

Abstract
The sleeping sickness parasite, Trypanosoma brucei, must differentiate in response to the changing environments that it encounters during its complex life cycle. One developmental form, the bloodstream stumpy stage, plays an important role in infection dynamics and transmission of the parasite. Recent advances have shed light on the molecular mechanisms by which these stumpy forms differentiate as they are transmitted from the mammalian host to the insect vector of sleeping sickness, tsetse flies. These molecular advances now provide improved experimental tools for the study of stumpy formation and function within the mammalian bloodstream. They also offer new routes to therapy via high-throughput screens for agents that accelerate parasite development. Here, we shall discuss the recent advances that have been made and the prospects for future research now available.
Keywords: Sleeping sickness, Trypanosoma brucei, Differentiation, Transmission, Parasite
Sleeping sickness: an ancient and current disease
The tropical parasite Trypanosoma brucei is the causative agent of both human African trypanosomiasis (HAT, or sleeping sickness) and the livestock disease, nagana. Over 50,000 people throughout 36 countries in sub-Saharan Africa are currently estimated to be infected [1]. The initial stage of disease, established when an individual is bitten by an infected tsetse fly, generates a local chancre at the bite site with this being followed by the proliferation of the parasite in the bloodstream resulting in fever and joint pain. At this stage, the disease is often undiagnosed and, as such, untreated, despite the availability of (albeit rather old) drugs such as suramin (for Trypanosoma brucei rhodesiense or acute sleeping sickness) and pentamidine (for Trypanosoma brucei gambiense or chronic sleeping sickness). Later, the parasites cross the blood–brain barrier to infect the central nervous system, causing disturbance to the patient's sleep patterns, as well as causing confusion and difficulty with coordination. The current drug treatments for the later stage of the disease are difficult to administer, requiring a rigorous hospital regime, and all have associated toxicity. For example, melarsoprol is an arsenic-based treatment developed in 1949 that kills 3–10% of patients and has increasing incidence of drug resistance in the parasite population. A more recently developed treatment for T. brucei gambiense only, eflornithine, is less toxic, but requires high doses and intravenous administration. Nonetheless, without treatment, trypanosome infection is always fatal. The clinical features and disease treatments of HAT are discussed in more detail in [1].
The ability of trypanosomes to infect both domestic and wild animals means that non-human hosts in affected regions can act as a reservoir for the parasite, confounding control of the disease. Moreover, vaccination approaches have not been considered feasible because these parasites evade mammalian immune responses by frequently changing the proteins on their surface in an extreme form of antigenic variation. Due to the severity and prevalence of the disease, as well as the lack of efficient disease management, African trypanosomiasis remains an important public health issue and is considered by the World Health Organisation to be a neglected tropical disease (http://www.who.int/en).
The biology of disease spread
Trypanosomes differentiate between distinct life stages in order to prepare for, and adapt to, the different environments they encounter during their life cycle [2, 3]. During the bloodstream stage of the life cycle, trypanosomes exist as either proliferative ‘slender forms’ or non-proliferative, transmissible, ‘stumpy forms’ [2, 3] (Fig. 1), with transitional forms between these two types being described as ‘intermediate’ forms. The proportion of these types changes during the course of infection, this apparently being governed by the density of parasites in the blood. The bloodstream trypanosome population is adapted to promote parasite maintenance in the mammalian blood and transmission to tsetse flies. Thus, the proliferation of slender forms contributes to the establishment of parasite numbers in the blood and, through antigen variation, to immune evasion. In contrast, the irreversible division-arrest of stumpy forms acts to control the expansion of parasite numbers in the mammalian bloodstream and so prolongs host survival. This, combined with the preferential survival and the adaptations for differentiation of stumpy forms upon tsetse uptake, increases the probability of disease transmission [4].
Fig. 1.
