

Summary
Accumulating evidence from murine and human studies supports a key role for interleukin-17 (IL-17) and IL-21 in the pathogenesis of inflammatory arthritis. The pathways and molecular mechanisms that underlie the production of IL-17 and IL-21 are being rapidly elucidated. This review focuses on interferon regulatory factor 4 (IRF4), a member of the IRF family of transcription factors, which has emerged as a crucial controller of both IL-17 and IL-21 production. We first outline the complex role of IRF4 in the function of CD4+ T cells and then discuss recent studies from our laboratory that have revealed a surprising role for components of Rho guanosine triphosphatase-mediated pathways in controlling the activity of IRF4. A better understanding of these novel pathways will hopefully provide new insights into mechanisms responsible for the development of inflammatory arthritis and potentially guide the design of novel therapeutic approaches.
Keywords: interferon regulatory factor 4, IL-17, IL-21, Def6
Introduction
While activation of CD4+ T helper (Th) cells is critical for an individual's ability to eliminate a wide array of pathogens, profound pathophysiological consequences can arise if the activation of CD4+ T cells is erroneously directed against self-antigens. Extensive investigations have provided crucial insights into the myriad defects in immunoregulatory mechanisms, which are presumed responsible for the development of autoimmune responses (1, 2). One of the major mechanisms employed by the immune system to avoid autoreactivity is to precisely gauge the strength of interactions between the antigen receptors and the antigens (3). Indeed, the avidity of the T-cell receptor (TCR) for self-major histocompatibility complex (MHC) ligands in the thymus is a key determinant of shaping the normal T-cell repertoire (4, 5). An accurate read-out of the strength of the TCR interaction with self-ligands is also crucial for the maintenance of T-cell tolerance in the periphery. The sensing of the potency of TCR engagement is dependent on a complex cascade of biochemical events initiated upon TCR triggering (6). Antigen recognition by T cells also results in a profound reorganization of the actin cytoskeleton, which is essential for the formation of the immunological synapse (IS) and other structures that facilitate the appropriate delivery of TCR-induced signals (7).
In addition to accurately sensing the strength of their interaction with peptide-MHC (pMHC) complexes, CD4+ T cells also need to precisely regulate their effector functions. Indeed there is mounting evidence that inappropriate regulation of Th17 cells, a novel Th effector subset, can play an important role in the pathogenesis of inflammatory arthritides such as rheumatoid arthritis (RA) (8-10). One of the crucial mechanisms by which Th17 cells exert their pathogenic effects is via production of cytokines like interleukin-17 (IL-17) and IL-21 that can drive both inflammatory and autoantibody responses.
Features and functions of IL-17 and IL-21
The IL-17 cytokine family
IL-17 cytokines constitute a unique family of cytokines, which are gaining increasing attention because of their powerful pro-inflammatory effects. The central role of this family of cytokines in inflammatory responses and the growing complexity of their biology has been the subject of excellent reviews (11-13). In addition to IL-17A (previously referred to as IL-17), which was the first member of this family to be identified, the family consists of five other members, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F. IL-17A and IL-17F are the best-characterized members. Although both IL-17A and IL-17F have been implicated in promoting inflammatory responses, they can exert distinct biological functions (14, 15). In particular, IL-17A has been shown to have a more potent role than IL-17F in driving autoimmune responses in various mouse models including models of arthritis (14, 15). Although IL-17B and IL-17C have not been fully characterized, they may also exert a proinflammatory role as evidenced by the finding that transfer of CD4+ T cells transduced with IL-17B or IL-17C exacerbated collagen-induced arthritis (16).
The IL-17R family (reviewed in 11) comprises of five receptor subunits, IL-17RA, IL-17RB, IL-17RC, IL-17RD, and IL-17RE. Although the precise composition of the receptor complexes has not been fully defined, interactions between IL-17RA and IL-17RC appear to mediate many of the biological effects of IL-17A and IL-17F. While the expression of IL-17RA is ubiquitous, IL-17RC is preferentially expressed in non-hematopoietic cells including synovial cells (17, 18). Consistent with the finding that the cytoplasmic domain of IL-17RA contains a region termed the SEFIR domain, which is homologous to the Toll/IL-1R (TIR) domain (19), one of the major signaling events triggered by engagement of IL-17RA is the activation of nuclear factor-κB (NF-κB) (20). Members of the CCAAT/enhancer-binding protein (C/EBP) family of transcription factors are also important downstream components of the signal transduction cascade activated by IL-17RA engagement (21, 22). IL-17RA signaling also leads to the activation of mitogen-activated protein kinase (MAPK) pathways (23). Although IL-17RC pairs with IL-17RA and plays an important role in IL-17A and IL-17F signaling, little is known about the mechanistic basis by which it participates in IL-17-mediated signaling. An important function may be to recruit critical signaling molecules to the receptor complex (11).
As outlined in detail in previous reviews (24-26), IL-17 exerts diverse effects on the development of the inflammatory lesions in RA, which are characterized by inflammatory cell infiltrates in the synovium, cartilage degradation, and bone erosions. Importantly, IL-17 stimulates the recruitment of inflammatory cells by inducing the production of chemokines and enhances inflammation by promoting the production of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α), IL̃-6, and IL-1. IL-17 can furthermore mediate cartilage destruction and bone erosions by upregulating the expression of RANK ligand, and by inducing nitric oxide production and expression of matrix metalloproteinases. Consistent with the idea that IL-17 can mediate functions relevant to the pathophysiology of RA, elevated production of IL-17 has been observed in murine models of RA as well as in patients affected by this disorder (27-29). Increased levels of IL-17 also correlate with more severe joint damage (30). Importantly, mice deficient in IL-17 are protected from the development of collagen-induced arthritis (CIA), a mouse model of inflammatory arthritis, and blockade of IL-17 via either antibodies or soluble IL-17 receptor can ameliorate symptoms in CIA even after arthritis is established (31-33).
IL-21
IL-21 is a member of the common receptor γ-chain (γc) family of cytokines, whose involvement in the pathophysiology of various diseases is becoming increasingly recognized (reviewed in 34, 35). Like other members of this family, which also includes IL-2, IL-4, IL-7, IL-9, and IL-15, IL-21 binds to a receptor complex composed of the γc subunit and a unique receptor chain, the IL-21R (reviewed in 36, 37). IL-21R is broadly expressed by both immune and non-immune cells and its expression can be regulated by the state of activation and/or differentiation of the cell. Similarly to other γc family members, engagement of the IL-21R leads to activation of the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) signaling cascade, specifically, of Jak1 and Jak3. While activation of these kinases leads to the subsequent phosphorylation of various STAT proteins including Stat1, Stat3, and Stat5, activation of Stat3, as discussed later, plays a major role in mediating the biological effects of IL-21. Activation of MAPKs and phosphoinositide 3-kinase (PI3K) is also believed to contribute to IL-21 signaling.
IL-21 exerts a broad range of biological effects many of which are relevant to RA pathophysiology. One of the earliest recognized functions of IL-21 was to be a major regulator of B-cell responses and immunoglobulin G (IgG) production (reviewed in 36-38). IL-21 is thus believed to play an important role in the aberrant humoral responses that develop in RA patients, characterized by the presence of autoantibodies like rheumatoid factor (RF) and anti-cyclic citrullinated peptide (CCP) antibodies (39). IL-21 can also contribute to the development of inflammatory lesions in arthritis via its ability to augment Th17 differentiation (as discussed later), upregulate the expression of RANK ligand and fuel the homeostatic expansion of autoreactive T cells (40). Consistent with the idea that IL-21 production plays a fundamental role in RA, blockade of the IL-21/IL-21R pathway has recently been reported to be efficacious in ameliorating disease in CIA (41). Furthermore, spontaneous development of arthritis in K/B×N mice is completely abrogated by knocking out the IL-21R (40). In addition to IL-21, IL-17 may also participate in driving autoantibody production by promoting the formation of autoreactive germinal centers and the survival and differentiation of B cells (42, 43). Given the importance of IL-17 and IL-21 in the pathogenesis of inflammatory arthritis, a detailed understanding of the mechanisms required for the proper control of the production of these potentially pathogenic cytokines will provide crucial information on the molecular networks that become deregulated in inflammatory arthritis.
Signaling and molecular pathways controlling the production of IL-17 and IL-21 by CD4+ T cells
Signaling pathways regulating the development of IL-17 and IL-21 producing CD4+ T cells
The cytokine environment of CD4+ T cells during the initial antigenic encounter can profoundly alter their ability to acquire specific effector functions (44). In the absence of a strong polarizing cytokine milieu, activation of a naive Th cell results in its differentiation into a Th0 cell, which produces low-levels of a broad range of cytokines. The presence of specific ‘polarizing’ cytokines, instead, leads to the development of effector T-cell subsets, termed Th1 and Th2 cells, that produce high-levels of restricted sets of cytokines and orchestrate different types of immune responses. In particular, presence of IL-12 will drive the differentiation of Th cells into Th1 cells, which secrete IFN-γ and have been implicated in the regulation of delayed type hypersensitivity responses. Exposure to IL-4, in contrast, will drive the development of Th2 cells, which produce IL-4, IL-5, and IL-13 and can direct B cells to mount strong humoral responses. Although IL-4 was initially believed to be the major cytokine responsible for the ability of Th2 cells to promote humoral responses, Th2 cells were later also found to secrete IL-21, which, as discussed above, was discovered by genetic studies to be a key regulator of B-cell expansion, differentiation, and IgG production (36, 37).
Differentiation of a Th cell toward either a Th1 or a Th2 phenotype in response to different types of pathogens (e.g. tuberculosis versus parasites) was deemed important for effectively combating these organisms. However, excessive or uncontrolled polarization of Th cells into Th1 or Th2 cells in response to self-antigens or allergens was believed to underlie the development, respectively, of autoimmune disorders like RA or of allergic responses. The paradigm that Th1 cells played an essential role in the development of autoimmunity was, however, challenged when mice deficient in the expression of components of the IL-12 and IFN-γ signaling pathways were found to still develop significant and, at times, even exacerbated autoimmune responses (45, 46). These findings coupled with the recognition that the expression of IL-17 could be detected in a variety of autoimmune diseases led to the realization that other effector Th subsets might exist. This notion was soon supported by elegant work demonstrating that IL-17 producing CD4+ T cells represented a subset of effector Th cells distinct from Th1 and Th2 cells and thus these cells became termed Th17 cells (47-49).
The cytokines regulating the development of Th17 cells were rapidly identified and found to be different from those controlling the differentiation of Th1 and Th2 cells. This topic has been the subject of extensive reviews (13, 46, 50, 51) and thus is only briefly outlined here. In the murine system, the critical cytokines regulating the initial commitment of a CD4+ Th cell to become a Th17 cell are TGF-β and IL-6. Since presence of TGF-β also regulates the development of Foxp3+ regulatory T cells (Tregs), there is a reciprocal relationship between Th17 cells and Tregs whereby, in the setting of a proinflammatory environment (i.e. IL-6), TGFβ will drive the development of an inflammatory Th cell subset (Th17 cells) rather than promote the generation of an immunosuppressive subset (Tregs). The dose of TGFβ appears to be important in this lineage decision, since lower doses of TGFβ favor commitment of Th cells toward the Th17 lineage, while higher doses of TGFβ skew differentiation of Th cells toward a regulatory fate (52). The cytokine milieu favoring the generation of human Th17 cells has been the subject of some controversy possibly due to confounding effects of cell culture conditions. It does, however, appear that TGFβ together with other inflammatory mediators, is also required for the generation of human Th17 cells (53-55).
