

Abstract
Background
The serine/threonine mammalian Ste-20 like kinases (MSTs) are key regulators of apoptosis, cellular proliferation as well as polarization. Deregulation of MSTs has been associated with disease progression in prostate and colorectal cancer. The four human MSTs are regulated differently by C-terminal regions flanking the catalytic domains.
Principal Findings
We have determined the crystal structure of kinase domain of MST4 in complex with an ATP-mimetic inhibitor. This is the first structure of an inactive conformation of a member of the MST kinase family. Comparison with active structures of MST3 and MST1 revealed a dimeric association of MST4 suggesting an activation loop exchanged mechanism of MST4 auto-activation. Together with a homology model of MST2 we provide a comparative analysis of the kinase domains for all four members of the human MST family.
Significance
The comparative analysis identified new structural features in the MST ATP binding pocket and has also defined the mechanism for autophosphorylation. Both structural features may be further explored for inhibitors design.
Enhanced version
This article can also be viewed as an enhanced version in which the text of the article is integrated with interactive 3D representations and animated transitions. Please note that a web plugin is required to access this enhanced functionality. Instructions for the installation and use of the web plugin are available in Text S1.
Introduction
The Sterile-20 protein kinase (Ste20) was first identified in yeast as a key molecule involved in pheromone signaling [1] and has since been found to be involved in the regulation of a large number of diverse cellular functions including cell polarization [2], [3], actin organization [4], [5], regulation of exit from mitosis [6] and apoptosis [7], [8].
Subsequently several mammalian Ste20-like (MST) kinases sharing homology with the yeast ste20 were identified and grouped into two structurally distinct families: p21-activated kinase (PAK) and the germinal centre kinase (GCK) [9]. The GCK group can be further subdivided into GCKI to GCKVIII [10], [11], [12], [13] and are characterized by the positioning of the kinase domain at the N-terminus, as opposed to the PAK group where the kinase domain can be found at the C-terminus. GCKs are also devoid of an N-terminal GTP-binding domain, found in the PAKs.
The subfamily GCKII includes MST1 and MST2, two upstream kinases implicated in cell growth and apoptosis. Both kinases can be activated via caspase-mediated cleavage of the C-terminal inhibitory domain [14]. Activation of MST1 induces activation of JNK and p38 during apoptosis events in certain cell types [15], [16], [17]. Additionally, MST1 can also induce apoptosis via phosphorylation of histone 2B [7], [18], promotion of chromatin condensation [19], [20] and FOXO3 nuclear translocation in neuronal cells [21]. It has been further found that MST1 acts as an essential mediator in apoptosis of K-ras transformed cells [22], [23] and that the Drosophila homologue Hippo phosphorylates the tumor suppressor protein Salvador and is involved in the regulation of expression of cyclin E and apoptosis inhibitor DIAP1 [24]. Similarly, the human orthologue of Salvador (hSav) can bind and be phosphorylated by both MST1 and 2 [25]. Additionally, MST2 is involved in the LATS tumor suppressor pathway via complexation with hSav, RASSF1A, Nore1 and LATS1, resulting in the phosphorylation of LATS1 and transcription of proapoptotic genes [26], [27], [28].
The subfamily GCKIII contains MST3 and MST4 which share almost 90% amino acid identity in the kinase domain but less than 20% in the C-terminal domain [9]. Despite being related to subfamily II (MST1 and MST2), members of these two groups have apparently different cellular functions. Neither MST3 nor MST4 are capable of activating JNK and p38 MAPK kinase activity [29], [30] and both were shown to activate ERK in different cell lines, albeit via different pathways [29], [31], [32]. In contrast to MST1 and MST2 the C-terminus of MST3 and MST4 stimulates kinase activity by promoting auto-phosphorylation. MST4 has been shown to phosphorylate the actin remodeler Ezrin at its regulatory residue T567 resulting in transmission of cell polarization signals [33].
All MSTs (1–4) are expressed almost ubiquitously in all tissues, with higher expression levels found in placenta (MST1, MST2 and MST4), skeletal tissues (MST1, MST2, MST3), kidney (MST1 and MST2), heart (MST3), pancreas (MST3), thymus, and peripheral blood leukocytes (MST4) [29], [30], [34], [35], [36], [37]. Decreasing levels of MST2 has been observed to correlate with loss of RASSF2 in human colorectal carcinomas [38]. Additionally, MST1 and MST4 have been implicated in prostate cancer, whereby MST1 protein levels declined with disease progression [39] and MST4 expression has been correlated with tumourigenicity of human prostate cancer cell lines [36]. Overexpression of MST4 induces anchorage-independent growth and increases in vitro proliferation as well as in vivo tumourigenesis in prostate cancer cell lines [40] suggesting a role in prostate cancer progression. MST4 may also be involved in regulation of cell migration via its interaction with the Golgi protein GM130. Binding to GM130 activates MST4 as well as YSK, a related kinase, by promoting autophosphorylation of a conserved threonine within the activation segment. Interference with the kinase function perturbs perinuclear Golgi organization, cell migration, and invasion into type I collagen [41].
Other physiological functions of the MSTs include their possible involvement in the immune system. MST1 is implicated in cardiac apoptosis events (e.g. cardiac dilation, apoptosis and fibrosis after myocardial infarction [42]). MST1 has also been suggested to regulate lymphocyte polarity and adhesion, somatic hypermutation, class switching in B cells and apoptosis of eosinophils (see [9] for review). MST3 has been described to use metal ions other than Mg2+ as cofactors for kinase activity, such as Mn2+, Zn2+ and Co2+. Interestingly a variant of MST3 is expressed uniquely in brain, where Mn2+ and Zn2+ are abundant [32], [43].