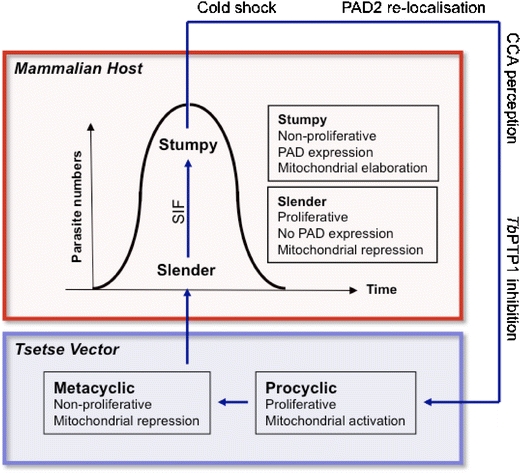
A simplified depiction of the T. brucei life cycle. In the bloodstream of the mammalian host, slender forms proliferate. As density increases, the parasites produce stumpy induction factor (SIF) which induces differentiation of a proportion of slender forms to stumpy forms. Stumpy forms are pre-adapted to life in the tsetse vector: they are cell cycle-arrested and have elaborated mitochondrial activity. Upon uptake in a tsetse blood-meal, stumpy forms differentiate into the proliferative procyclic forms in response to cold shock and cis-aconitate or citrate (CCA) in the tsetse midgut. Other signals such as proteases or pH stress may also contribute, as does inhibition of the tyrosine phosphatase, TbPTP1. Further differentiation events occur in the tsetse to produce mature metacyclic forms in the salivary glands. These are pre-adapted to life in the mammalian host
As the dynamic balance between slender and stumpy forms in mammalian hosts plays a pivotal role in trypanosome infection and transmission, understanding the molecular events implicit in the slender to stumpy form transition and their subsequent differentiation to tsetse midgut forms (procyclic forms) is of interest and importance, though poorly understood.
Adaptation of stumpy forms for parasite transmission
When a tsetse fly ingests a trypanosome-infected blood-meal, both slender and stumpy form parasites enter the tsetse midgut. Although subject to some controversy [5], it is generally thought that stumpy forms are pre-adapted to life in the tsetse midgut, and only they differentiate efficiently to the tsetse midgut procyclic form [3]. Cytological evidence based on in vitro differentiation experiments suggests that bloodstream forms can only transform to procyclic forms from a given point in the cell cycle, namely within the G1, or the quiescent G0, phase [6]. As stumpy forms are uniformly arrested as a population in G0/G1 and can accumulate to near homogeneity in experimental infections, this allows for their efficient and synchronous differentiation to procyclic forms. In contrast, proliferative slender cell populations are asynchronous in their cell cycle and must reach G0/G1 before differentiation [7]. Additionally, as cells enter the tsetse midgut, they are exposed to harmful proteases and changes in their environmental pH. The stumpy forms are more resistant than slender forms to the effects of such stresses [8] and, as such, are more likely to endure the transition from the bloodstream to the tsetse midgut. Moreover, in the glucose-rich mammalian bloodstream, slender cells obtain their energy entirely via glycolysis [9] and, as a result, the mitochondrion of slender cells is relatively inactive [3]. In intermediate and stumpy forms, however, the mitochondrion is elaborated and mitochondrial enzymes are partially expressed [3, 9]. This enables them to quickly begin oxidative phosphorylation as they enter the glucose-poor tsetse midgut [3, 9].
Stumpy cells also persist longer than slender cells in the face of the developing immune response in the mammalian bloodstream [10, 11], as well as in the tsetse midgut. This is probably not caused by antigenic variation: being irreversibly arrested in the bloodstream, stumpy forms are unlikely to be able to undergo the DNA recombination events important for antigen switching. Rather, they withstand antibody clearance more effectively than slender cells because they preferentially clear bound antibody by hydrodynamic flow, whereby parasite-bound immunoglobulin is swept backward by the swimming action of the trypanosome in the blood and is then internalised and degraded [12].