While exposure to TGFβ and IL-6 initiates the commitment of a naive Th cell to become a Th17 cell, additional stimuli are required for the full acquisition of the Th17 cell program. An important step in this process is the ability of a developing Th17 cell to attain the capacity of producing IL-21 (56-58). The acquisition of IL-21 production plays a key role in amplifying the generation of Th17 cells since IL-21 functions in an autocrine manner to reinforce the commitment of the newly differentiating Th17 cell towards the Th17 cell fate. Interestingly, although both Th2 and Th17 cells can produce IL-21, Th17 cells produce much higher levels of IL-21 than Th2 cells (57). IL-21 can also substitute for IL-6 in promoting the initial differentiation of murine Th17 cells (via what has been termed an alternative pathway) as well as in driving the differentiation of human Th17 cells (55). Another cytokine, IL-23, a member of the IL-12 family of cytokines, subsequently plays a crucial role in the expansion and terminal differentiation of Th17 cells (59, 60). Whereas IL-23 was originally thought to participate early in the process of Th cell differentiation toward the Th17 lineage, it was later found that naïve T cells are unresponsive to IL-23 and upregulate the expression of the IL-23R only during later stages of Th17 development.
Although IL-6 plays a unique role in the commitment of a naive Th cell to become a Th17 cell, the presence of other proinflammatory cytokines, like TNF-α and IL-1, also aids in the differentiation of Th17 cells (61). In particular, it was recently shown that developing Th17 cells upregulate IL-1R1 and that, under certain stimulatory conditions, IL-1 responsiveness by T cells is critical for early Th17 differentiation and the maintenance of polarized effector Th17 cells (62). Some of the Th17 promoting effects of IL-1 may be related to its ability to enhance the proliferation and survival of activated CD4+ T cells, an effect that, interestingly, is not restricted to Th17 cells but can also be observed in Th2 cells (63). Consistent with these findings, neutralization of IL-1 in IL-1 receptor antagonist-deficient mice, a spontaneous mouse model of inflammatory arthritis due to excessive IL-1 signaling, significantly reduces the expansion of Th17 cells in these mice (64).
In addition to cytokines, recent studies have also highlighted an important role for TCR signaling in the development of IL-17-producing CD4+ T cells. Indeed, high antigen concentrations enable naive CD4+ T cells to undergo a modest degree of Th17 differentiation even in the absence of polarizing cytokines (65). Strong TCR engagement furthermore synergizes with TGF-β and IL-6 in driving optimal Th17 differentiation. These effects have been ascribed to the induction of CD40L expression on T cells by strong antigenic stimulation, which, in turn, can promote CD40-induced IL-6 production by dendritic cells. It is, however, also possible that differences in the strength of TCR engagement can affect Th17 differentiation in a T-cell intrinsic manner. Indeed, raftlin, a lipid raft constituent, was recently shown to regulate the ability of CD4+ T cells to produce IL-17 via its effects on TCR signaling (66).
The generation of Th17 cells can be inhibited by several factors (13, 46, 50, 51). Importantly, cytokines produced by Th1 and Th2 cells, IFNγ and IL-4, respectively, interfere with the development of Th17 cells. IL-2 also blocks Th17 differentiation and instead favors the generation of a regulatory T-cell phenotype (67). Development of Th17 cells can also be suppressed by the presence of IL-27, a member of the IL-12 family of cytokines (68, 69). Consistent with these effects, administration of IL-27 in vivo attenuates pathology of CIA (70). Retinoic acid also inhibits Th17 development while promoting differentiation of regulatory T cells and it can provide beneficial effects in CIA (71-75). The ability of Th17 cells to mediate pathogenic effects can also be restrained after Th17 cells have already been generated. Indeed TGF-β and IL-6 upregulate not only the production of IL-17 but also that of IL-10, which can exert potent anti-inflammatory effects and thus can temper the pathogenic functions of IL-17 (76). Exposure to IL-23, however, inhibits the production of IL-10 by Th17 cells and fully unleashes their pathogenic potential.
Whereas Th17 cells were initially deemed to represent a completely distinct lineage from Th1 and Th2 cells, recent work has uncovered an increasingly complex relationship between Th17 cells and other T cell subsets. Indeed Th17 cells can convert to Th1 cells and simultaneous production of IL-17 and IFNγ by Th cells is frequently detected in inflammatory conditions (77, 78). Furthermore, Tregs stimulated with inflammatory cytokines can become IL-17 producing CD4+ T cells (79, 80). New effector Th subsets, such as follicular T helper cells (TFH), which are powerful mediators of humoral responses, have also been recently characterized (81). TFH cells produce high levels of IL-21 but have also been reported to produce IL-17 (82-84). These recent findings have led to the suggestion that there is an unanticipated plasticity in the system, which may enable CD4+ T cells to properly tailor their effector function in response to a dynamic and unpredictable environment (85). Importantly, these findings indicate that IL-17- and IL-21-producing CD4+ T cells consist of a heterogeneous group of effector Th cells, which may have attained this capability thru different routes and may not necessarily express identical transcriptional programs.
Molecular mechanisms controlling the development of CD4+ T cells producing IL-17 and IL-21
Rapid progress has been made in elucidating the molecular mechanisms that control Th17 differentiation and the production of IL-17 and IL-21 (13, 46, 50, 51). Here we wll briefly outline the major factors that regulate the production of these cytokines focusing, in particular, on transcriptional regulators that, as will be discussed later, may be relevant to the role of IRF4 in this process.
STAT proteins
Activation of STAT proteins is a critical initiating event in the signaling cascades of many cytokines (86). It is thus not surprising that these transcription factors play a crucial role in the differentiation of Th cells (87). Since distinct cytokines activate different STAT proteins, Th differentiation towards the various phenotypes is controlled by different STATs. Thus activation of Stat4 upon IL-12 stimulation will skew Th cells toward the Th1 phenotype while activation of Stat6 in response to IL-4 is critical for the generation of Th2 responses (87). In line with the finding that IL-6 plays a pivotal role in Th17 differentiation, Stat3, the major STAT protein activated in response to IL-6, plays a key role in the generation of Th17 cells and in the production of both IL-17 and IL-21 (67, 88, 89). Interestingly, IL-21 itself activates Stat3, and this effect is likely to play a major role in the ability of IL-21 to augment the commitment of a developing Th17 cell toward the Th17 phenotype or to substitute for IL-6 in the initial differentiation of Th17 cells (58, 88). Activation of Stat3 by IL-23 similarly plays an important role in the later stages of Th17 differentiation (89, 90). In contrast to Stat3, STAT proteins that are activated in response to IL-2 and IFNγ (Stat5 and Stat1, respectively) are crucial mediators of the inhibitory effects of these cytokines on Th17 differentiation (47, 67). Stat1 has also been implicated in the ability of IL-27 to suppress Th17 differentiation (68, 69).
Lineage-specific transcriptional regulators
Although activation of STATs is essential for the initial commitment toward specific Th effector subsets, full implementation of the transcriptional programs associated with each Th effector phenotype relies on the upregulation of specific lineage-specific transcriptional regulators like T-bet for Th1 and GATA3 for Th2 cells (91, 92). In the case of Th17 cells, this role is fulfilled by members of the retinoic-acid-receptor-related orphan nuclear hormone receptor family, RORγt, and, to a lesser extent, RORα (93, 94). Expression of both RORγt and RORα is upregulated upon exposure of naïve Th cells to TGFβ and IL-6 (or TGFβ and IL-21) in a Stat3-dependent manner (67, 89, 94). Overexpression of RORγt is sufficient to drive IL-17 production in TCR-stimulated cells while its deficiency results in impaired Th17 differentiation (93). Whereas the absence of RORγt by itself leads to profound impairments in IL-17 production, combined deficiencies in RORγt and RORα are required for a reduction in IL-21 synthesis indicating that RORα can compensate for RORγt in this process (94). Interestingly, recent studies have shown that the functions of RORγt can be inhibited by Foxp3 (52), the key master regulator of the transcriptional program of regulatory T cells suggesting that the balance between RORγt and Foxp3 plays a major role in the commitment of Th cells to become either a Th17 cell or a regulatory T cell.
Other transcription factors
An increasing number of transcription factors have been recently linked to the regulation of IL-17 and IL-21 production. In particular, nuclear factor of activated T cells (NFAT) proteins have been implicated in the TCR-mediated stimulation of these cytokines. Indeed, the human IL-17 promoter contains NFAT-binding sites, to which both NFATc1 and NFATc2 can bind (95). Consistent with these results the production of IL-17 can be inhibited by cyclosporine A (29). NFAT proteins can also regulate the production of IL-21. NFATc2 can bind to and transactivate the IL-21 promoter and, similarly to the case of IL-17, the production of IL-21 can also be inhibited by cyclosporine A (96, 97). Conflicting results have, however, been obtained regarding the ability of T cells deficient in NFATc2 to produce IL-21 (96, 97). Furthermore, recent studies have found that c-Maf, a transcription factor previously linked to Th2 differentiation and IL-10 production is expressed at high levels in both Th17 and TFH cells and participates in the regulation of IL-21 production (82). No effects of c-Maf on IL-17 production and the early stages of Th17 differentiation have, however, been observed. An AP-1 family member, BATF, was also recently shown to be a key regulator of Th17 differentiation (98). In addition to positive regulators of Th17 differentiation, inhibitors of this process have also been identified. For instance, growth factor independent 1 (Gfi-1), a transcriptional repressor whose presence is required for optimal Th2 expansion, was recently shown to inhibit IL-17 production (99). Interestingly TGF-β can repress Gfi-1 expression, suggesting that downregulation of Gfi-1 is an important step for the acquisition of the capability to produce IL-17 (99).
IRF4: a crucial controller of both IL-17 and IL-21 production
Among the newly identified transcription factors that participate in the regulation of IL-17 and IL-21, IRF4 (also known as Pip, MUM1, LSIRF, NFEM5, and ICSAT), plays a unique and integral role in the control of these two cytokines since it is absolutely required for the production of both IL-17 and IL-21 (100-102). Despite its fundamental role in the function of mature CD4+ T cells, IRF4 has not been the subject of extensive reviews. We thus first provide a broad overview of the essential features of IRF4 and later discuss its role in mature CD4+ T-cell function and, specifically, in the regulation of IL-17 and IL-21 production.
Structure and transcriptional activity
As its name implies, IRF4 belongs to the IRF family of transcription factors, which in mice and humans consists of nine members, IRF1 through IRF9 (103, 104). Like all members of the IRF family of transcription factors, IRF4 contains a highly conserved N-terminal DNA-binding domain (DBD) of ∼120 amino acids (Fig. 1). This DBD is characterized by 5 conserved tryptophan residues separated by 10-18 amino acids and forms a helix-turn-helix motif. The carboxy-terminal portion of IRF4 contains an IRF-association domain (IAD), which is also found in IRF3 thru IRF9 and regulates the transcriptional activity of IRFs by mediating protein-protein interactions. Structural studies of IRF3 and IRF5 have demonstrated that the IAD forms a β-sandwich core, which is flanked by N- and C-terminal α-helical regions (105-107). These α-helical regions can block access to the IAD and serve an autoinhibitory function. Serine phosphorylation of the C-terminal autoinhibitory region in IRF5 has been shown to trigger a conformational rearrangement that relieves the autoinhibition and enables the IAD to mediate dimerization and interaction with coactivators (105). Interestingly, despite sharing structural similarities, the C-terminal regions of different IRFs exhibit very low sequence similarity suggesting that autoinhibition in distinct IRFs is relieved by different mechanisms and that relief of this autoinhibition may lead to distinct functional outcomes.
Fig. 1. Schematic diagram of interferon regulatory factor 4 (IRF4).

DBD, DNA-binding domain; IAD, IRF-association domain.