Here we present the crystal structure of the unphosphorylated kinase domain of human MST4 refined at 2.35 Å resolution in complex with an ATP mimetic quinazoline inhibitor. We compare the crystal structures of all available MST kinase domains (MST1, 3 and 4) and a homology model (MST2) to derive information integrating structure and function.
Results and Discussion
Overall structures: kinase domain
The overall structure of the kinase domains of MST1, MST3 and MST4 are very similar, as expected for their high degree of sequence conservation, epitomized by 90% identity between the kinase domains of MST3 and MST4 (Figure 1). Nevertheless, structural differences resulted from differential phosphorylation states, complex with ligands and crystal forms provide information on the structural plasticity and regulation of this group of kinases (Figure 2).
Figure 1. Alignment of human MST kinase domains.

The secondary elements shown in the alignment were derived from the structure of MST4 (PDB ID 3GGF). The conserved Asp residue position that is involved in the coordination of ATP via its ribose oxygen atoms is marked with a yellow arrow.
Figure 2. Overall structure of the MST kinase domains.

Superimposition of the C-lobe from the three MST structures: Active apo MST1 (magenta, PDB ID: 3COM); Active MST3 (green, PDB ID: 3A7J) in complex with ADP (sticks) and Mn2+ ions (spheres); Inactive MST4 (blue, PDB ID: 3GGF) in complex with a quinazoline inhibitor (sticks) and Cd2+ (sphere). Phosphorylated threonine residues (Tpo) are shown as sticks.
The structures of MST1 and MST3 used for comparison with our experimental structure of MST4 are available in the protein data base whereas additional structures of MST3 have been reported recently [44].
The crystal structure of MST1 (PDB ID: 3COM) has been determined without bound cofactor (apo structure) whereas MST3 (PDB ID: 3CKX) has been co-crystallised with a non-selective inhibitor staurosporine. Both MST1 and MST3 structures assume an active conformation, with well-defined activation loops (A-loops) and the canonical salt-bridge between Lys59 and Glu73 (MST1 numbering), despite the difference in the number of phosphorylated sites. The A-loop of MST1 is diphosphorylated (Thr177, Thr183 - UniProt numbering for Q13043) while that of MST3 is monophosphorylated in the canonical position (Thr190 - UniProt numbering for Q9Y6E0-1; numbered as Thr163 in the structure PDB ID: 3CKX). The crystal structure of MST4 (PDB ID: 3GGF; data collection and refinement statistics in table 1) has been solved in complex with an ATP mimetic quinazoline inhibitor. Interestingly, this structure has captured MST4 in its inactive form with a partially disordered, unphosphorylated A-loop. Finally, the conformations adopted by the αC-helix from MST4 also differ from those seen in the structures of the other two MSTs.
Table 1. Data Collection and Refinement Statistics.
| Complex | MST4-quinazoline |
| PDB accession code | 3ggf |
| Wavelength (Å) | 0.8800 |
| Spacegroup | P212121 |
| Unit cell dimensions | a = 65.92, b = 91.25, c = 108.97 Åα = β = γ = 90.0° |
| Resolution rangea (Å) | 46.78-2.35 Å (2.48-2.35) |
| No. unique reflectionsa | 28094 (4028) |
| Completenessa (%) | 100.0 (100.0) |
| Average I/σIa | 9.0 (2.0) |
| Rmerge a | 0.139 (0.849) |
| Rpim a | 0.072 (0.432) |
| Redundancya | 4.6 (4.6) |
| Refinement | |
| No. Atoms P/L/Ob | 4295/58/153 |
| Rcryst/Rfree | 0.219/0.277 |
| rms. bond lengthc (Å) | 0.016 |
| rms. bond anglec (°) | 1.50 |
| B-factor P/L/Ob (Å2) | 21.8/24.5/32.2 |
| B-factor, Wilson plot (Å2) | 35.8 |
| Molprobity | |
| Ramachandran favour | 96.09% |
| Ramachandran allowed | 100.00% |
Values in brackets show the statistics for the highest resolution shell.
P/L/O represents protein/ligand/other (water, ion and solvent).
rms indicates root-mean-square.
The ATP binding site
Regardless of the presence of ligands, the structure of the ATP binding site is very similar for all three MST kinases (Figure 3A–D). In MST3 the bulk of the staurosporine ligand mimics the structure of the ATP adenine rings and sits in the flat cleft between the two lobes. As with MST1, hydrophobic regions above and below the ligand clamp it in place. A minor modification of the immediate pocket is seen in MST3: Ile15 replaces Leu36 in MST1. At the hinge backbone, hydrogen-bonding was observed between two main-chain functional groups; the amide of Leu87 and carbonyl of Glu85, and the staurosporine molecule.
Figure 3. ATP binding site.

Different kinases are color coded as follows: MST1: magenta; MST3: green; MST4: blue. Binding site surfaces are represented by semi-transparent orange skins. A. MST1 apo (PDB ID: 3COM); B. MST3 in complex with staurosporine (PDB ID: 3CKX); C. MST3 in complex with ADP (PDB ID: 3A7J); D. MST4 in complex with a quinazoline inhibitor (PDB ID: 3GGF); E. Fo-Fc difference map of the quinazoline inhibitor bound to MST4 (PDB ID: 3GGF). Isosurface contouring levels: 1σ (blue) and 3σ (red). This view is rotated 90° on the X-axis (as compared to the orientation shown in (D) and looks towards the C-lobe; F. Details of the active site of MST4 in complex with a quinazoline inhibitor (blue), superimposed to PAK6 (yellow; PDB ID: 2C30) and TAO2 in complex with ATP (grey; PDB ID: 1U5R).