The molecular mechanism by which bloodstream trypanosomes perceive their change in environment and initiate differentiation to procyclic forms once in the tsetse fly has been elusive, until recently. It has long been known that the use of high concentrations (>3 mM) of citrate or cis-aconitate (CCA) could induce differentiation of bloodstream form trypanosomes to procyclic forms in vitro [13–15]. The biological relevance of this signal was in doubt, however, as these levels exceed the concentrations of CCA that a trypanosome would be exposed to in the tsetse midgut [15]. However, in 2004, Engstler and Boshart discovered that a drop in temperature from 37°C to 20°C could significantly increase trypanosome sensitivity to CCA, potentially matching levels present within the tsetse blood-meal [16]. Since this temperature drop was compatible with that encountered by trypanosomes as they entered tsetse flies feeding at the cooler ambient temperatures of dusk or dawn, CCA could be considered physiologically relevant. Since temperature drop also allows some molecules regulated access to the parasite surface, Engstler and Boshart proposed a model whereby an unknown surface protein responsible for the reception of the differentiation signal would be retained within the cell in the bloodstream (i.e. when at 37°C) and then trafficked to the cell surface in response to the cooler temperatures associated with tsetse uptake [16, 17]. The molecular basis of this model, however, was unknown until a gene family encoding surface carboxylate transporters known as PAD proteins (proteins associated with differentiation) was recently identified [18]. Indeed, two of the encoded PAD proteins (PAD1 and PAD2) were shown to be expressed in stumpy forms but not in slender forms and were found to be required for perception of CCA at physiological concentrations [18]. Matching the predictions of Engstler and Boshart, PAD2 was also demonstrated to be confined to the parasite's flagellar pocket region at 37°C but released to the cell surface at 20°C, thereby demonstrating the redistribution of the transporter in response to cold shock [16, 18]. Such thermoregulatory events are increasingly recognised as being important in insect-borne parasites, including in leishmania and malaria [19, 20].
The differential expression and localisation of the PAD proteins provide two levels of control during the differentiation to procyclic forms (Fig. 2). First, differential expression of PAD proteins between slender and stumpy forms ensures only the pre-adapted transmissible stumpy forms are able to receive the differentiation signal. Second, the differential localisation of PAD2 between stumpy cells at 37°C and 20°C ensures that only cells in the appropriate environment (i.e. the tsetse midgut) are able to receive the signal. As citrate is present in blood at ∼130 µM [21], this secondary control point could be crucial to prevent premature differentiation to procyclic forms whilst within the host, this signal only being detected upon temperature reduction in the tsetse blood-meal.
Fig. 2.
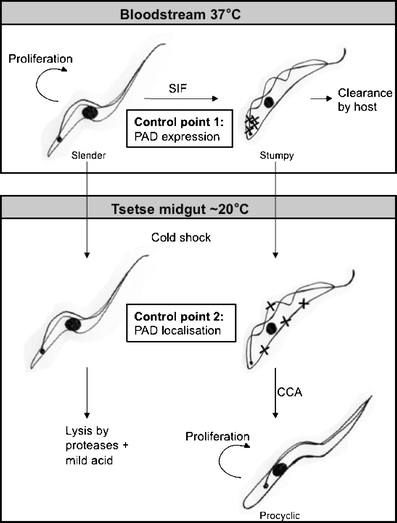
PAD protein expression and localisation act as checkpoints for differentiation. A summary of the expression of PAD proteins and the consequences for trypanosome differentiation events, based on the predictive model of Engstler and Boshart (2004) [16]. Slender cells proliferate in the bloodstream of the mammalian host. As parasite density increases, slender cells produce SIF which induces production of stumpy forms. Stumpy forms express PAD proteins (denoted by X), whereas slender forms do not. This ensures that only the transmissible stumpy forms are able to detect the differentiation signal. Upon ingestion in a tsetse blood-meal and exposure to a drop in temperature, there is up-regulation and a relocation of at least one PAD protein (PAD2) to the cell surface. Retention of PAD2 within the cell prior to cold shock ensures stumpy forms do not differentiate prematurely. Stumpy forms then differentiate to procyclic forms synchronously in response to CCA. Slender forms do not perceive the signal and are sensitive to proteolytic and potential pH stress in the tsetse midgut and therefore do not survive
Before the discovery of PAD proteins, it was already established that the activity of a protein tyrosine phosphatase, TbPTP1, inhibits the differentiation of stumpy forms whilst in the bloodstream, this block being removed upon uptake by the tsetse fly [22]. Hence, it may be that TbPTP1 acts downstream of the PAD proteins as an effector of the CCA signal (Fig. 1) or it may act via a parallel pathway, providing additional stringent control of differentiation. Similarly, exposure to proteases [15, 23] or pH stress [24] in the tsetse midgut might also trigger differentiation, either in a complementary manner or as independent cues [24]. Although the role of these other environmental cues is yet to be fully understood, the discovery of the PAD proteins provides the first molecular insight into the environmental sensing mechanisms used by the stumpy form to perceive transmission and so initiate differentiation.