Although the DBD of all IRFs recognizes 5′-GAAA-3′ as the core recognition sequence, the ability of IRFs to bind DNA can be further modulated by sequences flanking the core motif, by the capacity of distinct IRFs to homodimerize or heterodimerize, or by their interaction with other transcriptional comodulators. These additional interactions can have profound functional implications. Indeed IRF4 can function as an activator or a repressor depending on the regulatory region that it targets and the presence/absence of cofactors. For instance, IRF4 can cooperate with ETS proteins and act as a transactivator of the immunoglobulin light-chain enhancer (108). In this case IRF4 binds to a composite DNA element containing an IRF4 binding site adjacent to an ETS binding motif. Recruitment of IRF4 to this site is dependent on its interaction with the DNA bound ETS protein, which leads to a conformational change in IRF4 that relieves an intramolecular autoinhibitory interaction between the N-terminal DNA binding domain and the C-terminal regulatory region (109, 110). Binding of IRF4 to DNA can also occur independently of its interacting partner, as in the case of its cooperation with Stat6 in the regulation of CD23b (111). Presence of both IRF4 and Stat6 is, however, needed for optimal transactivation of this gene (111). IRF4, either by itself or in a complex with IRF8, can also bind to interferon-stimulated response elements (ISREs) and repress IRF1 mediated gene transcription and the expression of interferon-inducible genes (112-114). In B cells, IRF4 has also been shown to act as a repressor of BCL-6 gene expression (115). IRF4 is thus the quintessential ‘context-dependent’ transcription factor whose activity can be profoundly shaped by the precise molecular milieu of a cell.
Expression
In contrast to other members of the IRF family, the expression of IRF4 in both T and B cells is primarily regulated by pathways known to drive lymphocyte activation and not by type I or type II interferons (111, 116). Indeed, stimulation of T cells with either anti-CD3 antibodies or T-cell mitogens leads to a robust induction of IRF4 expression (116-119). Kinetic experiments have demonstrated that induction of murine IRF4 in response to TCR stimulation can be detected within a couple of hours, peaks at 6-9 h, then declines to much lower levels by 12-16 h (116). Interestingly, the kinetics and levels of IRF4 expression induced by TCR stimulation were recently shown to differ in naive versus effector/memory CD4+ T cells with naive CD4+ T cells exhibiting higher but more transient expression of IRF4 (120). Consistent with the upregulation of IRF4 expression upon T-cell activation, constitutive expression of IRF4 can be detected in HTLVI-transformed T cells (119). Since TCR engagement is the major pathway controlling the expression of IRF4, its expression is not restricted to a specific Th effector subset but can be detected in Th1, Th2, and Th17 cells as well as in Tregs (100, 121). Subtle differences in the precise patterns of IRF4 expression amongst different Th effector subsets, however, are beginning to be uncovered. Indeed, although all Th effector subsets upregulate IRF4 to a similar extent upon stimulation, the levels of IRF4 in resting Th2 cells are higher than those observed in resting Th1 or Th17 cells (100). Furthermore, a recent study has demonstrated that IL-1 signaling in T cells helps maintain high levels of IRF4 expression after stimulation and that this effect is critical for the differentiation of Th17 cells (62). Interestingly, the expression of IRF4 in CD4+ T cells cultured under Th17-skewing conditions can be decreased by exposure to retinoic acid (74).
The molecular mechanisms regulating the induction of IRF4 in response to TCR stimulation have been shown to involve the activation of NF-κB family members. c-Rel appears to play a particularly important role, since c-Rel-containing complexes can bind to NF-κB elements within the IRF4 promoter upon T-cell stimulation and c-Rel-deficient lymphocytes fail to upregulate IRF4 in response to various mitogenic stimuli (118). Activation of NF-κB also participates in the HTLVI-mediated upregulation of IRF4 (119). Activation of NFAT proteins has also been implicated in the regulation of IRF4 expression and consistent with this idea, the TCR-mediated upregulation of IRF4 can be blocked by cyclosporine A (116, 119). The IRF4 promoter also contains GAS elements, which can be bound by STAT proteins including Stat4 and Stat6 (122). While the role of STAT proteins in the regulation of IRF4 in T cells has not been directly investigated, it is intriguing to speculate that the enhanced expression of IRF4 in resting Th2 cells may be related to the presence of IL-4 in these cultures and may occur in a Stat6-dependent manner. IRF4 may also be able to regulate its own expression since the IRF4 promoter contains an IRF/Ets response element, which can be targeted by IRF4 containing complexes (122). Whether such autoregulation plays a role in controlling the expression of IRF4 in CD4+ T cells is, however, not known. Of interest, it has recently been shown that Foxp3 can also bind to the IRF4 promoter and regulate the expression of IRF4 (121).
Role in the function of mature CD4+ T cells
No significant effects of IRF4 deficiency on T-cell development have been observed, but IRF4 plays a major role in mature CD4+ T-cell function (123). Early studies, conducted on total T-cell populations, indicated that IRF4 is required for the optimal proliferation of CD4+ T cells in response to mitogenic stimuli as well as for the production of IL-2, IL-4, and IFN-γ (123). Follow-up studies confirmed that IRF4 plays an important role in the production of IL-2 and IFN-γ by naive cells and that it is also required for optimal production of IL-2 by effector/memory T cells (120). Investigations of the role of IRF4 in the production of IL-4 and in the differentiation of specific Th subsets, however, has revealed a much more intricate role for IRF4 in the effector function of Th cells. Since most of this information has emerged from studies on the role of IRF4 in the regulation of Th2 responses, we will first review its role in this lineage and then discuss more recent findings that implicate IRF4 as a key regulator of Th17 differentiation. Given that IRF4 does not appear to exert significant effects on Th1 differentiation (124, 125), its role in this subset will not be addressed. Although TFH cells are major producers of IL-21 and IRF4 is expressed in germinal center CD4+ T cells (126), the effects of IRF4 in TFH cells have not been specifically investigated, and thus this subset will also not be discussed.
Roles in IL-4 production and Th2 responses
IRF4 plays a diverse and complex role in the regulation of Th2 responses. Its effects have been shown to be highly dependent on the precise stage of differentiation of CD4+ T cells. Under neutral conditions, IRF4 inhibits the production of IL-4 by naive CD4+ T cells but promotes the production of IL-4 by effector/memory CD4+ T cells (120). When CD4+ T cells are exposed to Th2 skewing conditions, lack of IRF4 profoundly impairs the ability of CD4+ T cells to differentiate toward the Th2 cell lineage and produce IL-4, IL-5, IL-13, and IL-21 (102, 124, 125, 127). Consistent with in vitro findings, the development of Th2 responses in vivo is critically dependent on the presence of IRF4 (120, 125, 127). Interestingly, a lack of IRF4 results in enhanced IFN-γ production under Th2 conditions suggesting that IRF4 is critical for blocking the differentiation of developing Th2 cells toward the Th1 phenotype (124, 125). It is furthermore important to remember that IRF4 also plays a key role in non-T cells, especially in B cells (128, 129), and that its functions in these compartments could also impact its overall ability to control Th2 immune responses.
At the molecular level, IRF4 employs both direct and indirect mechanisms to control the production of IL-4 and the development of Th2 responses. Lack of IRF4 does not alter the early signaling events that occur in response to either TCR engagement or IL-4 stimulation, such as the activation of Stat6 (125, 127). Instead, we and others have shown that IRF4 can directly bind to and transactivate the IL-4 promoter (124, 130). Critical to this effect is its ability to cooperate with NFAT family members. Indeed the IL-4 promoter contains multiple IRF4 binding sites that are located adjacent to known NFAT binding sites and both NFATc1 and NFATc2 have been shown to cooperate with IRF4 in transactivating reporter constructs driven by the IL-4 promoter (124, 130). A physical interaction between NFATc2 and IRF4 has also been observed (124). Studies have also revealed that IRF4 and NFATc2 potently synergize with another Th2 transcription factor, c-Maf, in maximally inducing IL-4 expression (124). These studies are indicative of an important regulatory role of IRF4 in IL-4 synthesis after the initial activation events. IRF4 also controls the expression of GATA3, the master regulator of Th2 differentiation, and can thus further amplify the commitment of Th cells toward the Th2 lineage (125, 127). IRF4 can furthermore regulate the expression of Gfi-1 and thus control the expansion of committed Th2 cells (125).
The understanding of the role of IRF4 in the regulation of Th2 responses recently took an unexpected turn when Zheng et al. (121) employed a conditional approach to selectively delete IRF4 within the regulatory T-cell compartment. Surprisingly, these mice spontaneously develop a disorder marked by high levels of Th2 cytokines, by elevated levels of IgG1 and IgE, and by plasma cell infiltration of the pancreas, stomach, and kidneys. Since Foxp3 can upregulate the expression of IRF4, these findings led the authors to propose that, once upregulated, IRF4 controls the expression of a subset of Foxp3 target genes that function to restrain excessive Th2 responses. A couple of key IRF4 targets in Tregs have been identified (121). One of these targets is inducible costimulator (ICOS), which is required by Tregs for optimal suppressive activity. Interestingly, while IRF4 is necessary for ICOS expression in Tregs, our group has found that the induction of high levels of ICOS expression upon TCR stimulation in Th cells does not require the presence of IRF4 (102). Since the IRF4-binding site in the ICOS promoter colocalizes with a Foxp3-binding site and Foxp3 and IRF4 can physically interact, this differential regulation suggests that presence of Foxp3 may be necessary for the recruitment of IRF4 to the ICOS promoter. In addition, IL-10, another key effector of Treg suppressive ability, is also regulated by IRF4. Interestingly, similarly to the IL-4 promoter, the IL-10 promoter contains clustered NFAT and IRF4-binding motifs, and cooperation between NFAT and IRF4 has been shown to be important for the control of IL-10 gene expression (130, 131). Given that c-Maf is also an IRF4 target (121) and that lack of c-Maf leads to impaired IL-10 synthesis (132, 133), IRF4 may also regulate IL-10 production indirectly by inducing c-Maf expression. Production of IL-10 by Th2 cells has also recently been shown to be regulated by IRF4 (134).
Roles in Th17 responses
In addition to being a major regulator of Th2 responses, recent work has brought to light a fundamental role for IRF4 in the control of Th17 differentiation. Indeed CD4+ T cells from mice deficient in IRF4 are completely defective in their ability to differentiate into Th17 cells and to produce IL-17 whether they are cultured under classical (i.e. TGF-β plus IL-6) or alternative (i.e. TGF-β plus IL-21) Th17-inducing conditions (100-102). This defect is intrinsic to CD4+ T cells and is not secondary to any developmental alterations. The ability of IRF4 to act as an essential controller of Th17 differentiation has profound consequences in vivo. Indeed, IRF4-deficient mice are totally resistant to the development of a Th17-mediated disease [experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis] (100). The degree of protection conferred by an absence of IRF4 is even greater than that imparted by the lack of RORγt, which is considered the master regulator of Th17 differentiation (93). Although the effects of IRF4 deficiency in mouse models of inflammatory arthritis have not yet been investigated, given the complete absence of Th17 differentiation in these mice, it is reasonable to expect that a similar protective effect will be observed.