The glycine-rich loop was well ordered in MST3 structures as expected in kinases with a bound ligand. However, residues of the glycine-rich loop have high B-factors in comparison with the rest of the molecule: both structures (in complex with staurosporine in 3CKX and ADP in 3A7J) are lacking the additional stabilization of the glycine-rich loop attributed to the gamma phosphate of the ATP. This can be illustrated by a comparison with the structure of related ser/thr kinase TAO2 which has been co-crystallised with ATP (PDB ID: 1U5R)(Figure 3F).
The binding of the quinazoline inhibitor in MST4 exhibits some interesting features not present in the MST1 or MST3 structures (Figure 3D–F). The presence of a pyrazole group in the ligand introduces the possibility of another H-bond with the hinge region. In addition to the two hydrogen bonds observed in the structure of MST3 in complex with staurosporine, the glutamic acid carbonyl (Glu100) is involved in a third hydrogen bond to the pyrazole group of the quinazoline inhibitor in MST4 (Figure 3D). Additionally electron density data has identified the positions of seven cadmium ions in the asymmetric unit in the crystal structure of MST4, two of which participates in the binding of the ligand in each monomer (Figure 3D). A four-coordinate Cd2+ ion (distorted tetrahedral geometry) sits in a location towards the open face of the ligand pocket, close to site occupied by ATP ribose 2′ and 3′ oxygen atoms. It is coordinated by two water molecules, the carboxy group of Asp109, and by a nitrogen atom of the adenine ring. Interestingly, despite the partially disordered A-loop, the geometry of the MST4 ATP pocket has undergone only limited change when compared to MST1 and 3 in the region occupied by the quinazoline ligand.
Superposition of the MSTs with the TAO2 kinase in complex with ATP suggests that the ribose group of ATP does not form any direct interaction with any of the residues lining the ATP binding pocket (Figure 3F). This is further contrasted when other kinases (e.g. PAK kinases) are superimposed – where conserved residues (PAK6 Asp530; PDB ID: 2C30) can interact directly with the oxygen atoms from the ribose group, enforcing the correct geometry for catalysis. Alternatively, an absolutely conserved Asp residue in MSTs (Asp109 in MST4) is found in a position allowing indirect interaction to the ATP ribose. This interaction could be mediated through a water molecule as seen in the structure of MST3 (PDB ID: 3A7J) or a cadmium ion as seen in MST4 (PDB ID: 3GFF). Additionally, in the STE kinase TAO2 (PDB ID: 1U5R) the aspartate residue coordinates a lysine, which in turn interacted with the ribose group. This observation indicates that the positioning of the ribose group through its oxygen atoms by the conserved aspartate requires the presence of a bridging element, which could be charged – a feature that could be explored for future inhibitor design.
We have also performed screening of MST4 against a panel of 1457 inhibitors, using thermal stability shift assay (Figure 4). Several compounds have been identified as strong binders, however most are non-specific kinase inhibitors that were previously described in the literature. This is also the case of the quinazoline inhibitor in the structure of MST4. The screening experiments were performed without adding cadmium to the buffer. The observed strong interactions between inhibitors and MST4 suggest that the bridging between Asp109 and ligand could be performed by other molecules, such as water, as observed in the structure of MST3 in complex with ADP (PDB ID: 3A7J).
Figure 4. Selected hits from screen of potential MST4 inhibitors against a panel of 1457 compounds.

α-helix C in MST4 shows an alternative conformation
The position of the αC-helix in MST1 and 3 is typical for an active conformer of a kinase. Lys59 and Glu73 (MST1) are orientated towards each other with geometry indicative of salt-bridge formation between the residues, with the αC-helix, well-positioned against the N-terminal lobe (N-lobe). The conformation of active MST structures (MST1 and MST3) is similar to conformations observed in other kinase structures especially in PAKs, which also belong to the STE family. It should be mentioned that the helix αA is present in the structures of PAKs and TAO2, stabilizing αC in an active conformation. Thus, the lack of the αA helix in the structures of active MST1 and MST3 does not seem to impact on the αC conformation (Figure 5).
Figure 5. Positioning of αC helix in the structure of three MSTs.

The kinase domains were superimposed at the N-lobe. MST1 (magenta), MST3 (green) and MST4 (blue). PAK6 (yellow wireframe) has been added to the superimposition to highlight the position typically occupied by the αC helix, in an active kinase.
Surprisingly, in the inactive structure of MST4 the αC helix is adopting a ‘swung-up’ conformation, bringing the N-terminal region of the αC helix closer to the N-terminus of the kinase domain. The approximately 13 degrees swinging motion (when compared to the same helix in MST3) allows the A-loop to also swing towards the N-lobe, resulting in the conformation captured in the crystal of MST4.
Dimerization through activation loop exchange as a model for MST autophosphorylation
The A-loop in both MST1 and MST3 are well-defined and in an active conformation. Interestingly it is diphosphorylated at both Thr177 and Thr183 in MST1 while MST3 is phosphorylated only at Thr178 (structure numbering, PDB ID: 3A7J). Structural superimposition shows that the canonical position (MST1: Thr177, MST3: Thr178) is well conserved, and so are the conformation adopted by the structures of MST1 and MST3 (Cα RMSD: 0.4 Å). In these structures, the canonical phosphorylated sites are coordinated by two conserved arginine residues (MST1: Arg148, Arg181; MST3: Arg143, Arg176).
In MST4, the unphosphorylated A-loop is positioned in a vastly different orientation when compared to MST1 and MST3 (Figure 2). It is poorly defined, where the visible portions belonging to the N-terminal section of the A-loop are seen adopting a conformation reaching towards the N-lobe, relative to the active form. The C-terminal section is seen extending away from the main core of the kinase domain and reaching into a neighboring kinase molecule, thus correlated with the dimerization seen in this crystal form (Figure 6A).