Molecules, models and medicines
One of the key reasons that the differentiation from bloodstream forms to procyclic forms has been so well-studied is that there are clear molecular markers to distinguish each developmental stage. Furthermore, the development between the bloodstream and procyclic forms can be easily mimicked in vitro. Contrasting with this, the absence of markers distinguishing slender from stumpy forms has hindered our understanding of the transmission biology of trypanosomes, as has the difficulty in successfully generating stumpy forms in culture. In earlier studies, various markers have been used to identify stumpy forms, including morphology, mitochondrial activation, cell cycle arrest and the capacity of cells to differentiate synchronously to the procyclic form [25–29]. These assays, however, are far from ideal. For example, analysis by morphology is subjective and time-consuming and is complicated by the presence of morphologically intermediate forms in the population. Similarly, differentiation analyses are time-dependent and require population assays, rather than the analysis of individual cells. Finally, mitochondrial activation, often visualised through the activity of dihydrolipoamide dehydrogenase in the diaphorase assay, may also be activated under non-physiological stress conditions. With the identification of PAD1 as a surface molecule that identifies cells that are functionally competent for transmission [18], as well as developments in parasite culture and transfection [30], the route has been opened up to dissect trypanosome transmissibility at the level of individual cells as well as in the infecting parasite population.
Differentiation of the individual parasite
Differentiation between life cycle stages and the progression of the cell cycle are intimately linked in trypanosomes [7, 22]. As mentioned above, stumpy forms are cell cycle-arrested in G0/G1 but derived from proliferative slender forms. This transition, therefore, requires exit from the cell cycle, morphological change and stumpy-specific gene expression and has been studied both experimentally and by mathematical modelling [7, 29, 31]. From these studies, it appears that the commitment of a slender cell to differentiate to a stumpy form precedes a final cell division and morphological change [7, 29]. However, the discovery of PAD1 as a molecular marker for stumpy forms enables the events of cell cycle exit and stumpy formation to be tracked at the level of an individual cell, providing a temporal map of the steps leading to transmission competence. Similarly, the signalling pathways that generate stumpy forms from slender forms can be investigated. This transition can occur independently of the host [32, 33] and correlates with increased cell density [28, 33, 34], with parasite–parasite signalling operating through a form of quorum sensing. This is proposed to occur through the production by slender forms of a signal, termed stumpy induction factor (SIF), which accumulates and stimulates stumpy formation as parasite numbers increase [33]. The identity of SIF and the molecular mechanisms of its reception are currently unknown. It was thought that the SIF signal was transmitted via the cAMP pathway, since cell-permeable cAMP can mimic the action of SIF (Fig. 1) [33]. However, more recent evidence suggests that hydrolysis products of cAMP also function, arguing against a conventional cAMP signalling pathway [27]. Additional compounds such as troglitazone [26] and Z-Phe-Ala-CHN2 [35] have also been proposed to mimic SIF activity in vitro, inhibiting cell growth and generating morphologically ‘stumpy-like’ forms. The biological significance of these different treatments, however, has been difficult to assess in the absence of a functional molecular marker for stumpy forms. Hence, the identification of PAD1 and its detection either by antibody or in transgenic reporter assays now enables SIF, or SIF mimics, to be tracked in high-throughput assays and identified.