The central role of IRF4 in the regulation of IL-17-producing CD4+ T cells is due to the ability of IRF4 to control multiple processes. The absence of IRF4 does not affect the early signaling events known to be critical for Th17 differentiation (100, 101). IRF4-deficient T cells indeed can activate Stat3 normally in response to IL-6 or IL-21 stimulation. Furthermore, upregulation of a subset of Stat3-dependent genes like SOCS3 is unaffected by the absence of IRF4. IRF4, however, is required for the induction of the Th17 lineage-specific regulators RORγt and RORα upon exposure to either TGFβ and IL-6 or TGF-β and IL-21 (100, 101). Interestingly, reintroduction of RORγt and RORα into IRF4-deficient CD4+ T cells only partially rescues the defects in IL-17 production, supporting the idea that IRF4 exert additional effects that are important for Th17 differentiation. Consistent with this notion, our laboratory has found that IRF4 has the potential to target the IL-17 promoter, which suggests that IRF4 might directly transactivate the IL-17 gene (102). IRF4 is also necessary for the IL-6- and IL-21-mediated suppression of Foxp3 expression since IRF4-deficient T cells are unable to downregulate Foxp3 upon exposure to either TGFβ and IL-6 or TGF-β and IL-21 (100, 101). Given that Foxp3 can interfere with the function of RORγt (52), the sustained expression of Foxp3 in IRF4-deficient T cells could also contribute to their inability of to produce IL-17. Upon exposure to Th17-skewing conditions, the lack of IRF4 also results in enhanced IFN-γ production (100, 102), an effect that can further inhibit differentiation toward the Th17 phenotype. Thus, similarly to its role in Th2 cells, one of the key functions of IRF4 in the differentiation of Th17 cells may be to block the development of alternative Th fates via its ability to interfere with Th1 and/or Treg commitment.
IRF4 is not only essential for the initial production of IL-17, but it is also absolutely required for the autocrine production of IL-21 by Th17 cells. Indeed, whether cultured under classical or alternative Th17-inducing conditions, IRF4-deficient CD4+ T cells fail to produce IL-21 and thus lack a critical amplification step for the development of Th17 cells (101, 102). Again multiple mechanisms underlie this profound defect. As mentioned above, IRF4 deficiency impairs the upregulation not only of RORγt but also of RORα whose expression is known to compensate for RORγt in the regulation of IL-21 (101). IRF4 can furthermore directly bind and transactivate the IL-21 promoter (102). As in the case of the IL-4 promoter, the IL-21 promoter contains clusters of potential IRF4 binding sites. Our laboratory has identified a functionally important IRF4 binding site within the IL-21 promoter, which is located adjacent to an NFAT-binding site and optimal transactivation of this promoter requires the presence of both NFAT and IRF4 (102). It is intriguing to speculate that IRF4 and NFAT may also cooperate with c-Maf in the regulation of IL-21, since c-Maf has been shown to be an important regulator of the late phases of IL-21 production (82). Since c-Maf is a potential IRF4 target, a plausible scenario is that IRF4 is not only a key regulator of the early IL-21 transcription but also upregulates the expression of additional regulators with which it needs to cooperate during the later stages of the production of this cytokine. Although an absence of IRF4 does not affect the expression of the IL-21R, IRF4-deficient T cells also fail to upregulate the expression of IL-23R in response to IL-21, indicating that a lack of IRF4 will impact the expansion and terminal differentiation of Th17 cells (101). The precise mechanisms by which IRF4 regulates the expression of IL-23R are, however, not known.
Summary and perspectives
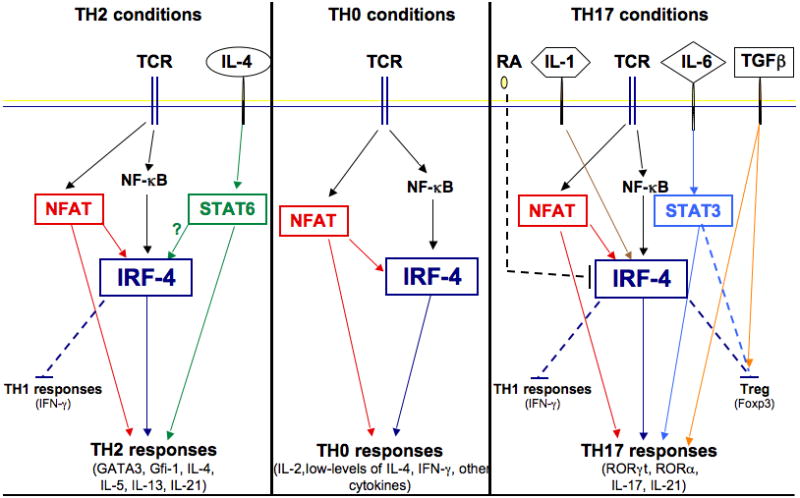
IRF4 thus appears to function as a critical molecular link between pathways triggered by TCR engagement and those induced by environmental clues such as cytokines (Fig. 2). In the absence of cytokine exposure, IRF4 acts as a central mediator of TCR signals and plays a key role in controlling the expression, albeit at a low-level, of a broad array of cytokines, likely via its ability to cooperate with NFAT and to directly target various cytokine promoters. In the presence of IL-4 or IL-6, instead, IRF4 acquires the ability to upregulate the expression of lineage-specific regulators such as GATA3 and RORγt/RORα, enabling the production of high-levels of selected cytokines as well as a whole array of additional lineage-specific functions. Given the known ability of IRF4 to cooperate with STATs in B cells, it is tempting to speculate that cooperation of IRF4 with either Stat6 or Stat3 is critical for the lineage-specific effects of IRF4 and contributes to the differential ability of IRF4 to regulate the differentiation of Th2 and Th17 cells, respectively. Cooperation with Foxp3 would instead enable IRF4 to function in a Treg-specific manner. The specific levels/kinetics of IRF4 expression coupled with the presence/absence of additional cofactors are also likely to participate in the precise regulation of the gamut of IRF4 targets. We previously proposed that IRF4 serves more as a ‘master integrator’ of lymphocyte responses rather than a ‘master regulator’ of specific differentiation programs (135). We believe that recent data further supports the contention that IRF4 functions as a molecular hub that can enable T cells to integrate the information provided to them by different pathways and fine-tune their effector functions in response to a complex and changing environment.
Fig. 2. Model of the regulation and role of IRF4 under distinct Th differentiation conditions.
Novel pathways regulating IRF4
As we have outlined above, the expression of IRF4 is upregulated upon TCR engagement and IRF4 has the potential to control a wide-range of effector Th responses. Regulatory mechanisms must, therefore, be in place to ensure that its function is tightly controlled and that the production of potentially pathogenic cytokines, like IL-17 and IL-21, only occurs in response to the appropriate antigen and in the setting of the proper inflammatory milieu. In this section, we describe our studies that have led us to uncover an unexpected link between IRF4 and Rho GTPase-mediated pathways as one of the key mechanisms that restrains the function of IRF4 under neutral conditions. We first briefly outline the basic scheme of Rho GTPase-mediated pathways and then focus our discussion on Def6, a novel component of these pathways, which plays a major role in the control of IRF4 function and the production of IL-17 and IL-21.
Rho GTPase-mediated pathways: the basic scheme
The Rho family of GTPases is a large family of proteins, which, in hematopoietic cells, includes RhoA, Rac1, Rac2, and Cdc42 (136-138). Like other small GTPases, Rho GTPases behave like ‘molecular switches’ that cycle between an inactive, GDP-bound, and an active, GTP-bound, state. Once they become activated, Rho GTPases control cytoskeletal dynamics as well as numerous signaling pathways such as the activation of MAPKs due to their capacity to bind to and activate a large number of downstream effector molecules, which include serine/threonine kinases, lipid kinases, and adapters. Rac and Cdc42 can bind to many of the same effectors such as the PAK kinases. In contrast, RhoA usually targets a different set of effectors, such as the Rho-associated coiled-coil-containing protein kinases. The activation of Rho GTPases is primarily controlled by a class of proteins termed guanine nucleotide exchange factors (GEFs) (139), which catalyze the release of GDP leading to the formation of active GTP-bound Rho GTPases. Most GEFs belong to the Dbl family of proteins, which are characterized by the presence of a catalytic Dbl-homology (DH) domain followed by a C-terminal PH domain necessary for proper intracellular localization and function. The ability of GEFs to activate Rho GTPases is itself strictly regulated since GEFs normally exist in an inactive state and become activated in response to extracellular stimuli via phosphorylation, changes in subcellular localization, or interaction with cofactors.
All the major classes of Rho GTPases are expressed in CD4+ T cells, and TCR engagement has been shown to lead to the activation of Rac1 and Rac2, Cdc42, and RhoA (140-144). TCR-induced activation of Rac proteins has been implicated in the activation of MAPKs and in the regulation of cytoskeletal processes essential for immunological synapse (IS) assembly, migration, and even apoptosis (145-148). Interestingly, CD4+ T cells from Rac2−/− mice have been found to exhibit defective TCR-mediated proliferation and diminished IL-2 and IFN-γ production (149, 150). The activation of Cdc42 has also been implicated in IS formation (143, 151), while activation of RhoA has been linked to ERK1/2 activation, calcium responses, and IL-2 production (140). It is, however, important to note that a precise understanding of the role of Rho GTPases in mature CD4+ T cells has not yet been attained due to the fact that many of these GTPases (e.g. Rac1 and Rac2) have redundant functions and that the loss of some of these GTPases leads to profound T-cell developmental defects (136).
Def6 (IRF-4 binding protein/SWAP-70 homologous T-cell adapter)
Although the best-characterized GEFs that mediate the activation of Rac proteins in response to TCR stimulation are the Vav proteins (152, 153), additional classes of Rac activators exist in CD4+ T cells. Here we focus on Def6, a novel type of Rac activator that has emerged as a critical controller of T-cell activation. Since Def6 is a relative newcomer in the world of T-cell biology, we first provide some background on this molecule and then discuss its role in T cells and its ability to act as a multifunctional protein that not only activates Rac but also regulates IRF4.
Nomenclature and expression
Def6 was first cloned during a search for genes that are downregulated during differentiation of a murine progenitor cell line toward either a myeloid or erythroid lineage (154). Our group independently cloned the human homologue of Def6 during a yeast two-hybrid screen aimed at identifying proteins interacting with IRF4, and hence we named it IRF4-binding protein (IBP) (155). Another group also independently cloned this molecule during a search for Th2-specific genes and termed it SWAP-70 homologous T-cell adapter (SLAT) (156). To simplify the terminology, we henceforth refer to this protein as Def6 according to the current official name of the gene. The human and murine Def6 cDNA sequences are highly homologous suggesting that Def6 is conserved evolutionarily (155). Def6 exhibits significant sequence homology to only one other molecule, SWAP-70, a novel type of Rac activator (157). The expression of Def6 in lymphoid tissues is broader and more abundant than that of SWAP-70 (155). Importantly, however, mature CD4+ T cells express only Def6 but not SWAP-70 (155, 158). Def6 is highly expressed in most T cells and equivalent levels of Def6 can be detected in all Th effector subsets (155, and unpublished observations) although upregulation of Def6 expression in Th2 cells after 4 days of culture has been reported (156). Interestingly, in B cells, Def6 expression appears to be confined to specific stages of B-cell differentiation, since Def6 is present in coronal or mantle zone B cells but largely absent in germinal center B cells, which instead strongly express SWAP-70 (155, 158). The human Def6 gene is located on chromosome 6p21.31, centromeric to the MHC complex (155).
Structure
Def6 and SWAP-70 share a similar molecular structure, which includes a putative N-terminal EF-hand motif and a central PH domain (Fig. 3). Between these two modules Def6 contains a series of tyrosine phosphorylation sites, which have been shown to be a target for Lck (159, 160). The C-terminal region of both proteins contains an α-helical region, which displays limited sequence homology to the Dbl-homology (DH) domain of classical GEFs and has thus been termed a DH-like (DHL) domain (157, 161). The unusual location of the DHL module at the C-terminus, rather than at the N-terminus, of the PH domain in both SWAP-70 and Def6 molecules, coupled with their low-degree of homology with other DH domain-containing proteins support the notion that these two proteins represent a distinctive class of activators for Rho GTPases. The DHL domain has been shown to be essential for the ability of Def6 to activate Rac and Cdc42 (160, 161). Interestingly, Def6 can also bind activated forms of Rho GTPases (162). This binding is mediated by the amino-terminus of Def6 rather than its C-terminus suggesting a complex interplay between Def6 and Rho GTPases. Def6 and SWAP-70 contain nuclear localization signals and both proteins can be detected not only in the cytoplasm but also in the nucleus (102, 163). As will be discussed below, Def6 not only acts as an activator for Rac but also physically interacts with IRF4 (102). This interaction primarily occurs in the nucleus and involves binding of the carboxy-terminus of Def6 to a region of IRF4 containing most of the IAD.