Figure 6. MST4 dimer seen in the asymmetric unit.

Chain A is shown in blue, chain B is shown in orange. A. Overall packing. Cadmium ions are shown as yellow spheres, ligands are shown as sticks; B. Molecular interactions seen in the A-loop. Residue Thr178 (to be phosphorylated) is shown with red labels; C. Surface electrostatics potential in the dimerization region – the segment involved in the interaction from the chain B is shown in sticks representation.
In the crystal of MST1, the kinase molecules are positioned roughly perpendicular to each other. In such orientation, the active site is out of reach to phosphorylate the A-loop residues Thr177 and Thr183. Similarly, in the structure of MST3, none of the symmetry-related neighbors are found to position the observed phosphorylation site close to the active site, suggesting that the crystal packing of neither MST1 nor MST3 reflect the kinase-kinase interaction positioning necessary for autophosphorylation.
Conversely, as described above, the asymmetric unit of MST4 contains a symmetry-related dimer where the visible C-terminal section of the A-loop of the molecules is contacting each other. Four residues (178-TFVGT-182) of the molecule A are packing against the same residues in molecule B in an anti-parallel disposition, forming a series of hydrogen bonds: each Thr178 main chain carbonyl interacts with Thr182-OG1 from the other molecule; likewise each Val180 main chain nitrogen hydrogen bonds to Gly181 main chain carbonyl from the neighboring molecule. Additionally, the side chains of Val180 are involved in hydrophobic contacts. More critically, the canonical Thr178 to be phosphorylated is found in close proximity to the phosphate transfer site of the neighboring molecule in the dimer (Figure 6C).
The combined observation of the dimer in the asymmetric unit and the A-loop conformation suggest that the kinase domain of MST4 can dimerize in a ‘face-to-face’ fashion. During that stage the inactive and partially disordered A-loop can reach satisfactorily into the substrate binding site of the other kinase in the dimer. In this process Thr178 can be positioned at the catalytic site to be phosphorylated. Similar autophosphorylation mechanism where the A-loops are exchanged in a ‘face-to-face’ kinase dimer has been described before for SLK, LOK and DAPK3 [45], [46].
The observed reciprocal interaction of A-loops in the MST4 dimer implies the availability of a site binding the swapped A-loops. The observed A-loop exchange also creates a distinction between the sequence requirement for the autophosphorylation mechanism and the physiological consensus sequence. Albeit the autophosphorylation consensus has been defined in all human MSTs as KRNT*(V/F)(I/V)GTPFWMAPEVI [9], the physiological consensus has not been determined so far. Ultimately, A-loop swapping allows a single kinase molecule to catalyze two different subsets of residues, which can be explored by designing of peptides (or peptidomimetics) targeting at either the autophosphorylation or physiological consensus binding sites.
While the structure of MST4 supports the A-loop exchange autophosphorylation model, it probably represents an intermediate, pre-catalytic state. The catalysis is dependent on the correct positioning of the αC helix and establishment of the activation salt-bridge, which are not present in the present structure of MST4.
It is important to emphasize that while autophosphorylation of MSTs seems to involve dimerization, it does not necessarily imply that this is the physiological dimer for the full-length protein. Previous reports have identified a C-terminal dimerization domain in MST1 and MST2, which is not required for kinase activity [47]. Additionally, MST2 lacking the dimerization domain can still autophosphorylate [48]. This suggests that although dimerization driven by the C-terminal domain (at least in MST1 and MST2) might be necessary for other biological/regulatory functions, autophosphorylation in trans will probably involve the face-to-face disposition seen in the asymmetric unit of MST4 crystals. We were interested to see if MST4 indeed dimerizes in solution. To this end we performed analytical ultracentrifugation sedimentation velocity experiments. Analysis of the sedimentation data collected on the unphosphorylated protein indicated clearly the presence of MST4 dimers in solution (Figure 7A). In addition, recombinant MST4 are autophosphorylated in solution at three sites, as shown by electrospray mass spectroscopy (Figure 7B).
Figure 7. Self association and autophosphorylation of MST4 in solution.

A: Self association studied by analytical ultracentrifugation. Shown are sedimentation velocity data. The sedimentation coefficients have been normalized to standard condition. Peaks corresponding to the MST4 monomer and dimer are indicated by “M and D” respectively. B: Autophosphorylation of MST4 studied by ESI-MS. The mass of the unmodified protein (33.818 Da) has been indicated. In addition to the three detected phosphorylation events the protein also oxidized (+16 mass shifts indicated by *).
C-terminal α-helix K
At the C-terminus of the full-length MSTs, we find a regulatory domain, which has been described to be involved in dimerization and not essential to the kinase activity [47]. It can be removed via cleavage with caspase - however the reported cleavage site is located around 30 residues from the last observable residue in the crystal structures [15], [49]. The regulatory domain is connected to the kinase domain via an α-helix (αK) which interacts with the C-terminal lobe (C-lobe) with a multitude of aromatic and hydrophobic interactions. The αK helix spans through the entire height of the C-lobe, packed against a cleft formed by helices αD/αE and the beta hairpin βHβI. Difference in sequences at the C-terminus end of αK between MST1/MST2 and MST3/MST4 (2-residue deletion) alters the αK packing angle. Interestingly, the fold in this region in MST3/MST4 is now similar to that adopted by PAKs whereas the fold adopted by MST1 is comparable to that adopted by TAO2 (Figure 8).
Figure 8. αK and αD conformations for the two subfamilies of MSTs.