Differentiation of the population
PAD1 also provides a route to analyse the proportions of slender and stumpy forms in infections and to understand how the dynamics of these change throughout the course of a classical chronic infection. As trypanosome infections last from months to years, each infected individual can act as a reservoir for the disease for a considerable period of time. However, the degree of periodicity in their transmission capacity is poorly understood but potentially vital in a context where tsetse bites are rare or show seasonality [17]. Furthermore, through mathematic modelling, it has been predicted that the production of stumpy forms is a key contributor to the ordered expression of distinct antigenic variants that sustain chronic trypanosome infections [36]. This, combined with the likelihood of co-infection with different trypanosome strains during the course of a chronic infection, generates the potential for complex infection dynamics with important possible implications for disease progression and transmission in endemic areas. With the availability of a marker for the transmission stage, quantitative models for the dynamics of slender and stumpy parasites as a proportion of the total parasite numbers over time can be generated and the consequences of potential therapeutic strategies predicted.
Accelerated parasite development as a therapeutic tool
The identification of a molecular marker that distinguishes the non-proliferative stumpy form provides the ability to develop high-throughput screens for compounds that accelerate stumpy formation at abnormally low parasite density. Genetically modified reporter lines could provide simple assays for enhanced stumpy formation accessible to screening with small molecule or natural product libraries. If administrable compounds were identified that are able to force the irreversible differentiation of slender forms to stumpy forms at low levels of parasitaemia, then this could have potential therapeutic consequences.
Although accelerated parasite development has the potential to reduce host parasitaemia, it also has the potential to increase the transmission potential of the population through increasing the proportion of stumpy forms. The extent of this risk depends on several factors: the density of parasites in the bloodstream, the number of parasites required to infect a tsetse fly and the efficiency of accelerated development. Assuming a typical parasitaemia in cattle of 105 parasites/ml [37] and an infective dose to tsetse under optimal conditions of one trypanosome [38], then a reduction in parasite numbers of at least three orders of magnitude would be required to minimise the probability of parasite transmission in a tsetse blood-meal. However, in the field, chronic infections of cattle can exhibit refractory periods when there are insufficient parasites for tsetse transmission, suggesting that the parasites may be on the threshold of transmissibility, particularly when compounded by seasonal effects. This might make transmission blocking feasible. Moreover, if stumpy formation could be driven to 100% efficiency, then the irreversible division-arrest of these forms would ensure complete parasite elimination through cell senescence within only a few days, limiting the risks of enhanced transmission and providing a curative regime.
Clearly, without better understanding of the transmission biology of trypanosomes in the field, the prospects for a transmission blocking approach are difficult to assess. It is satisfying, therefore, that the identification of stumpy marker proteins such as PAD offers the ability to both screen for new drugs to accelerate stumpy formation and to refine the transmission models required to optimise their potential deployment in the field.
Perspectives
Considerable progress has been made in recent years in understanding trypanosome transmission at the level of basic biology. However, these studies also have direct application in the search for new therapies for trypanosomes in the field. In particular, the ability of PAD proteins to discriminate stumpy forms enables high-throughput screens for compounds that accelerate stumpy formation at low parasite density. By inducing stumpy formation at low parasitaemia, the potential for parasite transmission may be reduced (Fig. 3). Furthermore, with a reduced parasite load, the host immune system may be able to control the infection. Importantly, genetically modified reporter lines could provide simple assays for enhanced stumpy formation accessible to screening with small molecule or natural product libraries. This offers great potential for disrupting the normal life cycle progression of the parasite and thereby restricting both pathogenicity and transmissibility of these important pathogens. As so often, an investment in basic knowledge has provided an unexpected opportunity for the rational screening for, and development of, novel therapeutic approaches.
Fig. 3.