Fig. 3. Schematic diagram of Def6.

PH, pleckstrin homology domain; DHL, Dbl homology-like domain; NLS, nuclear localization signal
Regulation
Similarly to what has been described for many classical GEFs, the activity of Def6 is under tight regulation. Indeed full-length Def6 does not exhibit any GEF activity due to an autoinhibitory interaction, which likely occurs between its N-terminus and its C-terminus. TCR engagement, via the activation of Lck, leads to the tyrosine phosphorylation of the N-terminus of Def6 (159, 160). This phosphorylation event is believed to disrupt this autoinhibitory interaction and enables Def6 to be recruited to the immunological synapse and to activate specific Rac- and Cdc42-mediated pathways (156, 159, 160). In addition to phosphorylation events, the Def6 PH domain has been shown to bind PI(3,4,5)P3 (160, 162) and this interaction may also participate in the recruitment of Def6 to the immunological synapse and in the control of its GEF activity although the requirement for this step is more variable (159, 160). These data are thus consistent with a model whereby Def6 normally exists in an “inactive” or dormant conformation, which is rendered active in response to TCR-mediated signals. Whether similar or different sets of regulatory conditions controls its nuclear function and/or its ability to interact with IRF4 is currently not known.
Spontaneous development of autoimmunity in the absence of Def6
The first clue that Def6 might play an important and unique immunoregulatory role in vivo came from studies of mice deficient in Def6 (164). Def6 deficient mice are viable and fertile and do not exhibit any major T or B cell developmental abnormalities except for a modest defect in the proliferation of DN thymocytes (164, 165). Beginning at 5 months of age, however, ≈60% of the Def6 deficient female mice on a mixed 129/BL6 background develop a lupus-like syndrome characterized by multiple enlarged lymph nodes, splenomegaly, hypergammaglobulinemia, anti-dsDNA autoantibodies, and glomerulonephritis (164). As observed in other spontaneous models of lupus, the lymphoproliferative disorder that develops in the absence of Def6 is characterized by a marked accumulation of effector CD4+ T cells, IgG1+ B cells, and plasma cells (164).
Surprisingly, when Def6 deficient mice are backcrossed onto the Balb/c background and then crossed to DO11.10 mice, which carry a transgenic T cell receptor for ovalbumin (166), absence of Def6 leads to the spontaneous development of a disorder that clinically resembles RA (102). Indeed beginning at about 2 months of age, Def6 deficient DO11.10 mice spontaneously develop symmetrical joint swelling with hyperemia. The symptoms are chronic and progressive and often lead to impaired mobility upon aging. Female mice tend to develop more severe symptoms than male mice indicating a sex bias in the development of this disorder albeit not to the same degree as that observed in the development of the lupus-like disorder. Histopathologically the lesions are characterized by severe synovitis with a striking accumulation of neutrophils, macrophages, lymphocytes, and plasma cells. Pannus formation with destruction of the adjacent cartilage and the subchondral bone is clearly evident. The serologic abnormalities in Def6 deficient DO11.10 mice are characterized by elevated titers of Rheumatoid Factor and anti-CCP antibodies, also highly reminiscent of the findings in RA patients. Interestingly, many Def6 deficient DO11.10 mice also develop severe inflammation of the large- and medium-sized elastic arteries and exhibit early mortality. Analysis of the lymphoid organs of these mice demonstrates a marked accumulation of effector CD4+ T cells that express the DO11.10 TCR transgene as well as high levels of ICOS and other activation markers. These results thus indicate that, in the absence of exposure to ovalbumin, the lack of Def6 leads DO11.10 TCR CD4+ T cells to become spontaneously activated likely in response to self-peptides derived from the connective tissue/extracellular matrix of the joints and blood vessels.
Molecular pathways controlled by Def6
Our laboratory has conducted an extensive analysis to delineate the mechanisms by which absence of Def6 leads to such pathophysiology. Given that transfer experiments supported the idea that CD4+ T cells play a key role in this pathophysiology (102), we focused our studies on this cellular compartment. We have found that the lack of Def6 does not affect central tolerance or the development/function of regulatory T cells (102). The absence of Def6, instead, profoundly affects two major aspects of peripheral CD4+ T-cell biology, their ability to properly respond to an antigenic encounter and their capacity to produce IL-17 and IL-21. Here we review the present knowledge regarding the role of Def6 in each of these two processes. Whereas Def6 also controls the elimination of activated T cells (164), much less is known about this aspect of Def6 function and thus a detailed discussion of the role of Def6 in T-cell apoptosis is not be included (although this effect also likely contributes to the emergence of autoimmunity in Def6-deficient mice).
Def6 and TCR responsiveness
Def6 plays a complex role in the regulation of TCR responsiveness. Consistent with their spontaneous activation in vivo, Def6-deficient DO11.10 T cells display enhanced proliferative responses in vitro in response to low levels of antigenic stimulation (102). In unpublished studies, we have observed, furthermore, that absence of Def6 leads to exaggerated homeostatic proliferation of T cells in response to lymphopenia, a process driven by interaction of the TCR with self-pMHC complexes (167-171). The immunopathology observed in Def6-deficient DO11.10, which occurs in the absence of any exposure to the exogenous antigen ovalbumin, is thus likely to be driven by a heightened recognition of the DO11.10 TCR for self-peptides, which might be expressed in the tissues where the inflammatory response is occurring. In support of this notion, immunoblot analysis with serum derived from Def6-deficient DO11.10 but not with serum from control mice revealed strong recognition of a 66 kDa band present in extracts from joints and elastic arteries but not in extracts obtained from spleens or lymph nodes (unpublished observations).
While Def6-deficient CD4+ T cells become hyperresponsive to low levels of antigenic stimulation, the lack of Def6 also renders CD4+ T cells less responsive to strong stimuli as evidenced by the defective proliferation of Def6-deficient DO11.10 CD4+ T cells to high doses of ovalbumin (102). In line with these findings, nontransgenic Def6-deficient CD4+ T cells exhibit impaired proliferative responses upon stimulation with αCD3 and αCD28 (164, 165). These studies thus raise the possibility that absence of Def6, while predisposing to autoimmunity, may also impair the ability of an individual to effectively clear infections. Thus Def6 may be a critical component of the machinery that enables CD4+ T cells to properly discriminate between low affinity interactions with self-peptides, which are required for their maintenance in the periphery, and high affinity interactions with foreign peptides that result in T-cell activation.
The mechanisms by which Def6 controls TCR responsiveness are still being investigated. Many of the early TCR-induced signaling events, such as the phosphorylation of Lck, ZAP-70, and AKT, are not affected by the absence of Def6 (164, 165). The lack of Def6, however, leads to abnormalities in TCR-mediated ERK1/2 activation, IS formation, and TCR-mediated actin polymerization (164, 165). The latter defect can be rescued by a constitutively active form of Rac or by wildtype Def6 but not by a Def6 mutant lacking the DHL domain (164). Given that Rho GTPases are critical regulators of both MAPK activation and IS assembly, and that these processes have previously been shown to underlie the ability of a T cell to properly sense the potency of TCR engagement (172, 173) these findings support the idea that the ability of Def6 to activate Rac and Cdc42 plays a critical role in its capacity to control TCR responsiveness. More work, however, is needed to fully define the molecular orchestration conducted by Def6 to control this process.
Def6 and CD4+ T-cell effector function
Def6 also controls the effector function of CD4+ T cells independently of its effects on TCR responsiveness. Importantly, Def6 regulates the production of IL-17 and IL-21 (102). Indeed, we have found that naïve Def6-deficient CD4+ T cells stimulated under neutral conditions exhibit an increased ability to produce IL-17 and IL-21 when compared to wt CD4+ T cells. Repeated stimulation of Def6-deficient T cells in vitro leads to even higher levels of IL-17 and IL-21 production (unpublished observations), suggesting amplification of this circuit upon chronic stimulation of these cells. The increased production of IL-17 is associated with enhanced expression of RORγt. Consistent with these findings increased levels of IL-17 and IL-21 can be observed in the sera, lymphoid organs, and joints of the Def6-deficient mice that develop RA-like arthritis (102, authors' unpublished observations). Interestingly, the absence of Def6 is also accompanied by a profound disorganization of the lymphoid architecture characterized by the accumulation of plasma cells within the T-cell zone suggesting that the deregulated Th effector function observed in the absence of Def6 might lead to aberrant T-cell interactions with B cells (102). Our laboratory has found that a key mechanism by which Def6 controls the production of IL-17 and IL-21 is to directly interact with IRF4 and prevent IRF4 from targeting and transactivating the regulatory regions of IL-17 and IL-21 (102). The deregulated production of IL-17 and IL-21 observed in the absence of Def6, indeed, is abolished by the concurrent lack of IRF4. Interestingly, the lack of Def6 does not lead to increased production of IL-17 and IL-21 when CD4+ T cells are cultured under Th17-skewing conditions (unpublished observations), indicating that the ability of Def6 to inhibit IRF4 function and the production of IL-17 and IL-21 is dependent on the presence/absence of an inflammatory milieu. While absence of Def6 leads to deregulated IL-17 and IL-21 production in vitro by CD4+ T cells derived from various strains (unpublished observations), the development of pathophysiology in vivo exhibits strain-dependent effects suggesting that, similarly to what is observed in human autoimmune disorders, the full enactment of the pathogenic potential of Def6-deficient CD4+ T cells is shaped by additional genetic and environmental interactions.
Def6 also controls the production of other cytokines, in particular IL-2 and IFN-γ (164, 165). In this case, however, the absence of Def6 leads to a decrease, rather than an increase, in the production of these cytokines. Effects on IL-4 production have also been reported although they tend to be more variable (164, 165, authors' unpublished observations). Th1 and Th2 responses in vivo upon immunization with either OVA-CFA or OVA-alum also appear to be affected by the absence of Def6 (165). The mechanism responsible for the effects of Def6 on the production of these other cytokines does not appear to be related to the Def6-IRF4 interaction but has instead been found to be due to the ability of Def6 to control calcium mobilization and NFAT translocation in a GEF-dependent manner (159, 165). Defective activation of AP-1 complexes may also contribute to these abnormalities (164). Interestingly, while the lack of Def6 in vivo leads to a marked upregulation of ICOS expression, no effects of Def6 deficiency can be observed on the TCR-mediated induction of ICOS in vitro, suggesting that this effect is not intrinsic to T cells but is likely regulated in vivo by interactions with other cellular compartments.
