

Abstract
Alzheimer’s disease is the fourth biggest killer in developed countries. Amyloid precursor protein (APP) plays a central role in the development of the disease, through the generation of a peptide called Aβ by proteolysis of the precursor protein. APP can function as a metalloprotein and modulate copper transport via its extracellular copper binding domain (CuBD). Copper binding to this domain has been shown to reduce Aβ levels and hence a molecular understanding of the interaction between metal and protein could lead to the development of novel therapeutics to treat the disease. We have recently determined the three-dimensional structures of apo and copper bound forms of CuBD. The structures provide a mechanism by which CuBD could readily transfer copper ions to other proteins. Importantly, the lack of significant conformational changes to CuBD on copper binding suggests a model in which copper binding affects the dimerisation state of APP leading to reduction in Aβ production. We thus predict that disruption of APP dimers may be a novel therapeutic approach to treat Alzheimer’s disease.
Keywords: Alzheimer’s disease, Amyloid precursor protein, Copper binding, Crystal structures, Receptor signalling
Introduction
Alzheimer’s disease (AD) is characterised pathologically by the presence of amyloid plaques, extensive neuronal death and shrinkage of the brain. The symptoms of the disease begin with short-term memory disturbances and disorientation, and as the disease progresses judgement and reasoning become impaired and the sufferer dies after several years of dementia. Currently there is no treatment for the disease, although acetylcholinesterase inhibitors have displayed some moderate benefit in treatment.
There is increasing evidence that the neurotoxic Aβ peptide can compromise neuronal functions and trigger cell death (Selkoe 2002). The peptide can aggregate into small soluble oligomers, eventually leading to the amyloid plaques observed in AD diseased brains. Small soluble oligomers of Aβ have been shown to cause damage to cultured neurones (Haass and Selkoe 2007). The peptide is derived from the cleavage of the amyloid precursor protein (APP) and is the main constituent of the amyloid plaques (Kang et al. 1987). APP belongs to a larger family of APP-like proteins (APLPs) and homologues have been identified in a range of higher organisms including worms, insects and rodents (Coulson et al. 2000). These proteins exhibit some functional redundancy but only APP cleavage gives rise to the Aβ peptide. The cleavage occurs during trafficking and maturation through the protein secretory pathway by membrane-bound proteases. Aβ arises through the sequential cleavage by the BACE enzyme (Sinha et al. 1999; Yan et al. 1999) at a site just outside the plasma membrane and the γ-secretase protein complex (Haass and Steiner 2002) within the membrane (Fig. 1). An alternative APP proteolytic pathway exists in which cleavage by α-secretase results in the release of soluble APP. The secreted APP has been shown to possess growth-promoting activities and can bind to extracellular matrix components (De Strooper and Annaert 2000). This major pathway is non-amyloidogenic since it cleaves close to the middle of the Aβ peptide and therefore prevents intact Aβ peptide formation.
Fig. 1.
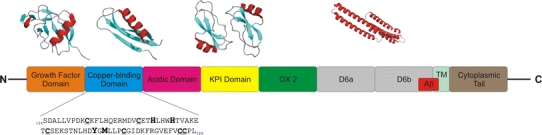
Domain arrangement of APP highlighting some important regions of the molecule. Cartoon ribbon pictures of known 3D structures of domains are shown at the top of the picture. The N-terminal growth factor domain (GFD; PDB id: 1MWP) is followed by CuBD (PDB ids: 1OWT, 2FKL, 2FJZ and 2FK3), an acidic-rich region, Kunitz-type protease inhibitor (KPI; PDB id: 1AAP) and OX2 domains that occur in some APP isoforms, a couple of glycosylated domains (D6a, sometimes called the E2 domain or CAPPD; PDB ids: 1TKN, 1RW6; followed by an unstructured domain we refer to as D6b), a transmembrane region (TM) and a cytoplasmic tail. The location of the Aβ region, a major component of Alzheimer’s disease plaques, is shown. The sequence of CuBD is shown with the copper binding ligands and Met170 in large bold type and the cysteines that are involved in disulfide bridges shown in underlinedbold type
APP as a signalling molecule
Amyloid precursor protein is a Type-I transmembrane protein with a large extracellular region that can be subdivided into several distinct domains (Fig. 1). At the N-terminus is a cysteine-rich region consisting of the growth factor domain (GFD), which binds heparin and can stimulate neurite outgrowth (Ohsawa et al. 1997; Rossjohn et al. 1999) and a copper-binding domain (CuBD) (Barnham et al. 2003). The cysteine-rich region is followed by an acidic-rich region, a Kunitz-type protease inhibitor (KPI) domain and an OX2 domain (Tanaka et al. 1988; Tanzi et al. 1988). The KPI and OX2 domains can be spliced out, to produce three main variants: APP770 (with 770 amino acid residues), APP751 and APP695. The latter is the most common isoform in the brain (Haass et al. 1991). Following these domains is a glycosylated domain referred to as E2 (Wang and Ha 2004) and an unstructured region immediately preceding the transmembrane domain. The E2 domain possesses a RERMS sequence that may contribute to APP’s growth-promoting properties and it also has a heparin binding site. The C-terminal cytoplasmic tail is involved in various cellular functions through interaction with a multitude of proteins (De Strooper and Annaert 2000).