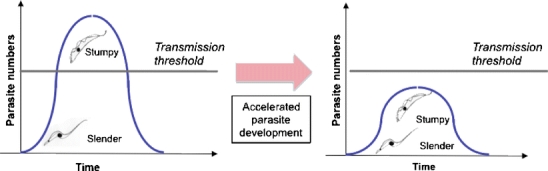
Accelerated parasite development as a route to limiting parasite transmission and parasitaemia. In the normal course of infection, slender cells generate stumpy forms once parasite numbers are sufficient to ensure transmission if taken up during a tsetse blood-meal. By accelerating stumpy formation at a lower parasitaemia, the parasite load in the host would be reduced, potentially reducing pathogenicity. Furthermore, the consequent limitation in parasite numbers would reduce the potential for parasite transmission during a tsetse blood-meal
Acknowledgements
This work is supported by a programme grant from the Wellcome Trust to KRM and a Wellcome Trust PhD studentship to PM. Work in the Matthews lab is carried out within the Centre for Immunity, Infection and Evolution, supported by a Strategic Award from the Wellcome Trust.
Conflict of interest declaration
None declared.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Brun R, Blum J, Chappuis F, Burri C. Human African trypanosomiasis. Lancet. 2010;375:148–159. doi: 10.1016/S0140-6736(09)60829-1. [DOI] [PubMed] [Google Scholar]
- 2.Vickerman K. Developmental cycles and biology of pathogenic trypanosomes. Brit Med Bull. 1985;41:105–114. doi: 10.1093/oxfordjournals.bmb.a072036. [DOI] [PubMed] [Google Scholar]
- 3.Vickerman K. Polymorphism and mitochondrial activity in sleeping sickness trypanosomes. Nature. 1965;208:762–766. doi: 10.1038/208762a0. [DOI] [PubMed] [Google Scholar]
- 4.Tyler KM, Higgs PG, Matthews KR, Gull K. Limitation of Trypanosoma brucei parasitaemia results from density-dependent parasite differentiation and parasite killing by the host immune response. Proc Roy Soc B. 2001;268:2235–2243. doi: 10.1098/rspb.2001.1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bass KE, Wang CC. The in vitro differentiation of pleomorphic Trypanosoma brucei from bloodstream into procyclic form requires neither intermediary nor short-stumpy stage. Mol Biochem Parasitol. 1991;44:261–270. doi: 10.1016/0166-6851(91)90012-U. [DOI] [PubMed] [Google Scholar]
- 6.Ziegelbauer K, Quinten M, Schwarz H, Pearson TW, Overath P. Synchronous differentiation of Trypanosoma brucei from bloodstream to procyclic forms in vitro. Eur J Biochem / FEBS. 1990;192:373–378. doi: 10.1111/j.1432-1033.1990.tb19237.x. [DOI] [PubMed] [Google Scholar]
- 7.Matthews KR, Gull K. Evidence for an interplay between cell cycle progression and the initiation of differentiation between life cycle forms of African trypanosomes. J Cell Biol. 1994;125:1147–1156. doi: 10.1083/jcb.125.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nolan DP, Rolin S, Rodriguez JR, Van Den Abbeele J, Pays E. Slender and stumpy bloodstream forms of Trypanosoma brucei display a differential response to extracellular acidic and proteolytic stress. Eur J Biochem / FEBS. 2000;267:18–27. doi: 10.1046/j.1432-1327.2000.00935.x. [DOI] [PubMed] [Google Scholar]
- 9.Priest JW, Hajduk SL. Developmental regulation of mitochondrial biogenesis in Trypanosoma brucei. J Bioenerg Biomem. 1994;26:179–191. doi: 10.1007/BF00763067. [DOI] [PubMed] [Google Scholar]
- 10.Balber AE. Trypanosoma brucei: fluxes of the morphological variants in intact and X-irradiated mice. Exp Parasitol. 1972;31:307–319. doi: 10.1016/0014-4894(72)90122-1. [DOI] [PubMed] [Google Scholar]