Summary and perspectives
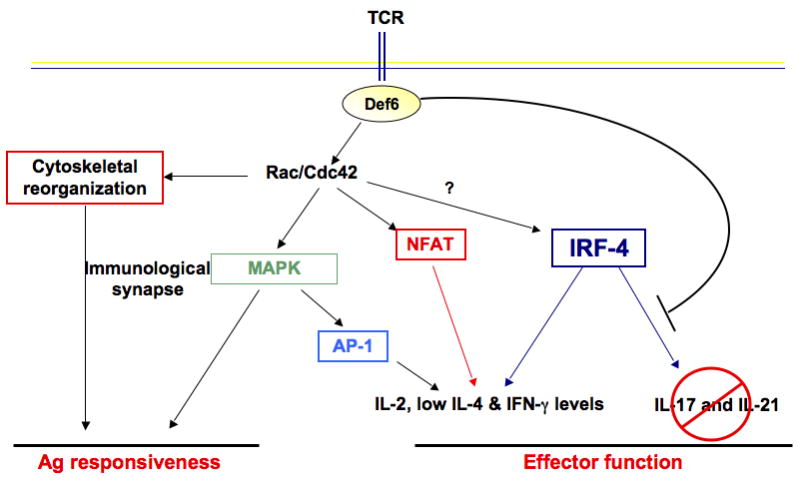
The described studies thus support a model whereby Def6, via its dual capacity to control TCR responsiveness and Th effector functions, is an important component of the molecular machinery that links TCR sensing to the accurate acquisition of Th effector functions thus enabling Def6 to play a key role in the prevention of T-cell-mediated autoimmunity (Fig. 4). Importantly, Def6 seems to function as a critical ‘brake’ that, under neutral conditions, restrains the ability of IRF4 to access a gene expression program (which includes RORγt, IL-17, and IL-21) that should be executed only under selected inflammatory conditions. One therefore would expect that when CD4+ T cells are exposed to the appropriate inflammatory milieu, this brake is released so that IRF4 can now implement this transcriptional program. The finding that, under Th17 skewing conditions, the absence of Def6 does not significantly affect Th17 differentiation indeed supports this idea. We have not detected any differences in the expression and/or nuclear localization of Def6 under Th0 versus Th17 condition, leading us to suspect that the assembly/disassembly of the Def6-IRF4 complex may be regulated via posttranslational modifications. We have furthermore recently observed that Def6 can also control the function of IRF4 in a manner dependent on its ability to influence the activation of Rho GTPases. Thus, both aspects of Def6 biology eventually converge in regulating IRF4 function.
Fig. 4. Model of the role of Def6 in Th cells.
This novel spontaneous model of arthritis can provide insights into the multiple pathways by which arthritogenic T cells may arise. Unlike induced models of arthritis like CIA, where the development of pathogenic Th17 cells is driven by providing strong inflammatory stimuli (i.e. CFA), spontaneous models like the Def6-deficient DO11.10 mice suggest that arthritogenic Th17-like cells can also arise in the absence of such strong inflammatory milieu. We favor a scenario whereby the enhanced responsiveness of Def6-deficient DO11.10 T cells to self-peptides derived from the connective tissue/extracellular matrix of the joints and blood vessels leads to a low-level of activation of these T cells, which is accompanied by the aberrant production of modest levels of IL-17 and IL-21. Due to the pervasive presence of these self-peptides we envision that repetitive stimulation of these cells will occur eventually leading to the production of sufficient levels of IL-17 and IL-21 to mediate pathogenic effects. Transient lymphopenic states may hasten the expansion of these arthritogenic T cells. It is intriguing to speculate that the IL-17 and IL-21-producing T cells that develop in the absence of strong inflammatory conditions may not fully complete the Th17 differentiation program and may instead represent only partially differentiated Th17-like cells. Since the transcriptional programs expressed by such partially differentiated Th17 cells may be different from those of fully differentiated Th17 cells, understanding the function and consequence of these heterogeneous IL-17 producing arthritogenic T-cell populations will have important clinical and therapeutic implications.
Acknowledgments
Work in our laboratory is supported by grants from the National Institutes of Health, the Alliance for Lupus Research, and the Lupus Research Institute.
References
- 1.Goodnow CC, Sprent J, de St Groth BF, Vinuesa CG. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature. 2005;435:590–597. doi: 10.1038/nature03724. [DOI] [PubMed] [Google Scholar]
- 2.Lohr J, Knoechel B, Nagabhushanam V, Abbas AK. T-cell tolerance and autoimmunity to systemic and tissue-restricted self-antigens. Immunol Rev. 2005;204:116–127. doi: 10.1111/j.0105-2896.2005.00241.x. [DOI] [PubMed] [Google Scholar]
- 3.Germain RN. Ligand-dependent regulation of T cell development and activation. Immunol Res. 2003;27:277–286. doi: 10.1385/IR:27:2-3:277. [DOI] [PubMed] [Google Scholar]
- 4.Love PE, Chan AC. Regulation of thymocyte development: only the meek survive. Curr Opin Immunol. 2003;15:199–203. doi: 10.1016/s0952-7915(03)00002-5. [DOI] [PubMed] [Google Scholar]
- 5.Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol. 2003;21:139–176. doi: 10.1146/annurev.immunol.21.120601.141107. [DOI] [PubMed] [Google Scholar]
- 6.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dustin ML. T-cell activation through immunological synapses and kinapses. Immunol Rev. 2008;221:77–89. doi: 10.1111/j.1600-065X.2008.00589.x. [DOI] [PubMed] [Google Scholar]
- 8.McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- 9.Toh ML, Miossec P. The role of T cells in rheumatoid arthritis: new subsets and new targets. Curr Opin Rheumatol. 2007;19:284–288. doi: 10.1097/BOR.0b013e32805e87e0. [DOI] [PubMed] [Google Scholar]
- 10.Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–467. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 14.Ishigame H, et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 15.Yang XO, et al. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamaguchi Y, et al. IL-17B and IL-17C are associated with TNF-alpha production and contribute to the exacerbation of inflammatory arthritis. J Immunol. 2007;179:7128–7136. doi: 10.4049/jimmunol.179.10.7128. [DOI] [PubMed] [Google Scholar]
- 17.Kuestner RE, et al. Identification of the IL-17 receptor related molecule IL-17RC as the receptor for IL-17F. J Immunol. 2007;179:5462–5473. doi: 10.4049/jimmunol.179.8.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zrioual S, et al. IL-17RA and IL-17RC receptors are essential for IL-17A-induced ELR+ CXC chemokine expression in synoviocytes and are overexpressed in rheumatoid blood. J Immunol. 2008;180:655–663. doi: 10.4049/jimmunol.180.1.655. [DOI] [PubMed] [Google Scholar]
- 19.Novatchkova M, Leibbrandt A, Werzowa J, Neubuser A, Eisenhaber F. The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem Sci. 2003;28:226–229. doi: 10.1016/S0968-0004(03)00067-7. [DOI] [PubMed] [Google Scholar]
- 20.Yao Z, et al. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811–821. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 21.Ruddy MJ, et al. Functional cooperation between interleukin-17 and tumor necrosis factor-alpha is mediated by CCAAT/enhancer-binding protein family members. J Biol Chem. 2004;279:2559–2567. doi: 10.1074/jbc.M308809200. [DOI] [PubMed] [Google Scholar]
- 22.Shen F, Hu Z, Goswami J, Gaffen SL. Identification of common transcriptional regulatory elements in interleukin-17 target genes. J Biol Chem. 2006;281:24138–24148. doi: 10.1074/jbc.M604597200. [DOI] [PubMed] [Google Scholar]
- 23.Martel-Pelletier J, Mineau F, Jovanovic D, Di Battista JA, Pelletier JP. Mitogen-activated protein kinase and nuclear factor kappaB together regulate interleukin-17-induced nitric oxide production in human osteoarthritic chondrocytes: possible role of transactivating factor mitogen-activated protein kinase-activated proten kinase (MAPKAPK) Arthritis Rheum. 1999;42:2399–2409. doi: 10.1002/1529-0131(199911)42:11<2399::AID-ANR19>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 24.Gaffen SL. Biology of recently discovered cytokines: interleukin-17--a unique inflammatory cytokine with roles in bone biology and arthritis. Arthritis Res Ther. 2004;6:240–247. doi: 10.1186/ar1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koenders MI, Joosten LA, van den Berg WB. Potential new targets in arthritis therapy: interleukin (IL)-17 and its relation to tumour necrosis factor and IL-1 in experimental arthritis. Ann Rheum Dis. 2006;65(Suppl):iii29–iii33. doi: 10.1136/ard.2006.058529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lubberts E. IL-17/Th17 targeting: on the road to prevent chronic destructive arthritis? Cytokine. 2008;41:84–91. doi: 10.1016/j.cyto.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 27.Chabaud M, et al. Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999;42:963–970. doi: 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 28.Lubberts E, et al. IL-1-independent role of IL-17 in synovial inflammation and joint destruction during collagen-induced arthritis. J Immunol. 2001;167:1004–1013. doi: 10.4049/jimmunol.167.2.1004. [DOI] [PubMed] [Google Scholar]
- 29.Ziolkowska M, et al. High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol. 2000;164:2832–2838. doi: 10.4049/jimmunol.164.5.2832. [DOI] [PubMed] [Google Scholar]
- 30.Kirkham BW, et al. Synovial membrane cytokine expression is predictive of joint damage progression in rheumatoid arthritis: a two-year prospective study (the DAMAGE study cohort) Arthritis Rheum. 2006;54:1122–1131. doi: 10.1002/art.21749. [DOI] [PubMed] [Google Scholar]
- 31.Bush KA, Farmer KM, Walker JS, Kirkham BW. Reduction of joint inflammation and bone erosion in rat adjuvant arthritis by treatment with interleukin-17 receptor IgG1 Fc fusion protein. Arthritis Rheum. 2002;46:802–805. doi: 10.1002/art.10173. [DOI] [PubMed] [Google Scholar]
- 32.Lubberts E, et al. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004;50:650–659. doi: 10.1002/art.20001. [DOI] [PubMed] [Google Scholar]
- 33.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 34.Ettinger R, Kuchen S, Lipsky PE. The role of IL-21 in regulating B-cell function in health and disease. Immunol Rev. 2008;223:60–86. doi: 10.1111/j.1600-065X.2008.00631.x. [DOI] [PubMed] [Google Scholar]
- 35.Monteleone G, Pallone F, Macdonald TT. Interleukin-21 (IL-21)-mediated pathways in T cell-mediated disease. Cytokine Growth Factor Rev. 2009;20:185–191. doi: 10.1016/j.cytogfr.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 36.Spolski R, Leonard WJ. Interleukin-21: basic biology and implications for cancer and autoimmunity. Annu Rev Immunol. 2008;26:57–79. doi: 10.1146/annurev.immunol.26.021607.090316. [DOI] [PubMed] [Google Scholar]
- 37.Mehta DS, Wurster AL, Grusby MJ. Biology of IL-21 and the IL-21 receptor. Immunol Rev. 2004;202:84–95. doi: 10.1111/j.0105-2896.2004.00201.x. [DOI] [PubMed] [Google Scholar]
- 38.Konforte D, Simard N, Paige CJ. IL-21: an executor of B cell fate. J Immunol. 2009;182:1781–1787. doi: 10.4049/jimmunol.0803009. [DOI] [PubMed] [Google Scholar]
- 39.Imboden JB. The immunopathogenesis of rheumatoid arthritis. Annu Rev Pathol. 2009;4:417–434. doi: 10.1146/annurev.pathol.4.110807.092254. [DOI] [PubMed] [Google Scholar]
- 40.Jang E, Cho SH, Park H, Paik DJ, Kim JM, Youn J. A positive feedback loop of IL-21 signaling provoked by homeostatic CD4+CD25- T cell expansion is essential for the development of arthritis in autoimmune K/BxN mice. J Immunol. 2009;182:4649–4656. doi: 10.4049/jimmunol.0804350. [DOI] [PubMed] [Google Scholar]
- 41.Young DA, et al. Blockade of the interleukin-21/interleukin-21 receptor pathway ameliorates disease in animal models of rheumatoid arthritis. Arthritis Rheum. 2007;56:1152–1163. doi: 10.1002/art.22452. [DOI] [PubMed] [Google Scholar]
- 42.Hsu HC, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 43.Doreau A, et al. Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat Immunol. 2009;10:778–785. doi: 10.1038/ni.1741. [DOI] [PubMed] [Google Scholar]
