominant epitope in MS. Previous low-resolution electron paramagnetic resonance measurements on the V83-T92 fragment, with Cys-mutations and spin-labeling that scanned the epitope, were consistent with it being a membrane-associated amphipathic α-helix. Pseudodeimination at several sites throughout the protein, all distal to the central segment, disrupted the α-helix at its amino-terminus and exposed it to proteases, representing a potential mechanism in the autoimmune pathogenesis of MS. Here, we have used magic-angle spinning solid-state NMR spectroscopy to characterize more precisely the molecular conformation and dynamics of this central immunodominant epitope of MBP in a lipid milieu, without Cys-substitution. Our solid-state NMR measurements have revealed that the α-helix present within the immunodominant epitope is shorter than originally modeled, and is independent of the pseudodeimination, highlighting the importance of the local hydrophobic effects in helix formation and stability. The main effect of pseudodeimination is to cause the cytoplasmic exposure of the fragment, potentially making it more accessible to proteolysis. These results are the first, to our knowledge, to provide atomic-level detail of a membrane-anchoring segment of MBP, and direct evidence of decreased MBP-membrane interaction after posttranslational modification.
Introduction
The myelin sheath is a multilamellar lipid membrane surrounding nerve axons in both the central and peripheral nervous systems (Fig. 1). Myelin basic protein (MBP) is a major protein component of the sheath in the brain and spinal cord. Its primary role is to maintain the compaction of myelin through peripheral association with the apposing cytoplasmic faces of the oligodendrocyte membrane (1–3) (Fig. 1). It is an intrinsically disordered protein which also interacts with many other proteins and ligands such as actin, tubulin, calmodulin, and SH3-domain containing proteins, thus potentially acting as a linker in structural and signaling networks (1,4–7).
Figure 1.
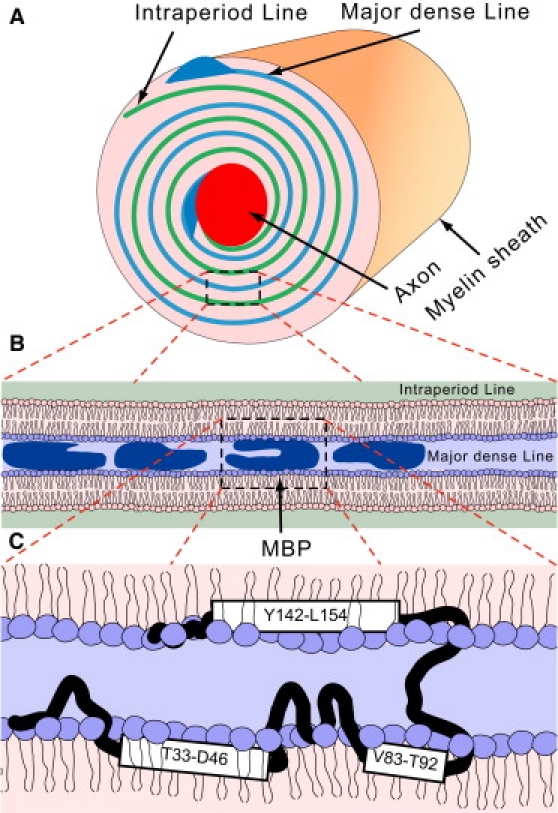
(A) Structure of the myelin sheath. (B) The myelin sheath consists of stacked lipid bilayers. Myelin basic protein peripherally binds the adjacent leaflets on the cytoplasmic side and acts as an adhesive protein. (C) There are three potential amphipathic helices in MBP shown in the figure, which may peripherally interact with lipids and represent putative lipid binding domains. The details of protein-lipid binding are not known, and the arrangement shown in this figure is one out of many possibilities.
The 18.5 kDa splice isoform of MBP is the most abundant in the adult human brain, and exists, due to a diversity of posttranslational modifications, as a number of charge components known as C1–C8, with net positive charge decreasing from +19 to <+13 at pH 7.0. Component C1 is the least modified, most cationic, and the most abundant variant in healthy adult myelin. Component C8 is characterized primarily by the enzymatic deimination of arginine to citrulline, reducing its net positive charge. The C8 component is found in elevated levels both in developing myelin in infants and in adults with the human inflammatory demyelinating disease multiple sclerosis (MS) (8,9). Our previous studies have used recombinant murine variants of these components, namely, unmodified rmC1 and pseudodeiminated rmC8 (Fig. 2), which are almost identical to the human forms (6,7), and which have properties similar to the natural proteins' forms C1 and C8 (10,11).
Figure 2.

Amino-acid sequences of recombinant murine 18.5 kDa rmC1 and rmC8 MBP variants. (Dashed rectangles) Gln substitutions (pseudodeimination) performed to produce rmC8. The highlighted region is the fragment examined here using MAS-SSNMR.
MBP adopts an extended conformation in aqueous solution, but gains local secondary structure upon interacting with binding partners, and in particular with lipids, which interaction is the focus of this article. The current portrayal of membrane-associated MBP derived from electron paramagnetic resonance (EPR) and solid-state NMR (SSNMR) measurements is depicted in Fig. 1. Hydrophobic moment analysis of the amino-acid sequence of MBP suggests the existence of three strongly amphipathic α-helices in the protein (12), located in the N- and C-termini and in the central region (Fig. 1 C). EPR measurements of the rmC1 variant (with single Cys-substitutions and spin labels distributed across the protein) have indicated that these regions are, indeed, embedded into the lipid membrane (13). Detailed EPR measurements of the central region comprising residues Pro82-Pro93 (murine 18.5 kDa sequence numbering), which represents the primary immunodominant epitope, were indeed consistent with this segment being an amphipathic α-helix lying on the membrane surface (14). Subsequent EPR measurements conducted on a pseudodeiminated MBP variant (1) further confirmed that the rmC8 variant was less-strongly associated with membranes along its C-terminal and central regions (13). In particular, the central fragment formed a two-residues' shorter α-helix comprising residues H85-T92 that was also more exposed (to proteases such as cathepsin D, for example) (15,16).