- 11.McLintock LM, Turner CM, Vickerman K. Comparison of the effects of immune killing mechanisms on Trypanosoma brucei parasites of slender and stumpy morphology. Parasite imm. 1993;15:475–480. doi: 10.1111/j.1365-3024.1993.tb00633.x. [DOI] [PubMed] [Google Scholar]
- 12.Engstler M, Pfohl P, Herminghaus S, et al. Hydrodynamic flow-mediated protein sorting on the cell surface of trypanosomes. Cell. 2007;131:505–515. doi: 10.1016/j.cell.2007.08.046. [DOI] [PubMed] [Google Scholar]
- 13.Brun R, Schonenberger M. Stimulating effect of citrate and cis-aconitate on the transformation of Trypanosoma brucei bloodstream forms to procyclic forms in vitro. Z Parasitenk (Berlin, Germany) 1981;66:17–24. doi: 10.1007/BF00941941. [DOI] [PubMed] [Google Scholar]
- 14.Czichos J, Nonnengaesser C, Overath P. Trypanosoma brucei: cis-aconitate and temperature reduction as triggers of synchronous transformation of bloodstream to procyclic trypomastigotes in vitro. Exp Parasitol. 1986;62:283–291. doi: 10.1016/0014-4894(86)90033-0. [DOI] [PubMed] [Google Scholar]
- 15.Hunt M, Brun R, Kohler P. Studies on compounds promoting the in vitro transformation of Trypanosoma brucei from bloodstream to procyclic forms. Parasitol Res. 1994;80:600–606. doi: 10.1007/BF00933009. [DOI] [PubMed] [Google Scholar]
- 16.Engstler M, Boshart M. Cold shock and regulation of surface protein trafficking convey sensitization to inducers of stage differentiation in Trypanosoma brucei. Genes Dev. 2004;18:2798–2811. doi: 10.1101/gad.323404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baylis M. The daily feeding rate of tsetse (Diptera: Glossinidae) on cattle at Galana Ranch, Kenya and comparison with trypanosomiasis incidence. Acta Trop. 1997;65:81–96. doi: 10.1016/S0001-706X(97)00655-4. [DOI] [PubMed] [Google Scholar]
- 18.Dean SD, Marchetti R, Kirk K, Matthews K. A surface transporter family conveys the trypanosome differentiation signal. Nature. 2009;459:213–217. doi: 10.1038/nature07997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zilberstein D, Shapira M. The role of pH and temperature in the development of leishmania parasites. Ann Rev Microbiol. 1994;48:449–470. doi: 10.1146/annurev.mi.48.100194.002313. [DOI] [PubMed] [Google Scholar]
- 20.Fang J, McCutchan TF. Thermoregulation in a parasite's life cycle. Nature. 2002;418:742. doi: 10.1038/418742a. [DOI] [PubMed] [Google Scholar]
- 21.Jacobs SL, Lee ND. Determination of citric acid in serum and urine using Br82. J Nucl Med. 1964;5:297–301. [PubMed] [Google Scholar]
- 22.Szoor B, Wilson J, McElhinney H, Tabernero L, Matthews KR. Protein tyrosine phosphatase TbPTP1: a molecular switch controlling life cycle differentiation in trypanosomes. J Cell Biol. 2006;175:293–303. doi: 10.1083/jcb.200605090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sbicego S, Vassella E, Kurath U, Blum B, Roditi I. The use of transgenic Trypanosoma brucei to identify compounds inducing the differentiation of bloodstream forms to procyclic forms. Mol Biochem Parasitol. 1999;104:311–322. doi: 10.1016/S0166-6851(99)00157-7. [DOI] [PubMed] [Google Scholar]
- 24.Rolin S, Hancocq-Quertier J, Paturiaux-Hanocq F, Nolan DP, Pays E. Mild acid stress as a differentiation trigger in Trypanosoma brucei. Mol Biochem Parasitol. 1998;93:251–262. doi: 10.1016/S0166-6851(98)00046-2. [DOI] [PubMed] [Google Scholar]