The function of normal APP remains largely unknown. There is appreciable evidence that APP acts as a cell surface receptor. It shares a similar architecture, cellular orientation and localisation to known (Type-1) cell surface receptors (Kang et al. 1987; Weidemann et al. 1989; Schubert et al. 1991). The APP cytoplasmic domain has been shown to bind to Fe65 protein, a protein related to oncogenic signal transducers, implicating a role for APP as a cell surface signalling receptor (Cao and Südhof 2001). Mouse knockouts of either APP, APLP1, APLP2 or their binding partner Fe65 cause the same phenotype of cortical dysplasias suggesting the phenotype is caused by decreased transmission of an APP-dependent signal through the Fe65 protein (Guenette et al. 2006). APP hereditary mutations that are linked with Alzheimer’s disease, can cause constitutive activation of Go, a member of the heteromeric G protein family that serve as signal tranducers of cell surface receptors (Okamoto et al. 1995). An antibody directed towards the APP N-terminal domain stimulates G protein and MAP kinase activity (Okamoto et al. 1995). The antibody is presumably mimicking the action of a still-to-be-identified physiological ligand. There is evidence that APP exists in a monomer to dimer equilibrium in the membrane (Scheuermann et al. 2001; Soba et al. 2005) and the binding of high molecular weight heparin induces dimerisation (Gralle et al. 2006). Dimerisation may cause an initiation of signalling events (Sondag and Combs 2004) and play a role in modulating APP cleavage as the production of Aβ is increased when APP is forcibly dimerised (Scheuermann et al. 2001).
APP as a copper binding molecule
Copper is essential to life but is also toxic and as a result cells have developed elaborate systems for copper storage and transport (Bush 2000). The CuBD of APP can strongly bind Cu(II) (KdAPP ≈ 10 nM) and reduce it to Cu(I) in vitro (Hesse et al. 1994; Multhaup et al. 1996). This can lead to increased copper mediated neurotoxicity in cultured neurones (White et al. 1999a), presumably through increased oxidative stress or the generation of reactive oxygen species (Multhaup et al. 1997). APP appears to play a role in copper transport. In particular, a role for APP in copper efflux has been supported by a series of experiments which demonstrated an increased intracellular accumulation of copper in neuronal cultures (Bellingham et al. 2004) and in the brains of APP/APLP2 double knockout mice (White et al. 1999b) whilst transgenic over-expression of APP had the opposite effects in cultured neurones and mice (Maynard et al. 2002; Bellingham et al. 2004). The involvement of CuBD in copper efflux has been demonstrated in yeast expressing an APP ectodomain fragment. Mutating the copper binding residues in CuBD significantly increases intracellular copper levels compared to yeast expressing a wild-type fragment (Treiber et al. 2004).
The interaction between copper ions and CuBD can modulate Aβ production. The treatment of CHO cells over-expressing APP with extracellular Cu(II) leads to reduced Aβ production and a shift of the cleavage equilibrium away from the amyloidgenic pathway (Borchardt et al. 1999). The effects are abolished when the copper binding residues of CuBD are mutated (Borchardt et al. 2000). The possible beneficial outcomes of Cu(II) binding to APP have been suggested in two independent transgenic mouse studies. The survival of APP23 mice (which overproduce Aβ) was improved along with reduced Aβ-levels in the brain when their drinking water was supplemented with Cu(II) ions (Bayer et al. 2003). Similar effects were achieved by crossing TgCRND8 mice (that overproduce Aβ) with mice that have raised brain copper levels due to the expression of a mutant copper transporter CuATPase7b. These mice had decreased amyloid plaque and plasma Aβ levels (Phinney et al. 2003).