In this study, we have used magic-angle spinning (MAS) solid-state NMR (SSNMR) spectroscopy to determine the molecular conformation and to characterize the dynamics of the central immunodominant epitope of MBP in lipid milieu. Our SSNMR measurements did not require site-specific Cys-substitution and labeling, and have confirmed that the immunodominant epitope adopts an α-helical conformation, but that the helix appears to be approximately one-turn shorter than originally believed. Pseudodeimination does not affect the helical structure. However, it does cause the exposure of the epitope to solvent, making it more accessible to proteolysis.
Materials and Methods
Protein overexpression, isotopic labeling, and purification
Materials were as described previously (17) and in the Supporting Material. The unmodified classic 18.5 kDa recombinant murine myelin basic protein (rmC1), and its pseudodeiminated variant (rmC8), were expressed in Escherichia coli BL21-CodonPlus(DE3)-RP cells (Stratagene, La Jolla, CA) and purified as described previously (10,11). The 18.5 kDa isoform of MBP contains only three valines at the positions V83, V84, and V91, and two asparagines at N81 and N89. All these residues are located within the immunodominant epitope or in its immediate vicinity (Fig. 2). Detection of Val and Asn resonances can be used to study the secondary structure and dynamics of the epitope. The [U-13C,15N-Val,Asn, U-15N]-labeled recombinant murine C1 (rmC1-NV) protein for SSNMR spectroscopy was derived from cells grown in M9 medium supplemented with uniformly 13C,15N-labeled Asn and Val at 1 mM, along with 15NH4Cl and natural abundance glucose. The [U-13C,15N-Val,Asn, U-15N]-labeled recombinant murine C8 form (rmC8-NV) was prepared with the addition of labeled amino acids at 0.5 mM. Selective labeling of the Val and Asn residues in rmC1 and rmC8 was optimized as described in the Supporting Material. For both samples, the labeled amino acids were added to the growing bacterial culture 1 h before isopropyl-β-d-thiogalactopyranoside induction.
Preparation of samples for solid-state NMR spectroscopy
The procedures for making large unilamellar vesicles (LUVs) were adapted from previous investigations (13–15,17). Briefly, dry lipid powders were mixed at the desired molar ratios (see Table 1 for details on the samples used), and dissolved in a 2:1:1 (vol %) mixture of methanol/chloroform/ddH2O. The organic solvent was then dried under a stream of nitrogen gas, followed by an overnight drying under vacuum. The lipids were rehydrated by vigorous shaking overnight with two freeze-thaw cycles in a buffer containing 2 mM HEPES-NaOH, 100 mM NaCl, and 1 mM EDTA, at pH 7.6. A mini-extruder (Avanti Polar Lipids, Alabaster, AL) was used to extrude the hydrated lipid mixture through a 100-nm polycarbonate filter. To ensure a uniform size distribution of the lipid vesicles, a minimum of 35 extrusion cycles was employed, resulting in lipid vesicles of ∼100-nm diameter as confirmed by dynamic light scattering. The concentration of lipids in the final preparation after extrusion was estimated using a phosphorus assay (18,19).
Table 1.
Lipid compositions of LUVs and P/Ls of samples used for examination of protein-lipid vesicle aggregation conditions
| Sample | Protein | Lipid composition (molar ratio) | Initial P/L (mass ratio) | Final P/L (mass ratio) | mf (mg)∗ |
|---|---|---|---|---|---|
| A | rmC1 | 1:1 DMPG/DMPC | 1:1 | 1:1.7 | 0.8 |
| B | rmC1 | 1:4:5 DMPC/Chol/DMPG | 1:2 | 1:1.8 | 0.8 |
| C | rmC1 | 1:1 DMPG/DMPC | 1:2 | 1:1.6 | 4.5† |
| D | rmC8 | 1:1 DMPG/DMPC | 1:2 | 1:2.5 | 6.0† |
| E | rmC8 | 1.5:8.5 DMPG/DMPC | 1:6 | 1:6 | 0.8 |
LUVs, large unilamellar vesicles; P/L, protein/lipid ratio.
mf, final protein content in solid-state (SSNMR) samples.
For P/L aggregation and preliminary SSNMR testing, small-scale samples have been used; the final protein content reported in this table is for our final samples used throughout the remaining studies.
The typical reconstitution procedure was as follows. Protein and LUVs were diluted in the same buffer (2 mM HEPES-NaOH, 100 mM NaCl, and 1 mM EDTA at pH 7.6) to a final concentration of 0.5 mg/mL. Aliquots of protein and lipid stocks were loaded into a 2 mL microfuge tube, mixed by inversion, and incubated at room temperature for 2 h. The protein/lipid aggregates were then collected by centrifugation at 18,000 × g for 2 h. The supernatants and pellets were separated and the protein and lipid contents were estimated using the standard bicinchoninic acid and phosphorus assays (18–21), respectively, as described previously (17). The pellets obtained were then center-packed into a standard 3.2-mm MAS rotor for examination using SSNMR. 31P measurements conducted previously confirmed the lamellar phase of lipids (17).
After several SSNMR investigations on reconstituted MBP/LUV samples with different lipid compositions and initial protein/lipid mass ratios, as described in Results and Discussion, we determined that the epitope conformation was independent of the protein/lipid and 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) to 1,2-dimyristoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]) (DMPG) ratios. Our final rmC1 sample was prepared from 1:1 (molar ratio) DMPG/DMPC large unilamellar vesicles (LUVs) mixed with protein at a 1:2 initial protein/lipid mass ratio. The protein-lipid aggregate was collected by ultracentrifugation at 100,000 × g for 80 min. Approximately 4.5 mg of rmC1 (1:46 protein/lipid molar ratio) was thus packed in a 3.2-mm rotor.