- 44.Mosmann T, Coffman R. Th1 and Th2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 45.Harrington LE, Mangan PR, Weaver CT. Expanding the effector CD4 T-cell repertoire: the Th17 lineage. Curr Opin Immunol. 2006;18:349–356. doi: 10.1016/j.coi.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 46.Takatori H, Kanno Y, Chen Z, O'Shea JJ. New complexities in helper T cell fate determination and the implications for autoimmune diseases. Mod Rheumatol. 2008;18:533–541. doi: 10.1007/s10165-008-0099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harrington LE, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 48.Park H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Langrish CL, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature. 2008;453:1051–1057. doi: 10.1038/nature07036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong C. Th17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–348. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 52.Zhou L, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Volpe E, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650–657. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 55.Yang L, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454:350–352. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Korn T, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nurieva R, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 58.Zhou L, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 59.McGeachy MJ, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314–324. doi: 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stritesky GL, Yeh N, Kaplan MH. IL-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol. 2008;181:5948–5955. doi: 10.4049/jimmunol.181.9.5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 62.Chung Y, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ben-Sasson SZ, et al. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci USA. 2009;106:7119–7124. doi: 10.1073/pnas.0902745106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koenders MI, et al. Interleukin-1 drives pathogenic Th17 cells during spontaneous arthritis in interleukin-1 receptor antagonist-deficient mice. Arthritis Rheum. 2008;58:3461–3470. doi: 10.1002/art.23957. [DOI] [PubMed] [Google Scholar]
- 65.Iezzi G, Sonderegger I, Ampenberger F, Schmitz N, Marsland BJ, Kopf M. CD40-CD40L cross-talk integrates strong antigenic signals and microbial stimuli to induce development of IL-17-producing CD4+ T cells. Proc Natl Acad Sci USA. 2009;106:876–881. doi: 10.1073/pnas.0810769106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saeki K, et al. A major lipid raft protein raftlin modulates T cell receptor signaling and enhances th17-mediated autoimmune responses. J Immunol. 2009;182:5929–5937. doi: 10.4049/jimmunol.0802672. [DOI] [PubMed] [Google Scholar]
- 67.Laurence A, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 68.Batten M, et al. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. 2006;7:929–936. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- 69.Stumhofer JS, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7:937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 70.Niedbala W, et al. Interleukin 27 attenuates collagen-induced arthritis. Ann Rheum Dis. 2008;67:1474–1479. doi: 10.1136/ard.2007.083360. [DOI] [PubMed] [Google Scholar]
- 71.Elias KM, et al. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood. 2008;111:1013–1020. doi: 10.1182/blood-2007-06-096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mucida D, et al. Reciprocal Th17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 73.Schambach F, Schupp M, Lazar MA, Reiner SL. Activation of retinoic acid receptor-alpha favours regulatory T cell induction at the expense of IL-17-secreting T helper cell differentiation. Eur J Immunol. 2007;37:2396–2399. doi: 10.1002/eji.200737621. [DOI] [PubMed] [Google Scholar]
- 74.Xiao S, et al. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol. 2008;181:2277–2284. doi: 10.4049/jimmunol.181.4.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nozaki Y, Yamagata T, Sugiyama M, Ikoma S, Kinoshita K, Funauchi M. Anti-inflammatory effect of all-trans-retinoic acid in inflammatory arthritis. Clin Immunol. 2006;119:272–279. doi: 10.1016/j.clim.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 76.McGeachy MJ, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 77.Lee YK, et al. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30:92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nurieva R, Yang XO, Chung Y, Dong C. Cutting edge: in vitro generated Th17 cells maintain their cytokine expression program in normal but not lymphopenic hosts. J Immunol. 2009;182:2565–2568. doi: 10.4049/jimmunol.0803931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang XO, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Voo KS, et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci USA. 2009;106:4793–4798. doi: 10.1073/pnas.0900408106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.King C, Tangye SG, Mackay CR. T follicular helper (TFH) cells in normal and dysregulated immune responses. Annu Rev Immunol. 2008;26:741–766. doi: 10.1146/annurev.immunol.26.021607.090344. [DOI] [PubMed] [Google Scholar]
- 82.Bauquet AT, et al. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and Th-17 cells. Nat Immunol. 2009;10:167–175. doi: 10.1038/ni.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nurieva RI, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. 2008;29:138–149. doi: 10.1016/j.immuni.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vogelzang A, McGuire HM, Yu D, Sprent J, Mackay CR, King C. A fundamental role for interleukin-21 in the generation of T follicular helper cells. Immunity. 2008;29:127–137. doi: 10.1016/j.immuni.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 85.Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–655. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 86.Schindler C, Strehlow I. Cytokines and STAT signaling. Adv Pharmacol. 2000;47:113–174. doi: 10.1016/s1054-3589(08)60111-8. [DOI] [PubMed] [Google Scholar]
- 87.O'Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity. 2008;28:477–487. doi: 10.1016/j.immuni.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wei L, Laurence A, Elias KM, O'Shea JJ. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem. 2007;282:34605–3410. doi: 10.1074/jbc.M705100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang XO, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 90.Mathur AN, et al. Stat3 and Stat4 direct development of IL-17-secreting Th cells. J Immunol. 2007;178:4901–4907. doi: 10.4049/jimmunol.178.8.4901. [DOI] [PubMed] [Google Scholar]
- 91.Ansel KM, Djuretic I, Tanasa B, Rao A. Regulation of Th2 differentiation and Il4 locus accessibility. Annu Rev Immunol. 2006;24:607–656. doi: 10.1146/annurev.immunol.23.021704.115821. [DOI] [PubMed] [Google Scholar]
- 92.Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol. 2003;21:713–758. doi: 10.1146/annurev.immunol.21.120601.140942. [DOI] [PubMed] [Google Scholar]
- 93.Ivanov II, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 94.Yang XO, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu XK, Lin X, Gaffen SL. Crucial role for nuclear factor of activated T cells in T cell receptor-mediated regulation of human interleukin-17. J Biol Chem. 2004;279:52762–52771. doi: 10.1074/jbc.M405764200. [DOI] [PubMed] [Google Scholar]
- 96.Kim HP, Korn LL, Gamero AM, Leonard WJ. Calcium-dependent activation of interleukin-21 gene expression in T cells. J Biol Chem. 2005;280:25291–25297. doi: 10.1074/jbc.M501459200. [DOI] [PubMed] [Google Scholar]
- 97.Mehta DS, Wurster AL, Weinmann AS, Grusby MJ. NFATc2 and T-bet contribute to T-helper-cell-subset-specific regulation of IL-21 expression. Proc Natl Acad Sci USA. 2005;102:2016–2021. doi: 10.1073/pnas.0409512102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schraml BU, et al. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature. 2009;460:405–409. doi: 10.1038/nature08114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhu J, et al. Down-regulation of Gfi-1 expression by TGF-beta is important for differentiation of Th17 and CD103+ inducible regulatory T cells. J Exp Med. 2009;206:329–341. doi: 10.1084/jem.20081666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brustle A, et al. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol. 2007;8:958–966. doi: 10.1038/ni1500. [DOI] [PubMed] [Google Scholar]
- 101.Huber M, et al. IRF4 is essential for IL-21-mediated induction, amplification, and stabilization of the Th17 phenotype. Proc Natl Acad Sci USA. 2008;105:20846–20851. doi: 10.1073/pnas.0809077106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen Q, et al. IRF-4 Binding Protein inhibits interleukin-17 and interleukin-21 production by controlling the activity of IRF-4 transcription factor. Immunity. 2008;299:899–911. doi: 10.1016/j.immuni.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 104.Lohoff M, Mak TW. Roles of interferon-regulatory factors in T-helper-cell differentiation. Nat Rev Immunol. 2005;5:125–135. doi: 10.1038/nri1552. [DOI] [PubMed] [Google Scholar]
- 105.Chen W, et al. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nat Struct Mol Biol. 2008;15:1213–1220. doi: 10.1038/nsmb.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Qin BY, et al. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol. 2003;10:913–921. doi: 10.1038/nsb1002. [DOI] [PubMed] [Google Scholar]
- 107.Takahasi K, et al. X-ray crystal structure of IRF-3 and its functional implications. Nat Struct Biol. 2003;10:922–927. doi: 10.1038/nsb1001. [DOI] [PubMed] [Google Scholar]
- 108.Eisenbeis C, Singh H, Storb U. Pip, a novel IRF family member, is a lymphoid-specific, PU.1-dependent transcriptional activator. Genes Dev. 1995;9:1377–1387. doi: 10.1101/gad.9.11.1377. [DOI] [PubMed] [Google Scholar]
- 109.Brass AL, Zhu AQ, Singh H. Assembly requirements of PU.1-Pip (IRF-4) activator complexes: inhibiting function in vivo using fused dimers. EMBO J. 1999;18:977–991. doi: 10.1093/emboj/18.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Perkel JM, Atchison ML. A two-step mechanism for recruitment of Pip by PU.1. J Immunol. 1998;160:241–252. [PubMed] [Google Scholar]
- 111.Gupta S, Jiang M, Anthony A, Pernis A. Lineage-specific modulation of IL-4 signaling by interferon regulatory factor 4. J Exp Med. 1999;190:1837–1848. doi: 10.1084/jem.190.12.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Brass A, Kehrli E, Eisenbeis C, Storb U, Singh H. Pip, a lymphoid-restricted IRF, contains a regulatory domain that is important for autoinhibition and ternary complex formation with the Ets factor PU.1. Genes Dev. 1996;10:2335–2347. doi: 10.1101/gad.10.18.2335. [DOI] [PubMed] [Google Scholar]
- 113.Yamagata T, et al. A novel interferon regulatory factor family transcription factor, ICSAT/Pip/LSIRF, that negatively regulates the activity of interferon-regulated genes. Mol Cell Bio. 1996;16:1283–1294. doi: 10.1128/MCB.16.4.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rosenbauer F, Waring JF, Foerster J, Wietstruk M, Philipp D, Horak I. Interferon consensus sequence binding protein and interferon regulatory factor-4/Pip form a complex that represses the expression of the interferon-stimulated gene-15 in macrophages. Blood. 1999;94:4274–4281. [PubMed] [Google Scholar]
- 115.Saito M, et al. A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell. 2007;12:280–292. doi: 10.1016/j.ccr.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 116.Matsuyama T, et al. Molecular cloning of LSIRF, a lymphoid-specific member of the interferon regulatory factor family that binds the interferon-stimulated response element (ISRE) Nucl Acids Res. 1995;23:2127–2136. doi: 10.1093/nar/23.12.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Grossman A, et al. Cloning of human lymphocyte-specific interferon regulatory factor (hLSIRF/hIRF4) and mapping of the gene to 6p23-25. Genomics. 1996;37:229–233. doi: 10.1006/geno.1996.0547. [DOI] [PubMed] [Google Scholar]