- 25.Breidbach T, Ngazoa E, Steverding D. Trypanosoma brucei: in vitro slender-to-stumpy differentiation of culture-adapted, monomorphic bloodstream forms. Exp Parasitol. 2002;101:223–230. doi: 10.1016/S0014-4894(02)00133-9. [DOI] [PubMed] [Google Scholar]
- 26.Denninger V, Figarella K, Schonfeld C, et al. Troglitazone induces differentiation in Trypanosoma brucei. Exp Cell research. 2007;313:1805–1819. doi: 10.1016/j.yexcr.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Laxman S, Riechers A, Sadilek M, Schwede F, Beavo JA. Hydrolysis products of cAMP analogs cause transformation of Trypanosoma brucei from slender to stumpy-like forms. Proc Natl Acad Sci USA. 2006;103:19194–19199. doi: 10.1073/pnas.0608971103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reuner B, Vassella E, Yutzy B, Boshart M. Cell density triggers slender to stumpy differentiation of Trypanosoma brucei bloodstream forms in culture. Mol Biochem Parasitol. 1997;90:269–280. doi: 10.1016/S0166-6851(97)00160-6. [DOI] [PubMed] [Google Scholar]
- 29.Tyler KM, Matthews KR, Gull K. The bloodstream differentiation-division of Trypanosoma brucei studied using mitochondrial markers. Proc Roy Soc B. 1997;264:1481–1490. doi: 10.1098/rspb.1997.0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vassella E, Boshart M. High molecular mass agarose matrix supports growth of bloodstream forms of pleomorphic Trypanosoma brucei strains in axenic culture. Mol Biochem Parasitol. 1996;82:91–105. doi: 10.1016/0166-6851(96)02727-2. [DOI] [PubMed] [Google Scholar]
- 31.Savill NJ, Seed JR. Mathematical and statistical analysis of the Trypanosoma brucei slender to stumpy transition. Parasitology. 2004;128:53–67. doi: 10.1017/S0031182003004256. [DOI] [PubMed] [Google Scholar]
- 32.Seed JR, Sechelski J. Growth of pleomorphic Trypanosoma brucei rhodesiense in irradiated inbred mice. J Parasitol. 1988;74:781–789. doi: 10.2307/3282254. [DOI] [PubMed] [Google Scholar]
- 33.Vassella E, Reuner B, Yutzy B, Boshart M. Differentiation of African trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the cAMP pathway. J Cell Sci. 1997;110(Pt 21):2661–2671. doi: 10.1242/jcs.110.21.2661. [DOI] [PubMed] [Google Scholar]
- 34.Hesse F, Selzer PM, Muhlstadt K, Duszenko M. A novel cultivation technique for long-term maintenance of bloodstream form trypanosomes in vitro. Mol Biochem Parasitol. 1995;70:157–166. doi: 10.1016/0166-6851(95)00027-X. [DOI] [PubMed] [Google Scholar]
- 35.Scory S, Stierhof YD, Caffrey CR, Steverding D. The cysteine proteinase inhibitor Z-Phe-Ala-CHN2 alters cell morphology and cell division activity of Trypanosoma brucei bloodstream forms in vivo. Kinetoplastid Biol Dis. 2007;6:2. doi: 10.1186/1475-9292-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lythgoe KA, Morrison LJ, Read AF, Barry JD. Parasite-intrinsic factors can explain ordered progression of trypanosome antigenic variation. Proc Natl Acad Sci USA. 2007;104:8095–8100. doi: 10.1073/pnas.0606206104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van den Bossche P, Ky-Zerbo A, Brandt J, Marcotty T, Geerts S, De Deken R. Transmissibility of Trypanosoma brucei during its development in cattle. Trop Med Int Health. 2005;10:833–839. doi: 10.1111/j.1365-3156.2005.01467.x. [DOI] [PubMed] [Google Scholar]
- 38.Maudlin I, Welburn SC. A single trypanosome is sufficient to infect a tsetse fly. Ann Trop Med Parasitol. 1989;83:431–433. doi: 10.1080/00034983.1989.11812368. [DOI] [PubMed] [Google Scholar]