The less positively-charged rmC8 variant (+13 compared to +19 for rmC1) had a naturally reduced tendency to aggregate lipid vesicles, which resulted in the reduction of protein content in the lipid aggregate. The preparation procedure was similar to that of rmC1, but we used larger initial amounts of protein and lipid: 9 mg of protein dissolved in 18 mL of buffer, and 18 mg of lipids diluted in 36 mL of buffer, were mixed. Approximately 6 mg (1:72 protein/lipid molar ratio) of rmC8 was packed into a thin-walled 3.2-mm MAS rotor (Bruker BioSpin, Billerica, MA). Both samples were fully hydrated as evident, for example, from the presence of a strong water line in the proton NMR spectra (data not shown), as well from our previously published water-MBP correlation spectra (17).
Solid-state NMR spectroscopy and secondary structure propensities
All experiments were performed on Avance III spectrometers (Bruker Optics, Billerica, MA) operating at 600.13 and 800.23 MHz proton frequencies, and equipped with a triple-resonance 1H-13C-15N 3.2 mm E-Free MAS probe (Bruker BioSpin) (22). The one-dimensional 13C spectra were collected using either the ramped version of cross-polarization (CP) (23), followed by an SPC53-based double-quantum filter (DQF) (24,25) used to eliminate the natural abundance 13C signal arising both from lipids and unlabeled parts of the protein, or through-bond insensitive nuclear enhancement by polarization transfer (INEPT) (26) excitations.
The two-dimensional 13C-13C correlation spectra were recorded using through-space (dipolar-assisted-rotational resonance, or spin diffusion) transfer mechanisms (27). All 13C-13C correlation spectra were acquired at a MAS rate of 12 kHz and with a recycle delay of 1.9 s. All pulse sequences were modified to start with 1H/13C CP and a π/2 13C pulse to ensure that both mobile and immobile parts of the protein were excited with maximum efficiency. The one-dimensional (CON)CA and (CANCO)CA spectra and two-dimensional 13C-15N N(CA)CX and (CO)N(CO)CA correlation spectra were collected at 240 K. Typical field strengths were 80 kHz for small-phase-incremental-alteration-heteronuclear-dipolar-decoupling-scheme-with-64-composite-pulses (SPINAL-64) decoupling (28), and 42 kHz, 30 kHz, and 90 kHz for 13C, 15N, and 1H fields, respectively, during 15N/13C CP. Typical 90° pulse lengths used were 2.78 μs, 5 μs, and 6 μs for proton, carbon, and nitrogen, respectively. Additional experimental details are given in Table S1 and Table S2 in the Supporting Material.
All two-dimensional spectra were processed with NMRPipe (29) using π/2-shifted sine-squared apodization for both dimensions before Fourier transformation. Carbon chemical shifts were indirectly referenced to 2,2-dimethyl-2-silapentane-5-sulfonic acid through the 13C adamantane downfield peak resonating at 40.48 ppm (30). Nitrogen chemical shifts were referenced indirectly by using the ratio of γN/γC = 0.402979946 (31). All temperature values reported here represent sample temperatures.
The chemical shift deviations were used to assess the secondary structure propensities along the backbone of both rmC1 and rmC8 within the assigned residues (32,33). Random coil values (34) were corrected for sequence dependence (35). We used the Cα secondary shifts only, because they were the least sequence-dependent of all resonances.
Results and Discussion
Sample reconstitution—protein labeling and choice of lipid composition
Two samples were analyzed by one- and two-dimensional SSNMR spectroscopy:
-
1.
Unmodified [U-13C,15N-Val,Asn, U-15N]-MBP (rmC1-NV, recombinant murine C1 form).
-
2.
Pseudodeiminated [U-13C,15N-Val,Asn, U-15N]-MBP (rmC8-NV, recombinant murine C8 form).
The amino-acid sequences of both proteins are shown in Fig. 2. We have previously shown that rmC1 has similar properties to the natural brain-derived C1 form, in particular lipid aggregation properties (11). The rmC8 variant differs from rmC1 by six citrulline-mimicking substitutions (R23Q, R31Q, K119Q, R127Q, R157Q, and R168Q), which reduce the overall positive charge of the protein, and which have been shown to result in similar membrane- and protein-interaction properties to the natural C8 component (12,36).
Our previous SSNMR studies of MBP have shown an overall high degree of mobility of the protein in the lipid environment (17). In INEPT-based experiments specifically optimized for detection of mobile fragments, the central region (residues K68–K102) containing the lipid-bound central epitope could not be observed. As mentioned previously, both Asn and all three Val residues in the entire protein are in or adjacent to the immunodominant epitope (Fig. 2). We thus labeled these residues selectively with 13C, and the whole protein uniformly with 15N, after optimization as described in Supporting Material.
The myelin sheath in the central nervous system is a complicated mixture of lipids, and includes phospholipids with negatively-charged headgroups (13% of phosphatidylserine and 2% of phosphatidylinositol, molar ratio), with neutral headgroups (27% of phosphatidylethanolamine and 11% of phosphatidylcholine), sphingomyelin (3%), and cholesterol (44%) (37). Many of these lipids have unsaturated bonds, which are not stable on the timescale of a typical SSNMR experiment. Thus, we also evaluated the effects of lipid composition on protein conformation.
For initial evaluations of conditions, five LUV samples (A–E) were prepared with lipids of different types, varying proportion of charged lipids, and with various protein/lipid (P/L) ratios to test the effect of lipid composition and P/L on the MBP conformation (Table 1). For sensitivity considerations, we only collected one-dimensional spectra in these preliminary trials. Although these experiments did not permit detailed site-specific assessment, we expected that any spectral changes would serve as an indicator of large conformational rearrangements induced by lipids or by high protein/lipid ratio.