- 118.Grumont RJ, Gerondakis S. Rel induces interferon regulatory factor 4 (IRF-4) expression in lymphocytes: modulation of interferon-regulated gene expression by rel/nuclear factor kappaB. J Exp Med. 2000;191:1281–1292. doi: 10.1084/jem.191.8.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sharma S, et al. Activation and regulation of interferon regulatory factor 4 in HTLV type I-infected lymphocytes. AIDS research and human retroviruses. 2000;16:1613–1622. doi: 10.1089/08892220050193047. [DOI] [PubMed] [Google Scholar]
- 120.Honma K, Kimura D, Tominaga N, Miyakoda M, Matsuyama T, Yui K. Interferon regulatory factor 4 differentially regulates the production of Th2 cytokines in naive vs. effector/memory CD4+ T cells. Proc Natl Acad Sci USA. 2008;105:15890–15895. doi: 10.1073/pnas.0803171105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zheng Y, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. 2009;458:351–356. doi: 10.1038/nature07674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lehtonen A, et al. Differential expression of IFN regulatory factor 4 gene in human monocyte-derived dendritic cells and macrophages. J Immunol. 2005;175:6570–6579. doi: 10.4049/jimmunol.175.10.6570. [DOI] [PubMed] [Google Scholar]
- 123.Mittrucker H, et al. Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science. 1997;275:540–543. [PubMed] [Google Scholar]
- 124.Rengarajan J, Mowen KA, Mcbride KD, Smith ED, Singh H, Glimcher LH. Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J Exp Med. 2002;195:1003–1012. doi: 10.1084/jem.20011128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tominaga N, Ohkusu-Tsukada K, Udono H, Abe R, Matsuyama T, Yui K. Development of Th1 and not Th2 immune responses in mice lacking IFN-regulatory factor-4. Int Immunol. 2003;15:1–10. doi: 10.1093/intimm/dxg001. [DOI] [PubMed] [Google Scholar]
- 126.Falini B, et al. A monoclonal antibody (MUM1p) detects expression of the MUM1/IRF4 protein in a subset of germinal center B cells, plasma cells, and activated T cells. Blood. 2000;95:2084–2092. [PubMed] [Google Scholar]
- 127.Lohoff M, et al. Dysregulated T helper cell differentiation in the absence of interferon regulatory factor 4. Proc Natl Acad Sci USA. 2002;99:11808–11812. doi: 10.1073/pnas.182425099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Klein U, Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol. 2008;8:22–33. doi: 10.1038/nri2217. [DOI] [PubMed] [Google Scholar]
- 129.Shaffer AL, Emre NC, Romesser PB, Staudt LM. IRF4: Immunity. Malignancy! Therapy? Clin Cancer Res. 2009;15:2954–2961. doi: 10.1158/1078-0432.CCR-08-1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hu CM, Jang SY, Fanzo JC, Pernis AB. Modulation of T cell cytokine production by interferon regulatory factor-4. J Biol Chem. 2002;277:49238–49246. doi: 10.1074/jbc.M205895200. [DOI] [PubMed] [Google Scholar]
- 131.Lee CG, et al. A distal cis-regulatory element, CNS-9, controls NFAT1 and IRF4-mediated IL-10 gene activation in T helper cells. Mol Immunol. 2009;46:613–621. doi: 10.1016/j.molimm.2008.07.037. [DOI] [PubMed] [Google Scholar]
- 132.Cao S, Liu J, Song L, Ma X. The protooncogene c-Maf is an essential transcription factor for IL-10 gene expression in macrophages. J Immunol. 2005;174:3484–3492. doi: 10.4049/jimmunol.174.6.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Xu J, et al. c-Maf regulates IL-10 expression during Th17 polarization. J Immunol. 2009;182:6226–6236. doi: 10.4049/jimmunol.0900123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ahyi AN, Chang HC, Dent AL, Nutt SL, Kaplan MH. IFN regulatory factor 4 regulates the expression of a subset of Th2 cytokines. J Immunol. 2009;183:1598–1606. doi: 10.4049/jimmunol.0803302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pernis AB. The role of IRF-4 in B and T cell activation and differentiation. J Interf Cytok Res. 2002;22:111–120. doi: 10.1089/107999002753452728. [DOI] [PubMed] [Google Scholar]
- 136.Bustelo XR, Sauzeau V, Berenjeno IM. GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays. 2007;29:356–370. doi: 10.1002/bies.20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 138.Tybulewicz VL, Henderson RB. Rho family GTPases and their regulators in lymphocytes. Nat Rev Immunol. 2009;9:630–644. doi: 10.1038/nri2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 2002;16:1587–1609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- 140.Angkachatchai V, Finkel Th. ADP-ribosylation of rho by C3 ribosyltransferase inhibits IL-2 production and sustained calcium influx in activated T cells. J Immunol. 1999;163:3819–3825. [PubMed] [Google Scholar]
- 141.Sanui T, et al. DOCK2 is essential for antigen-induced translocation of TCR and lipid rafts, but not PKC-theta and LFA-1, in T cells. Immunity. 2003;19:119–129. doi: 10.1016/s1074-7613(03)00169-9. [DOI] [PubMed] [Google Scholar]
- 142.Genot EM, Arrieumerlou C, Ku G, Burgering BM, Weiss A, Kramer IM. The T-cell receptor regulates Akt (protein kinase B) via a pathway involving Rac1 and phosphatidylinositide 3-kinase. Mol Cell Biol. 2000;20:5469–5478. doi: 10.1128/mcb.20.15.5469-5478.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tskvitaria-Fuller I, Seth A, Mistry N, Gu H, Rosen MK, Wulfing C. Specific patterns of Cdc42 activity are related to distinct elements of T cell polarization. J Immunol. 2006;177:1708–1720. doi: 10.4049/jimmunol.177.3.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tharaux PL, et al. Rho kinase promotes alloimmune responses by regulating the proliferation and structure of T cells. J Immunol. 2003;171:96–105. doi: 10.4049/jimmunol.171.1.96. [DOI] [PubMed] [Google Scholar]
- 145.Bustelo XR. Understanding Rho/Rac biology in T-cells using animal models. Bioessays. 2002;24:602–612. doi: 10.1002/bies.10107. [DOI] [PubMed] [Google Scholar]
- 146.Cantrell DA. GTPases and T cell activation. Immunol Rev. 2003;192:122–130. doi: 10.1034/j.1600-065x.2003.00028.x. [DOI] [PubMed] [Google Scholar]
- 147.Vicente-Manzanares M, Sanchez-Madrid F. Role of the cytoskeleton during leukocyte responses. Nat Rev Immunol. 2004;4:110–122. doi: 10.1038/nri1268. [DOI] [PubMed] [Google Scholar]
- 148.Burkhardt JK, Carrizosa E, Shaffer MH. The actin cytoskeleton in T cell activation. Annu Rev Immunol. 2008;26:233–259. doi: 10.1146/annurev.immunol.26.021607.090347. [DOI] [PubMed] [Google Scholar]
- 149.Li B, et al. Role of the guanosine triphosphatase Rac2 in T helper 1 cell differentiation. Science. 2000;288:2219–2222. doi: 10.1126/science.288.5474.2219. [DOI] [PubMed] [Google Scholar]
- 150.Yu H, Leitenberg D, Li B, Flavell RA. Deficiency of small GTPase Rac2 affects T cell activation. J Exp Med. 2001;194:915–926. doi: 10.1084/jem.194.7.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cannon JL, et al. Wasp recruitment to the T cell:APC contact site occurs independently of Cdc42 activation. Immunity. 2001;15:249–259. doi: 10.1016/s1074-7613(01)00178-9. [DOI] [PubMed] [Google Scholar]
- 152.Cantrell D. Vav-1 and T cells. Eur J Immunol. 2003;33:1070–1072. doi: 10.1002/eji.200323942. [DOI] [PubMed] [Google Scholar]
- 153.Tybulewicz VL. Vav-family proteins in T-cell signalling. Curr Opin Immunol. 2005;17:267–274. doi: 10.1016/j.coi.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 154.Hotfilder M, Baxendale S, Cross MA, Sablitzky F. Def-2, -3, -6, -8, novel mouse genes differentially expressed in the hematopoietic system. Br J Haematol. 1999;106:335–344. doi: 10.1046/j.1365-2141.1999.01551.x. [DOI] [PubMed] [Google Scholar]
- 155.Gupta S, et al. Molecular cloning of IBP, a SWAP-70 homologous GEF, which is highly expressed in the immune system. Human Immunol. 2003;64:389–401. doi: 10.1016/s0198-8859(03)00024-7. [DOI] [PubMed] [Google Scholar]
- 156.Tanaka Y, et al. SWAP-70-like adapter of T cells, an adapter protein that regulates early TCR-initiated signaling in Th2 lineage cells. Immunity. 2003;18:403–414. doi: 10.1016/s1074-7613(03)00054-2. [DOI] [PubMed] [Google Scholar]
- 157.Shinohara M, et al. SWAP-70 is a guanine-nucleotide-exchange factor that mediates signalling of membrane ruffling. Nature. 2002;416:759–763. doi: 10.1038/416759a. [DOI] [PubMed] [Google Scholar]
- 158.Borggrefe T, Masat L, Wabl M, Riwar B, Cattoretti G, Jessberger R. Cellular, intracellular, and developmental expression patterns of murine SWAP-70. Eur J Immunol. 1999;29:1812–1822. doi: 10.1002/(SICI)1521-4141(199906)29:06<1812::AID-IMMU1812>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 159.Becart S, Balancio AJ, Charvet C, Feau S, Sedwick CE, Altman A. Tyrosine-phosphorylation-dependent translocation of the SLAT protein to the immunological synapse is required for NFAT transcription factor activation. Immunity. 2008;29:704–719. doi: 10.1016/j.immuni.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gupta S, et al. T cell receptor engagement leads to the recruitment of IBP, a novel guanine nucleotide exchange factor, to the immunological synapse. J Biol Chem. 2003;278:43451–43459. doi: 10.1074/jbc.M308960200. [DOI] [PubMed] [Google Scholar]
- 161.Mavrakis KJ, McKinlay KJ, Jones P, Sablitzky F. DEF6, a novel PH-DH-like domain protein, is an upstream activator of the Rho GTPases Rac1, Cdc42, and RhoA. Exp Cell Res. 2004;294:335–344. doi: 10.1016/j.yexcr.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 162.Oka T, Ihara S, Fukui Y. Cooperation of DEF6 with activated Rac in regulating cell morphology. J Biol Chem. 2007;282:2011–2018. doi: 10.1074/jbc.M605153200. [DOI] [PubMed] [Google Scholar]
- 163.Masat L, et al. Association of SWAP-70 with the B cell antigen receptor complex. Proc Natl Acad Sci USA. 2000;97:2180–2184. doi: 10.1073/pnas.040374497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Fanzo JC, et al. Loss of IRF-4-binding protein leads to the spontaneous development of systemic autoimmunity. J Clin Invest. 2006;116:703–714. doi: 10.1172/JCI24096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Becart S, et al. SLAT regulates Th1 and Th2 inflammatory responses by controlling Ca2+/NFAT signaling. J Clin Invest. 2007;117:2164–2175. doi: 10.1172/JCI31640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Murphy KM, Heimberger AB, Loh DY. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science. 1990;250:1720–1723. doi: 10.1126/science.2125367. [DOI] [PubMed] [Google Scholar]
- 167.Baccala R, Theofilopoulos AN. The new paradigm of T-cell homeostatic proliferation-induced autoimmunity. Trends Immunol. 2005;26:5–8. doi: 10.1016/j.it.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 168.Hickman SP, Turka LA. Homeostatic T cell proliferation as a barrier to T cell tolerance. Philos Trans R Soc Lond B Biol Sci. 2005;360:1713–1721. doi: 10.1098/rstb.2005.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jameson SC. T cell homeostasis: keeping useful T cells alive and live T cells useful. Semin Immunol. 2005;17:231–237. doi: 10.1016/j.smim.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 170.Krupica T, Jr, Fry TJ, Mackall CL. Autoimmunity during lymphopenia: a two-hit model. Clin Immunol. 2006;120:121–128. doi: 10.1016/j.clim.2006.04.569. [DOI] [PubMed] [Google Scholar]
- 171.Marleau AM, Sarvetnick N. T cell homeostasis in tolerance and immunity. J Leukoc Biol. 2005;78:575–584. doi: 10.1189/jlb.0105050. [DOI] [PubMed] [Google Scholar]
- 172.Huppa JBGM, Sumen C, Davis MM. Continuous T cell receptor signaling required for synapse maintenance and full effector potential. Nat Immunol. 2003;4:749–755. doi: 10.1038/ni951. [DOI] [PubMed] [Google Scholar]
- 173.Tskvitaria-Fuller I, Rozelle AL, Yin HL, Wulfing C. Regulation of sustained actin dynamics by the TCR and costimulation as a mechanism of receptor localization. J Immunol. 2003;171:2287–2295. doi: 10.4049/jimmunol.171.5.2287. [DOI] [PubMed] [Google Scholar]