A. MST1 (magenta) adopts a conformation closer to that seen in TAO2 (red); B. MST3 and 4 (green and blue, respectively) adopts a similar fold to that of PAKs (yellow).
Surface properties and substrate selectivity
To assess the surface properties, in addition to the three human MST crystallographic structures available at the PDB, we have generated two models by homology: MST2 in the active conformation and MST4 also in the active conformation.
The distribution of electrostatic potential on the surface of the four human MSTs in their active conformations is remarkably similar despite the occurrence of several patches of low residue conservation (Figure 9). As expected, the surface electrostatic potential of the crystal structure of MST4 (i.e. inactive conformation) was significantly different from those in the active state. This however only highlights the advantages and versatility of the A-loop exchange autophosphorylation model: (i) that the sequence to be autophosphorylated does not need to reflect the kinase's natural substrate selectivity and (ii) that the domain swapping, due to the sequence specific packing, will be also highly specific to the sequence to be autophosphorylated. Additionally, the lack of variability in surface electrostatic potentials reinforces the regulatory role of the divergent C-terminal domain of different MSTs.
Figure 9. Surface electrostatic potential of MSTs.

Crystallographic models are labeled in blue; homology models are labeled in orange. The surface electrostatic potential is represented as a color ramp, ranging from −5 kcal/e.u. charge (red) to blue +5 kcal/e.u. charge (blue).
Materials and Methods
Cloning, Expression and Purification
The MST4 gene corresponding to residues 1–300 was PCR amplified from a Mammalian gene collection library clone and cloned into the expression vector pLIC-SGC1 (SGC) using ligase independent cloning. The MST4 gene sequence of the constructs was verified by DNA sequencing. MST4 protein was expressed in a BL-21(DE3)-R3-pRARE2 cells. The protein was induced by the addition of IPTG to cells growing in LB ampicillin media. Cells were harvested following overnight growth and were stored as cell pellets at −20°C. Approximately 5 grams of cell pellet was resuspended in 25 ml of 50 mM HEPES pH 7.5, 300mM NaCl, 20 mM imidazole. The resuspended cells were lysed by sonication. The lysate was cleared of DNA and cell debris by centrifugation at 20,000 rpm (4°C). Five (5) ml of 50% Ni-NTA slurry (Qiagen) was applied to a 1.5×10 cm gravity column. The column was equilibrated with 100 ml binding buffer. The lysate was applied to the column and was subsequently washed with a minimum of 50 ml of lysis buffer. MST4 was eluted with 25 ml of lysis buffer containing 100 mM Imidazole. DTT was added to the eluted MST4 protein to a final concentration of 5 mM. The N-terminal his6-tag was removed by the addition of approximately 100 mg of TEV protease and incubated at 4°C overnight. Kinase phosphorylation was removed by λ-phosphatase in the presence of 50 mM MnCl2. The protein was concentrated using a 10kDa cutoff ultrafiltration unit and further purified by fractionation on a size exclusion chromatography HiLoad 16/60 Superdex 200 column in 50 mM Hepes, pH 7.5, 300 mM NaCl, 5 mM DTT. The protein was diluted to 50 mM HEPES pH 7.5, 50 mM NaCl and further purified by fractionation on a MonoQ column using a 50 mM to 1 M NaCl gradient. The purity of the protein was confirmed by SDS-PAGE and the correct molecular weight was observed by ESI Mass spectroscopy. The collected protein was concentrated by ultrafiltration (10kDa cutoff) to 11.2 mg/ml (stored in 50 mM HEPES pH 7.5, 200 mM NaCl), snap frozen in liquid nitrogen and stored at −80°C.
Analytical Ultracentrifugation
Sedimentation velocity experiments were carried out on a Beckman Optima XL-I Analytical Ultracentrifuge (Beckman Instruments, Palo Alto, CA) equipped with an AnTi-50 rotor and cells with double sector centerpieces. Protein samples were studied at a concentration of 30 µM in 50 mM HEPES pH 7.5, 100 mM NaCl employing a rotor speed of 45,000 rpm. Radial absorbance scans were collected using absorbance optics at a wavelength of 280 nm in continuous scan mode, in 2 minute intervals. Data were analyzed using the SEDFIT software package [50] whereby differential sedimentation coefficient distributions, c(s) distributions, were obtained by direct boundary modeling to Lamm Equation solutions. Sedimentation coefficients, s, were obtained by integration of individual peaks in the calculated c(s) distributions, after fitting of the frictional ratio, in order to allow these distributions to be corrected for the effects of diffusion. The software package SEDNTERP was used in order to convert the obtained sedimentation coefficient values to the equivalent values in water at 20°C (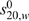 ,).
,).
Thermal stability shift assay
Thermal melting experiments were carried out using the Mx3005p real-time PCR machine (Agilent) and employing a protein concentration of 2 µM and inhibitor concentration of 10 µM. Buffer conditions were 10 mM HEPES, pH 7.5, 500 mM NaCl and 1∶1000 dilution of SyproOrange (Invitrogen, CA). The assay and data evaluation were carried out as described [51].