Fig. 3 A compares a series of DQF spectra of rmC1 obtained in different lipid compositions (Table 1, samples A–C). In these spectra, we observed signals from labeled amino acids in the protein, primarily asparagines and valines located in the immunodominant epitope of MBP, as well as contributions from scrambled amino acids (only pairs of directly bonded 13C atoms contributed to the DQF signal). All DQ-filtered spectra collected at −25°C were practically identical for all three systems, implying that the change of lipid composition and protein/lipid had minimal effect on the conformation of membrane-associated rmC1.
Figure 3.

The one-dimensional 13C spectra for rmC1 and rmC8 reconstituted in membranes of different lipid compositions and at different P/Ls. (A) The CPMAS DQF spectra for three rmC1 samples A–C (Table 1). (B) The CPMAS DQF spectra for two rmC8 samples D and E (Table 1). All spectra were acquired at −25°C and processed with a 100-Hz exponential window function before Fourier transformation. The spectra are scaled as indicated by the multiplication factors in both panels to bring them to equal heights for better visual comparison.
Fig. 3 B examines the effect of lipid composition and of protein/lipid ratios on rmC8 (Table 1, samples D and E). The appearance of the DQF spectrum is again identical for both samples, and is also similar to the DQF spectra of rmC1 in Fig. 3 A. Thus, these spectra indicate that at low temperature the conformations of labeled residues, primarily Val and Asn, are independent of the lipid composition, protein/lipid ratio, and the overall charge of the protein. In the following experiments, we have chosen samples with 1:1 DMPG/DMPC LUVs as a stable model (on the timescale of SSNMR experiments) to mimic the myelin sheath composition.
SSNMR spectra identify the presence of a short α-helical structure within the epitope in both unmodified and pseudodeiminated forms
Site-specific assignments of backbone 13C and 15N resonances in Val and Asn residues were obtained using a combination of spectral-edited one-dimensional spectra and two-dimensional 13C-13C correlation spectroscopy. The initial set of measurements was conducted at −25°C. Possible conformational changes induced by temperature-dependent lipid phase transition were studied by following a gradual change in peak positions in the two-dimensional 13C-13C spectra.
The Cα resonances of Val83 and Val84 were assigned using one-dimensional edited experiments (CON)CA and (CANCO)CA, in which polarization was transferred in a number of selective transfer steps between two sequentially labeled residues. Because Val83-Val84 was the only pair of sequentially labeled amino acids (Fig. 2), the (CON)CA and (CANCO)CA experiments allowed selective observation of the Val83Cα and Val84Cα peaks. The corresponding spectra are shown in Fig. 4 A, where the Val83Cα and Val84Cα peaks are clearly visible above the background of resonances from residually labeled carbon atoms.
Figure 4.

(A) One-dimensional 13C (CON)CA and (CANCO)CA spectra used to assign the Cα peaks of both Val83 and Val84 in membrane-associated rmC1. Both spectra were acquired at −25°C and processed with 100-Hz exponential line broadening before Fourier transformation. Because there is only one isotopically labeled pair of adjacent residues in the rmC1 and rmC8 sequences, both spectra should ideally display only one peak each. Because of scrambling during selective-labeling, there are many other overlapping residues that show up in the range from 52 to 62 ppm. (B) Two-dimensional (CO)NCOCA correlation spectrum allowing identification of the Asn81Cα chemical shift. (C) Two-dimensional 600 MHz 13C-13C correlation spectrum of membrane-associated rmC1 recorded at −25°C. (D) Sequence-corrected chemical shift deviations of Cα atoms of membrane-associated rmC1 and rmC8, at −25°C (shown in black). (Dark shaded bars) One standard deviation of α-helical ΔδCα shifts. (Light shaded bars) One standard deviation of β-sheet ΔδCα shifts.
Of the two asparagines in the MBP sequence, only Asn81 precedes proline (Fig. 2). The Asn81Cα peak was assigned from the two-dimensional (CO)N(CO)CA spectrum. The H-C′-N excitation was found to be more efficient for the excitation of the proline backbone 15N resonance than the direct H-N transfer. The (CO)N(CO)CA spectrum in Fig. 4 B shows a pronounced peak at 137.6/52.1 ppm, which corresponds to a Pro82N/Asn81Cα correlation.
The assignments for the remaining Val91 and Asn89 were obtained from the two-dimensional 13C-13C dipolar-assisted-rotational resonance correlation spectrum shown in Fig. 4 C. The spectrum displays three peaks in a valine Cα/Cβ fingerprint region centered at ∼F1/F2 of 64/32 ppm. The already assigned Val83 and Val84 Cα resonances result in two overlapping peaks at F1/F2 of 66.6/31.5 ppm. The third valine correlation at F1/F2 of 62.3/32.1 ppm corresponds to Val91. Because of the low degree of 13C labeling of proline (∼18% as evident from solution NMR results discussed in Supporting Material), we ruled out the possibility that these cross-peaks could also be due to Pro Cα/Cβ correlations.
Similarly, the correlation at F1/F2 at ∼52.7:40.5 ppm corresponds to the asparagine Cα/Cβ correlations. The volume of this peak is approximately equal to that of the Val83/Val84 Cα/Cβ overlapping correlations, and is approximately two-times larger than the assigned Val91 Cα/Cβ cross-peak. Thus, despite its appearance as a single peak, it likely results from two asparagines, Asn81 and Asn89. One of these resonances has already been assigned to Asn81 from the (CO)N(CO)CA experiment, and the other must be due to Asn89, as both Leu and Asp (the other possible candidates) showed very low levels of labeling at Cα positions.
A similar set of experiments on the reduced charge variant rmC8 resulted in nearly identical chemical shifts for Asn81, Val83, Val84, Asn89, and Val91, in complete agreement with nearly identical DQF one-dimensional spectra of these two samples. Thus, our results indicate that at low temperature, the Asn81-Thr92 fragment adopts a very similar overall conformation in both rmC1 and rmC8.
We finally note that the two-dimensional 13C-13C correlation spectrum (Fig. 4 C) shows a large number of additional cross-peaks between residually labeled carbon atoms of most of the residues. The most pronounced are those corresponding to Leu side-chain Cβ/Cγ correlations at ∼F1/F2 of 41.4/26.6 ppm, Cγ/Cδ correlations at F1/F2 of 26.6/22.7 ppm, and Cβ/Cδ correlations at F1/F2 of 41.4/22.7 ppm. As discussed in the Supporting Material, we do not expect any Leu correlations involving Cα, as these atoms remain unlabeled. Some Val Cα/Cγ and Thr Cβ/Cγ correlations are also detected at ∼F1/F2 of 67.1/22.4 ppm. These residual cross-peaks do not interfere with the Val and Asn correlations of interest.
NMR chemical shifts are sensitive reporters on local secondary structure. We used Cα chemical shifts of the assigned residues to compare the backbone conformation of the immunodominant epitope for both rmC1 and pseudodeiminated rmC8 reconstituted systems. The Cα secondary shifts (i.e., the differences between the experimentally determined chemical shifts and their random coil values) for both rmC1 and rmC8 at −25°C are shown in Fig. 4 C. Overall, the plot demonstrates clearly that the immunodominant epitope adopts a very similar conformation in both protein variants. The Cα secondary shifts of both Val83 and Val84 are strongly positive, of ∼5 ppm, indicating their strongly helical conformations. The Val83-Val84 pair is only three residues' away from a strongly hydrophobic Phe86-Phe87 pair, and both pairs likely face the hydrophobic core of the bilayer (14). On the other hand, Val91 and both Asn81 and Asn89 have only slightly positive secondary chemical shifts, and define the helical boundaries for the epitope. We therefore conclude that the α-helix likely comprises residues Val83-Lys88 in both rmC1 and rmC8 variants, and its structure in this environment is unaffected by the pseudodeimination at sites distal to it.
Secondary structure and dynamics of the epitope in the lipid fluid phase
The depth of penetration of MBP fragments into a lipid bilayer may depend not only on protein properties such as local hydrophobicity and overall charge, but also on the state of the lipids (1,13). In particular, MBP may preferentially bind the fluid domains of the membrane (38), and can be squeezed out of the membrane when lipids are in the gel phase. To obtain structural and dynamic information on the behavior of the epitope region of the protein at different lipid states, we collected a series of two-dimensional 13C-13C correlation spectra in the temperature range from −25°C to 35°C. The resonance assignments of all 13Cα shifts were obtained by tracking the gradual change in cross-peak positions as a function of temperature. As we reported previously, solvent-exposed fragments of MBP are highly dynamic. Although the membrane-associated immunodominant epitope is significantly less mobile than the solvent-exposed parts, it too experiences dynamic motions, which result in effective attenuation of dipole-dipole interactions and consequently in the reduction of cross-peaks intensities in the dipolar based correlation spectra. Nonetheless, the observed correlations allow us to draw conclusions about the temperature-induced effects.
The overall structure of carbon-carbon correlation spectra remained unchanged: both Asn and Val Cα/Cβ correlations remained pronounced, albeit of reduced amplitudes. The ratio between Val and Asn Cα/Cβ peak volumes was ∼3:2 throughout the entire probed temperature range in accordance with the expected number of correlations, suggesting that all the observed residues in the epitope are subjected to the same dynamics. Although these observations are of a semiquantitative nature, they indicate that all three valines and two asparagines can be observed in the spectra.
On the other hand, while an overall two-dimensional shape of Val Cα/Cβ cross peaks does not change (Fig. 5 A), we have observed significant redistribution of peak intensities in this region in both rmC1 and rmC8 samples. Specifically, the intensity of the Val83/Val84 overlapping correlation decreases, while the intensity of Val91 increases (Fig. 5 B), indicating possible conformational rearrangements within the epitope—for example, changes in conformation of V83 and/or V84. Although the assignment of Val83 and Val84 Cα resonances is ambiguous at high temperature, we note that Val83 is more likely to undergo stronger changes, as it is located at the N-terminal end of the helix.
Figure 5.
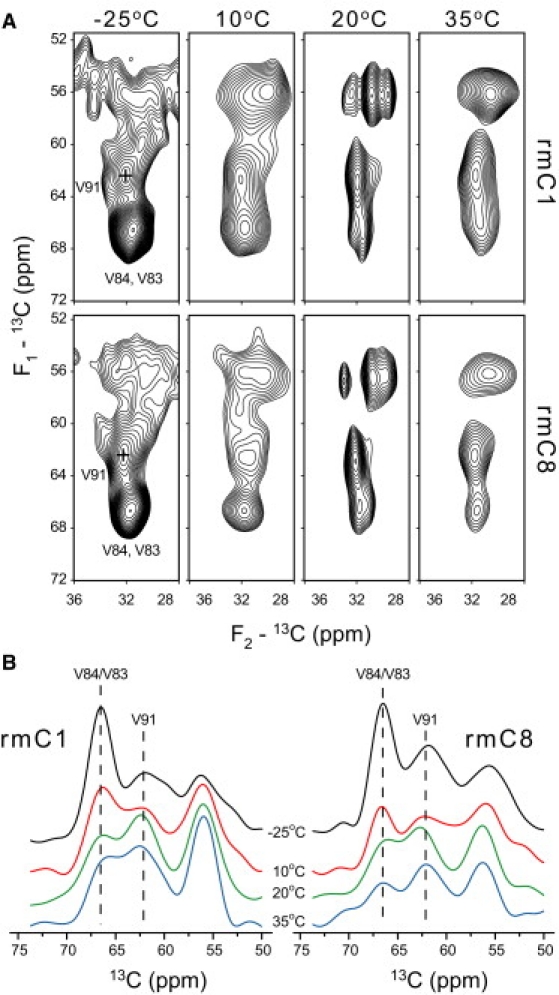
(A) The Val Cα/Cβ correlation region of a 13C-13C correlation spectrum as a function of temperature. V91 and V83 positions and V84 correlations are shown in the low temperature spectrum. (B) Projections of carbon correlations in the Val Cα/Cβ region for both rmC1 and rmC8 at all four temperatures, showing redistribution of intensities from the Val83/Val84 Cα to the Val91 Cα region.