Crystallization and Structure determination
Crystallization of MST4 was achieved by sitting drop vapor diffusion at 4°C. MST4 (stored in 50 mM HEPES pH 7.5, 200 mM NaCl) was pre-incubated with 1mM quinazoline inhibitor which was added from a 50 mM stock solution in DMSO at a protein concentration of 5.9 mg/ml. The protein was crystallized using 12% PEG 3350, 5mM CdCl2, and 0.1M HEPES, pH 7.0. Viable crystals of the complex were obtained after microseeding in a 300 nl drop, mixing protein and precipitant at 2∶1 volume ratio. Diffraction data were collected at Swiss Light Source station X10SA from crystals cryoprotected with a solution composed of mother liquor and 20% PEG 400 and flash cooled to 100°K. The data were processed with MOSFLM [52] and subsequently scaled with SCALA (as implemented in the CCP4 suite [53]). Structure of MST4 was solved by molecular replacement by using the program PHASER [54] and the structure of MST3 (PDB ID: 3CKX) as a model. Initial structure was obtained by automated model building using the Buccaneer program [55] supplemented with an improved phase output from DM (as implemented in the CCP4 suite [53]). Further iterative cycles of manual rebuilding in COOT [56] and refinement with REFMAC5 [57] were performed. Appropriate TLS groups calculated by the TLSMD program [58] were applied to a final round of TLS refinement. Completed structure was verified for geometric correctness with MolProbity [59]. Data collection and refinement statistics are summarized in Table 1.
Homology modeling
Homology modeling was performed using the ICM software package [60]. The modeling procedure was performed for the full-length of the MST2 kinase domain, since no crystal structure was available. We used the structure of the closely related MST1 (PDB ID: 3COM; sequence identity: 93% for the kinase domain) as the modeling template. Homology modeling, global minimization to solve potential clashes derived from the method (e.g. substitutions, loop modeling/grafting) as well as hydrogen bonding network optimization were performed using the protocols implemented in the program ICM [61].
Data deposition
The atomic coordinates and structure factors have been deposited with the Protein Data Bank, www.rcsb.org (PDB ID: 3GGF [MST4]).
Supporting Information
Standalone iSee datapack - contains the enhanced version of this article for use offline. This file can be opened using free software available for download at http://www.molsoft.com/icm_browser.html.
(ICB)
Instructions for installation and use of the required web plugin (to access the online enhanced version of this article).
(PDF)
Acknowledgments
We would like to thank Prof. Kevan Shokat (Department of Cellular and Molecular Pharmacology, University of California San Francisco) for providing several inhibitors used in this study.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research and the Wellcome Trust. The authors also thank the Oxford University Nuffield Department of Medicine Summer Internship Program 2009 for CJR's sponsorship. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Leberer E, Dignard D, Harcus D, Thomas DY, Whiteway M. The protein kinase homologue Ste20p is required to link the yeast pheromone response G-protein beta gamma subunits to downstream signalling components. EMBO J. 1992;11:4815–4824. doi: 10.1002/j.1460-2075.1992.tb05587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goehring AS, Mitchell DA, Tong AH, Keniry ME, Boone C, et al. Synthetic lethal analysis implicates Ste20p, a p21-activated potein kinase, in polarisome activation. Mol Biol Cell. 2003;14:1501–1516. doi: 10.1091/mbc.E02-06-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holly SP, Blumer KJ. PAK-family kinases regulate cell and actin polarization throughout the cell cycle of Saccharomyces cerevisiae. J Cell Biol. 1999;147:845–856. doi: 10.1083/jcb.147.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eby JJ, Holly SP, van Drogen F, Grishin AV, Peter M, et al. Actin cytoskeleton organization regulated by the PAK family of protein kinases. Curr Biol. 1998;8:967–970. doi: 10.1016/s0960-9822(98)00398-4. [DOI] [PubMed] [Google Scholar]
- 5.Wu C, Lytvyn V, Thomas DY, Leberer E. The phosphorylation site for Ste20p-like protein kinases is essential for the function of myosin-I in yeast. J Biol Chem. 1997;272:30623–30626. doi: 10.1074/jbc.272.49.30623. [DOI] [PubMed] [Google Scholar]
- 6.Hofken T, Schiebel E. A role for cell polarity proteins in mitotic exit. EMBO J. 2002;21:4851–4862. doi: 10.1093/emboj/cdf481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahn SH, Cheung WL, Hsu JY, Diaz RL, Smith MM, et al. Sterile 20 kinase phosphorylates histone H2B at serine 10 during hydrogen peroxide-induced apoptosis in S. cerevisiae. Cell. 2005;120:25–36. doi: 10.1016/j.cell.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 8.Ahn SH, Henderson KA, Keeney S, Allis CD. H2B (Ser10) phosphorylation is induced during apoptosis and meiosis in S. cerevisiae. Cell Cycle. 2005;4:780–783. doi: 10.4161/cc.4.6.1745. [DOI] [PubMed] [Google Scholar]
- 9.Ling P, Lu TJ, Yuan CJ, Lai MD. Biosignaling of mammalian Ste20-related kinases. Cell Signal. 2008;20:1237–1247. doi: 10.1016/j.cellsig.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 10.Boomer JS, Tan TH. Functional interactions of HPK1 with adaptor proteins. J Cell Biochem. 2005;95:34–44. doi: 10.1002/jcb.20401. [DOI] [PubMed] [Google Scholar]