Mobility of the immunodominant epitope in rmC1 and rmC8
The line widths of Cα and Cβ resonances of the epitope residues remained heterogeneously broadened on the order of 2 ppm in both rmC1 and rmC8. Thus, the conformational motions were unable to average this heterogeneity. Additional evidence of strong motions in rmC1 and rmC8 was derived from proton-carbon heteronuclear chemical-shift correlation spectra recorded at 35°C using INEPT (through-bond) and ramped CP (through-space) as mixing methods. Both methods yielded similar correlation patterns as shown for the case of INEPT in Fig. 6 for both Asn and Val-labeled rmC1 and rmC8 samples, along with the spectra of samples prepared with natural abundance rmC1 and rmC8 proteins. The correlations from natural abundance protein samples result only from lipids and help identify the protein-specific signals, which could be assigned by simply matching 13C shifts. In particular, the Val91 Cα/Hα correlation is clearly distinguished in both samples. The proton line-width of this peak in rmC1 is ∼570 Hz (Fig. 6), dramatically reduced compared to a typical 1H line-width in rigid solids where dipolar broadening is approximately tens of kilohertz (39). This line-width reduction indicates the presence of extensive molecular motions. The proton line-width of Val91 in rmC8 becomes even smaller than in rmC1, ∼260 Hz, indicating a further increase in mobility. Thus, the immunodominant epitope appears to be less motionally restricted in rmC8, despite having a very similar molecular conformation.
Figure 6.
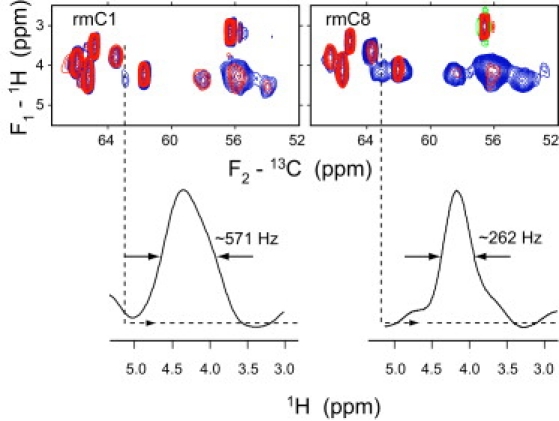
Two-dimensional 1H-13C INEPT heteronuclear chemical-shift correlation spectra in [13-C-Asn,Val,U-15N]-labeled rmC1 and rmC8. (Red spectra) Samples with unlabeled protein, showing lipid correlations. The one-dimensional 1H traces at the position of V91Cα are shown at the bottom for both spectra.
Significance and Conclusions
Multiple sclerosis (MS) is a human demyelinating disease of multifactorial origin. The severity of the disease has been correlated with elevated levels of the C8 component of MBP, which is the least cationic of all MBP variants. An overall charge reduction in deiminated forms of MBP has an effect on the protein/lipid interactions strengths, and may be responsible for modulating the compactness of the myelin sheath. The importance of understanding of MBP-lipid interactions has prompted numerous structural studies of MBP and of its lipid-binding domains. Extensive biochemical and biophysical studies have revealed that the least-modified C1 component of MBP contains three lipid-binding fragments: in addition to the immunodominant epitope studied in this work, two additional fragments are located in the C- and N-termini, while the remaining parts of the protein remain solvent-exposed, mobile, and unstructured.
The main finding of this study, shown in Fig. 7, is that the immunodominant epitope contains a short α-helix encompassing residues Val83-Lys88 in both rmC1 and rmC8 MBP variants (which may even be shorter at physiologically relevant temperatures), and is not sensitive to the overall electrostatic charge of the protein in this reconstituted system. Indeed, the nearest deimination site to the immunodominant epitope is at the position of 119 where Lys119 in rmC1 is replaced by Q119 in rmC8 (Fig. 2). This site is 25 residues away from the epitope. Given that MBP remains mobile and largely unstructured in lipids, we expect that the K119Q substitution would have a minimal effect on the helix. Instead, the secondary structure is governed by the strong local hydrophobic interactions between the core of the lipid bilayer and the nonpolar face of the helix (V83–V84 and F86–F87 pairs) (14). On the other hand, the increased dynamics of the epitope in rmC8 points to the importance of electrostatic interactions, and is consistent with it being more exposed to the exterior of the membrane, although this exposure has no significant effect on the local secondary structure.
Figure 7.
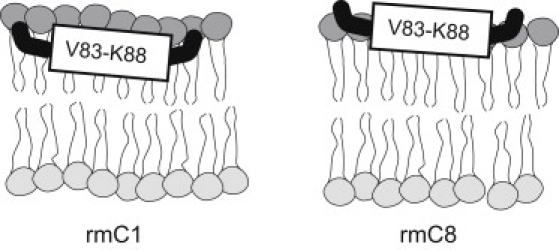
The immunodominant epitope adopts a similar α-helical molecular conformation in both rmC1 and rmC8 proteins. The α-helix is more exposed in rmC8, as follows from the dynamic information derived from the measurements of proton line widths.
Our observation of only a short helical six-residue-long structure is somewhat in disagreement with previous site-directed spin-labeling (SDSL)/electron-paramagnetic resonance (EPR) measurements, which were consistent with the presence of a longer helix within the epitope. Site-directed mutagenesis to Cys required for SDSL/EPR may introduce some structural distortions to the protein in its membrane-associated form. Furthermore, ambiguity of the cysteine side-chain conformation and preferential partitioning of the paramagnetic label into a lipid bilayer or solvent may introduce additional complication to the interpretation of SDSL/EPR data. Our results, on the other hand, are based on the measurements conducted in intact rmC1 and rmC8.
Another possible reason for disagreement between EPR and NMR results may be related to the difference in lipid composition and protein/lipid used in the measurements. The EPR experiments were performed on proteins reconstituted at a 10-fold lower protein/lipid, with lipid vesicles comprising roughly 18% (mol) charged lipids. In this study, the higher percentage of charged lipids (50 mol %) may have resulted in a stronger overall electrostatic interaction of both the rmC1 and rmC8 variants with the membrane. Although our control experiments indicate that these factors play a minor role in the formation of the epitope secondary structure at low temperature (Fig. 3), they will certainly affect the dynamic properties of the proteins and may become important at higher temperatures. Investigations of the effects of lipid composition on the MBP dynamics and structure are currently underway.
Acknowledgments
We are grateful to Ms. Valerie Robertson and Mr. Peter Scheffer of the University of Guelph NMR Centre for technical support, and to Drs. Joan Boggs and Leonid Brown for helpful discussions and comments on the manuscript.
This work was supported by the Canadian Institutes of Health Research (operating grant No. MOP 74468 to G.H. and V.L.), the Canada Foundation for Innovation, and the Ontario Innovation Trust. V.L. is a Tier II Canada Research Chair in Biophysics, and a recipient of an Early Researcher Award from the Ontario Ministry of Research and Innovation. M.A. was initially a recipient of a Doctoral Studentship from the Ministry of Higher Education and Scientific Research of Egypt. V.V.B. was the recipient of a Postdoctoral Fellowship from the Multiple Sclerosis Society of Canada.
Footnotes
Mumdooh A. M. Ahmed's present address is Department of Physics, Faculty of Science at Suez, Suez Canal University, Suez, Egypt.
Contributor Information
George Harauz, Email: gharauz@uoguelph.ca.
Vladimir Ladizhansky, Email: vladizha@uoguelph.ca.
Supporting Material
One figure and three tables are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00734-4.
Supporting Material
References
- 1.Boggs J.M., Bates I.R., Harauz G. Interactions of the 18.5 kDa myelin basic protein (MBP) with lipid bilayers—studies by electron paramagnetic resonance (EPR) spectroscopy and implications for generation of autoimmunity in multiple sclerosis. In: Boggs J.M., editor. Myelin Basic Protein. Nova Science; New York: 2008. [Google Scholar]
- 2.Hu Y., Doudevski I., Israelachvili J. Synergistic interactions of lipids and myelin basic protein. Proc. Natl. Acad. Sci. USA. 2004;101:13466–13471. doi: 10.1073/pnas.0405665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Min Y., Kristiansen K., Israelachvili J. Interaction forces and adhesion of supported myelin lipid bilayers modulated by myelin basic protein. Proc. Natl. Acad. Sci. USA. 2009;106:3154–3159. doi: 10.1073/pnas.0813110106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boggs J.M. Myelin basic protein: a multifunctional protein. Cell. Mol. Life Sci. 2006;63:1945–1961. doi: 10.1007/s00018-006-6094-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boggs J.M. Myelin basic protein interactions with actin and tubulin in vitro—binding, assembly, and regulation. In: Boggs J.M., editor. Myelin Basic Protein. Nova Science; New York: 2008. [Google Scholar]
- 6.Harauz G., Ishiyama N., Farès C. Myelin basic protein-diverse conformational states of an intrinsically unstructured protein and its roles in myelin assembly and multiple sclerosis. Micron. 2004;35:503–542. doi: 10.1016/j.micron.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 7.Harauz G., Libich D.S. The classic basic protein of myelin-conserved structural motifs and the dynamic molecular barcode involved in membrane adhesion and protein-protein interactions. Curr. Protein Pept. Sci. 2009;10:196–215. doi: 10.2174/138920309788452218. [DOI] [PubMed] [Google Scholar]
- 8.Moscarello M.A., Mastronardi F.G., Wood D.D. The role of citrullinated proteins suggests a novel mechanism in the pathogenesis of multiple sclerosis. Neurochem. Res. 2007;32:251–256. doi: 10.1007/s11064-006-9144-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mastronardi F.G., Moscarello M.A. Deimination of MBP by PAD enzymes, and their role in multiple sclerosis. In: Boggs J.M., editor. Myelin Basic Protein. Nova Science; New York: 2008. [Google Scholar]
- 10.Bates I.R., Libich D.S., Harauz G. An Arg/Lys→Gln mutant of recombinant murine myelin basic protein as a mimic of the deiminated form implicated in multiple sclerosis. Protein Expr. Purif. 2002;25:330–341. doi: 10.1016/s1046-5928(02)00017-7. [DOI] [PubMed] [Google Scholar]
- 11.Bates I.R., Matharu P., Harauz G. Characterization of a recombinant murine 18.5-kDa myelin basic protein. Protein Expr. Purif. 2000;20:285–299. doi: 10.1006/prep.2000.1307. [DOI] [PubMed] [Google Scholar]
- 12.Harauz G., Ladizhansky V., Boggs J.M. Structural polymorphism and multifunctionality of myelin basic protein. Biochemistry. 2009;48:8094–8104. doi: 10.1021/bi901005f. [DOI] [PubMed] [Google Scholar]
- 13.Bates I.R., Boggs J.M., Harauz G. Membrane-anchoring and charge effects in the interaction of myelin basic protein with lipid bilayers studied by site-directed spin labeling. J. Biol. Chem. 2003;278:29041–29047. doi: 10.1074/jbc.M302766200. [DOI] [PubMed] [Google Scholar]