- 11.Dan I, Watanabe NM, Kusumi A. The Ste20 group kinases as regulators of MAP kinase cascades. Trends Cell Biol. 2001;11:220–230. doi: 10.1016/s0962-8924(01)01980-8. [DOI] [PubMed] [Google Scholar]
- 12.Pombo CM, Force T, Kyriakis J, Nogueira E, Fidalgo M, et al. The GCK II and III subfamilies of the STE20 group kinases. Front Biosci. 2007;12:850–859. doi: 10.2741/2107. [DOI] [PubMed] [Google Scholar]
- 13.Strange K, Denton J, Nehrke K. Ste20-type kinases: evolutionarily conserved regulators of ion transport and cell volume. Physiology (Bethesda) 2006;21:61–68. doi: 10.1152/physiol.00139.2005. [DOI] [PubMed] [Google Scholar]
- 14.Feig LA, Buchsbaum RJ. Cell signaling: life or death decisions of ras proteins. Curr Biol. 2002;12:R259–261. doi: 10.1016/s0960-9822(02)00787-x. [DOI] [PubMed] [Google Scholar]
- 15.Graves JD, Gotoh Y, Draves KE, Ambrose D, Han DK, et al. Caspase-mediated activation and induction of apoptosis by the mammalian Ste20-like kinase Mst1. EMBO J. 1998;17:2224–2234. doi: 10.1093/emboj/17.8.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kakeya H, Onose R, Osada H. Caspase-mediated activation of a 36-kDa myelin basic protein kinase during anticancer drug-induced apoptosis. Cancer Res. 1998;58:4888–4894. [PubMed] [Google Scholar]
- 17.Reszka AA, Halasy-Nagy JM, Masarachia PJ, Rodan GA. Bisphosphonates act directly on the osteoclast to induce caspase cleavage of mst1 kinase during apoptosis. A link between inhibition of the mevalonate pathway and regulation of an apoptosis-promoting kinase. J Biol Chem. 1999;274:34967–34973. doi: 10.1074/jbc.274.49.34967. [DOI] [PubMed] [Google Scholar]
- 18.Cheung WL, Ajiro K, Samejima K, Kloc M, Cheung P, et al. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell. 2003;113:507–517. doi: 10.1016/s0092-8674(03)00355-6. [DOI] [PubMed] [Google Scholar]
- 19.Ura S, Masuyama N, Graves JD, Gotoh Y. Caspase cleavage of MST1 promotes nuclear translocation and chromatin condensation. Proc Natl Acad Sci U S A. 2001;98:10148–10153. doi: 10.1073/pnas.181161698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ura S, Masuyama N, Graves JD, Gotoh Y. MST1-JNK promotes apoptosis via caspase-dependent and independent pathways. Genes Cells. 2001;6:519–530. doi: 10.1046/j.1365-2443.2001.00439.x. [DOI] [PubMed] [Google Scholar]
- 21.Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villen J, et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 22.Oh HJ, Lee KK, Song SJ, Jin MS, Song MS, et al. Role of the tumor suppressor RASSF1A in Mst1-mediated apoptosis. Cancer Res. 2006;66:2562–2569. doi: 10.1158/0008-5472.CAN-05-2951. [DOI] [PubMed] [Google Scholar]
- 23.Teraishi F, Guo W, Zhang L, Dong F, Davis JJ, et al. Activation of sterile20-like kinase 1 in proteasome inhibitor bortezomib-induced apoptosis in oncogenic K-ras-transformed cells. Cancer Res. 2006;66:6072–6079. doi: 10.1158/0008-5472.CAN-06-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harvey KF, Pfleger CM, Hariharan IK. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell. 2003;114:457–467. doi: 10.1016/s0092-8674(03)00557-9. [DOI] [PubMed] [Google Scholar]
- 25.Callus BA, Verhagen AM, Vaux DL. Association of mammalian sterile twenty kinases, Mst1 and Mst2, with hSalvador via C-terminal coiled-coil domains, leads to its stabilization and phosphorylation. FEBS J. 2006;273:4264–4276. doi: 10.1111/j.1742-4658.2006.05427.x. [DOI] [PubMed] [Google Scholar]
- 26.Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–434. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 27.Matallanas D, Romano D, Yee K, Meissl K, Kucerova L, et al. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol Cell. 2007;27:962–975. doi: 10.1016/j.molcel.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Neill EE, Matallanas D, Kolch W. Mammalian sterile 20-like kinases in tumor suppression: an emerging pathway. Cancer Res. 2005;65:5485–5487. doi: 10.1158/0008-5472.CAN-05-1453. [DOI] [PubMed] [Google Scholar]
- 29.Qian Z, Lin C, Espinosa R, LeBeau M, Rosner MR. Cloning and characterization of MST4, a novel Ste20-like kinase. J Biol Chem. 2001;276:22439–22445. doi: 10.1074/jbc.M009323200. [DOI] [PubMed] [Google Scholar]
- 30.Schinkmann K, Blenis J. Cloning and characterization of a human STE20-like protein kinase with unusual cofactor requirements. J Biol Chem. 1997;272:28695–28703. doi: 10.1074/jbc.272.45.28695. [DOI] [PubMed] [Google Scholar]
- 31.Dan I, Ong SE, Watanabe NM, Blagoev B, Nielsen MM, et al. Cloning of MASK, a novel member of the mammalian germinal center kinase III subfamily, with apoptosis-inducing properties. J Biol Chem. 2002;277:5929–5939. doi: 10.1074/jbc.M110882200. [DOI] [PubMed] [Google Scholar]
- 32.Zhou TH, Ling K, Guo J, Zhou H, Wu YL, et al. Identification of a human brain-specific isoform of mammalian STE20-like kinase 3 that is regulated by cAMP-dependent protein kinase. J Biol Chem. 2000;275:2513–2519. doi: 10.1074/jbc.275.4.2513. [DOI] [PubMed] [Google Scholar]
- 33.ten Klooster JP, Jansen M, Yuan J, Oorschot V, Begthel H, et al. Mst4 and Ezrin induce brush borders downstream of the Lkb1/Strad/Mo25 polarization complex. Dev Cell. 2009;16:551–562. doi: 10.1016/j.devcel.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 34.Creasy CL, Chernoff J. Cloning and characterization of a member of the MST subfamily of Ste20-like kinases. Gene. 1995;167:303–306. doi: 10.1016/0378-1119(95)00653-2. [DOI] [PubMed] [Google Scholar]