- 14.Bates I.R., Feix J.B., Harauz G. An immunodominant epitope of myelin basic protein is an amphipathic α-helix. J. Biol. Chem. 2004;279:5757–5764. doi: 10.1074/jbc.M311504200. [DOI] [PubMed] [Google Scholar]
- 15.Musse A.A., Boggs J.M., Harauz G. Deimination of membrane-bound myelin basic protein in multiple sclerosis exposes an immunodominant epitope. Proc. Natl. Acad. Sci. USA. 2006;103:4422–4427. doi: 10.1073/pnas.0509158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Musse A.A., Harauz G. Molecular “negativity” may underlie multiple sclerosis: role of the myelin basic protein family in the pathogenesis of MS. Int. Rev. Neurobiol. 2007;79:149–172. doi: 10.1016/S0074-7742(07)79007-4. [DOI] [PubMed] [Google Scholar]
- 17.Zhong L., Bamm V.V., Ladizhansky V. Solid-state NMR spectroscopy of 18.5 kDa myelin basic protein reconstituted with lipid vesicles: spectroscopic characterization and spectral assignments of solvent-exposed protein fragments. Biochim. Biophys. Acta. 2007;1768:3193–3205. doi: 10.1016/j.bbamem.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen P.S., Toribara T.Y., Warner H. Microdetermination of phosphorus. Anal. Chem. 1956;28:1756–1758. [Google Scholar]
- 19.Fiske C.H., Subbarow Y. The colorimetric determination of phosphorus. J. Biol. Chem. 1925;66:375–400. [Google Scholar]
- 20.Smith P.K., Krohn R.I., Klenk D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 21.Wiechelman K.J., Braun R.D., Fitzpatrick J.D. Investigation of the bicinchoninic acid protein assay: identification of the groups responsible for color formation. Anal. Biochem. 1988;175:231–237. doi: 10.1016/0003-2697(88)90383-1. [DOI] [PubMed] [Google Scholar]
- 22.Gor'kov P.L., Chekmenev E.Y., Brey W.W. Using low-E resonators to reduce RF heating in biological samples for static solid-state NMR up to 900 MHz. J. Magn. Reson. 2007;185:77–93. doi: 10.1016/j.jmr.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 23.Pines A., Gibby M.G., Waugh J.S. Proton-enhanced NMR of dilute spins in solids. J. Chem. Phys. 1973;59:569–590. [Google Scholar]
- 24.Hohwy M., Rienstra C.M., Griffin R.G. Band-selective homonuclear dipolar recoupling in rotating solids. J. Chem. Phys. 2002;117:4973–4987. [Google Scholar]
- 25.Hohwy M., Rienstra C.M., Griffin R.G. Fivefold symmetric homonuclear dipolar recoupling in rotating solids: application to double quantum spectroscopy. J. Chem. Phys. 1999;110:7983–7992. [Google Scholar]
- 26.Morris G.A., Freeman R. Enhancement of nuclear magnetic resonance signals by polarization transfer. J. Am. Chem. Soc. 1979;101:760–762. [Google Scholar]
- 27.Takegoshi K., Nakamura S., Terao T. C13-H1 dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 2001;344:631–637. [Google Scholar]
- 28.Fung B.M., Khitrin A.K., Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000;142:97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- 29.Delaglio F., Grzesiek S., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 30.Morcombe C.R., Zilm K.W. Chemical shift referencing in MAS solid state NMR. J. Magn. Reson. 2003;162:479–486. doi: 10.1016/s1090-7807(03)00082-x. [DOI] [PubMed] [Google Scholar]
- 31.Markley J.L., Bax A., Wüthrich K. Recommendations for the presentation of NMR structures of proteins and nucleic acids—IUPAC-IUBMB-IUPAB Inter-Union Task Group on the standardization of data bases of protein and nucleic acid structures determined by NMR spectroscopy. Eur. J. Biochem. 1998;256:1–15. doi: 10.1046/j.1432-1327.1998.2560001.x. [DOI] [PubMed] [Google Scholar]
- 32.Luca S., Filippov D.V., Baldus M. Secondary chemical shifts in immobilized peptides and proteins: a qualitative basis for structure refinement under magic angle spinning. J. Biomol. NMR. 2001;20:325–331. doi: 10.1023/a:1011278317489. [DOI] [PubMed] [Google Scholar]
- 33.Wishart D.S., Sykes B.D. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR. 1994;4:171–180. [Google Scholar]
- 34.Wang Y.J., Jardetzky O. Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 2002;11:852–861. doi: 10.1110/ps.3180102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwarzinger S., Kroon G.J., Dyson H.J. Sequence-dependent correction of random coil NMR chemical shifts. J. Am. Chem. Soc. 2001;123:2970–2978. doi: 10.1021/ja003760i. [DOI] [PubMed] [Google Scholar]
- 36.Libich D.S., Ahmed M.A.M., Harauz G. Fuzzy complexes of myelin basic protein: NMR spectroscopic investigations of a polymorphic organizational linker of the central nervous system. Biochem. Cell Biol. 2010;88:143–155. doi: 10.1139/o09-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Farès C., Libich D.S., Harauz G. Solution NMR structure of an immunodominant epitope of myelin basic protein. Conformational dependence on environment of an intrinsically unstructured protein. FEBS J. 2006;273:601–614. doi: 10.1111/j.1742-4658.2005.05093.x. [DOI] [PubMed] [Google Scholar]
- 38.Boggs J.M., Moscarello M.A., Papahadjopoulos D. Structural organization of myelin: role of lipids-protein interactions determined in model systems. In: Jost P.C., Griffith O.H., editors. Lipids-Protein Interactions. Wiley-Interscience; New York: 1982. [Google Scholar]
- 39.Filip C., Hafner S., Spiess H.W. Solid-state nuclear magnetic resonance spectra of dipolar-coupled multi-spin systems under fast magic angle spinning. J. Chem. Phys. 1999;110:423–440. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.