- 35.Creasy CL, Chernoff J. Cloning and characterization of a human protein kinase with homology to Ste20. J Biol Chem. 1995;270:21695–21700. doi: 10.1074/jbc.270.37.21695. [DOI] [PubMed] [Google Scholar]
- 36.Lin JL, Chen HC, Fang HI, Robinson D, Kung HJ, et al. MST4, a new Ste20-related kinase that mediates cell growth and transformation via modulating ERK pathway. Oncogene. 2001;20:6559–6569. doi: 10.1038/sj.onc.1204818. [DOI] [PubMed] [Google Scholar]
- 37.Taylor LK, Wang HC, Erikson RL. Newly identified stress-responsive protein kinases, Krs-1 and Krs-2. Proc Natl Acad Sci U S A. 1996;93:10099–10104. doi: 10.1073/pnas.93.19.10099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cooper WN, Hesson LB, Matallanas D, Dallol A, von Kriegsheim A, et al. RASSF2 associates with and stabilizes the proapoptotic kinase MST2. Oncogene. 2009;28:2988–2998. doi: 10.1038/onc.2009.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cinar B, Fang PK, Lutchman M, Di Vizio D, Adam RM, et al. The pro-apoptotic kinase Mst1 and its caspase cleavage products are direct inhibitors of Akt1. EMBO J. 2007;26:4523–4534. doi: 10.1038/sj.emboj.7601872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sung V, Luo W, Qian D, Lee I, Jallal B, et al. The Ste20 kinase MST4 plays a role in prostate cancer progression. Cancer Res. 2003;63:3356–3363. [PubMed] [Google Scholar]
- 41.Preisinger C, Short B, De Corte V, Bruyneel E, Haas A, et al. YSK1 is activated by the Golgi matrix protein GM130 and plays a role in cell migration through its substrate 14-3-3zeta. J Cell Biol. 2004;164:1009–1020. doi: 10.1083/jcb.200310061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Odashima M, Usui S, Takagi H, Hong C, Liu J, et al. Inhibition of endogenous Mst1 prevents apoptosis and cardiac dysfunction without affecting cardiac hypertrophy after myocardial infarction. Circ Res. 2007;100:1344–1352. doi: 10.1161/01.RES.0000265846.23485.7a. [DOI] [PubMed] [Google Scholar]
- 43.Lu TJ, Huang CY, Yuan CJ, Lee YC, Leu TH, et al. Zinc ion acts as a cofactor for serine/threonine kinase MST3 and has a distinct role in autophosphorylation of MST3. J Inorg Biochem. 2005;99:1306–1313. doi: 10.1016/j.jinorgbio.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 44.Ko T-P, Jeng W-Y, Liu C-I, Lai M-D, Wu C-L, et al. Structures of human MST3 kinase in complex with adenine, ADP and Mn2+. Acta Crystallogr. 2010;D66:145–154. doi: 10.1107/S0907444909047507. [DOI] [PubMed] [Google Scholar]
- 45.Pike AC, Rellos P, Niesen FH, Turnbull A, Oliver AW, et al. Activation segment dimerization: a mechanism for kinase autophosphorylation of non-consensus sites. EMBO J. 2008;27:704–714. doi: 10.1038/emboj.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oliver AW, Knapp S, Pearl LH. Activation segment exchange: a common mechanism of kinase autophosphorylation? Trends Biochem Sci. 2007;32:351–356. doi: 10.1016/j.tibs.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 47.Creasy CL, Ambrose DM, Chernoff J. The Ste20-like protein kinase, Mst1, dimerizes and contains an inhibitory domain. J Biol Chem. 1996;271:21049–21053. doi: 10.1074/jbc.271.35.21049. [DOI] [PubMed] [Google Scholar]
- 48.Deng Y, Pang A, Wang JH. Regulation of mammalian STE20-like kinase 2 (MST2) by protein phosphorylation/dephosphorylation and proteolysis. J Biol Chem. 2003;278:11760–11767. doi: 10.1074/jbc.M211085200. [DOI] [PubMed] [Google Scholar]
- 49.Lee WS, Hsu CY, Wang PL, Huang CY, Chang CH, et al. Identification and characterization of the nuclear import and export signals of the mammalian Ste20-like protein kinase 3. FEBS Lett. 2004;572:41–45. doi: 10.1016/j.febslet.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 50.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bullock AN, Debreczeni JE, Fedorov OY, Nelson A, Marsden BD, et al. Structural basis of inhibitor specificity of the human protooncogene proviral insertion site in moloney murine leukemia virus (PIM-1) kinase. J Med Chem. 2005;48:7604–7614. doi: 10.1021/jm0504858. [DOI] [PubMed] [Google Scholar]
- 52.Leslie AGW. The integration of macromolecular diffraction data. Acta Crystallogr. 2006;D62:48–57. doi: 10.1107/S0907444905039107. [DOI] [PubMed] [Google Scholar]
- 53.CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 54.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, et al. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cowtan K. Buccaneer software for automated model building. Acta Crystallogr. 2006;D62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 56.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 57.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 58.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. 2006;D62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 59.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abagyan R, Totrov M, Kuznetsov DA. ICM: A New Method For Protein Modeling and Design: Applications To Docking and Structure Prediction From The Distorted Native Conformation. J Comp Chem. 1994;15:488–506. [Google Scholar]
- 61.Cardozo T, Totrov M, Abagyan R. Homology modeling by the ICM method. Proteins. 1995;23:403–414. doi: 10.1002/prot.340230314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Standalone iSee datapack - contains the enhanced version of this article for use offline. This file can be opened using free software available for download at http://www.molsoft.com/icm_browser.html.
(ICB)
Instructions for installation and use of the required web plugin (to access the online enhanced version of this article).
(PDF)