A molecular target for the APP:Cu or APLP2:Cu complexes is the proteoglycan molecule glypican-1 (Cappai et al. 2005). APP and APLP2 can regulate the non-enzymatic metabolism of the glypican-1 heparin sulfate side-chains in a copper-dependent manner in vitro and in vivo. The APP : Cu complex stimulates glypican-1 autodegradation in the presence of either Cu(II) and Zn(II) ions, whereas the Cu(I) form of APP and the Cu(II) and Cu(I) forms of APLP2 inhibit autodegradation (Cappai et al. 2005).
In addition to copper binding to CuBD, copper is also known to bind the Aβ peptide fragment of APP, inducing peptide aggregation and leading to the production of reactive oxygen species via Fenton chemistry (Smith et al. 2007). The interested reader is referred to more in-depth reviews on this topic found elsewhere in this special issue.
CuBD structure
To understand the interaction of copper with APP at the molecular level, we initially determined the three-dimensional structure of CuBD (APP residues 124–189) by nuclear magnetic resonance spectroscopy (NMR) (PDB id: 1OWT; Barnham et al. 2003). This work revealed that the CuBD structure consists of an α-helix (residues 147–159) packed against a triple-stranded β-sheet (residues 133–139, 162–167 and 181–188) (Fig. 2a). A Cys144–Cys174 disulfide bond connected two loops at one end of the molecule. On the other end a disulfide bond between Cys133 and Cys187 linked strands β1 and β3 and another between Cys158 and Cys186 linked the α-helix to strand β3. The surface of CuBD is highly charged with several areas of high negative (Glu156, Glu160, Glu183, Asp167 and Asp131) and positive (Lys132, Lys134, Lys161, His147, His151 and Lys155) charge. CuBD belongs to the class of mixed α/β proteins, displays an architecture of a two-layered sandwich, with a topology that can be classified as either α-β plaits or defensin A-like (Pearl et al. 2005). Proteins that exhibit the latter topology include copper transport proteins such as yeast metallochaperone Atx1 (Arnesano et al. 2001), human metallochaperone HAH1 (Anastassopoulou et al. 2004) and the metal-binding domains of Menkes Protein ATP7A (Banci et al. 2004, 2005). These proteins contain an additional α-helix and β-strand after the βαββ motif of CuBD and display no sequence similarity to CuBD. Their copper binding sites are located on the surface, but unlike CuBD, in the loop joining strand β1 and helix α1, and use two cysteines in a CXXC motif as copper ligands.
Fig. 2.
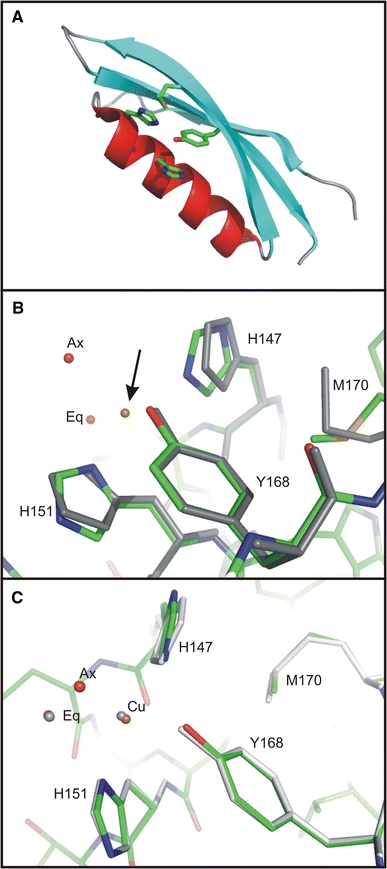
Three-dimensional structure of CuBD (Kong et al. 2007a). a Ribbon picture of apo CuBD (PDB ids: 2FKL, 2FJZ and 2FK3) highlighting some of the side-chains that contribute to the copper binding site. b The Cu(II) coordination geometry in CuBD (PDB id: 2FK1) and comparison of the copper binding residues in the Cu(II)-bound and apo structures. The Cu(II)-bound structure is displayed in standard atomic colouring and the apo structure is in grey. The Cu(II) ion is indicated by the arrow, while ‘eq’ and ‘ax’ denote the equatorial and axial water molecules, respectively. C The Cu(I) coordination geometry in CuBD (PDB id: 2FK2) and a comparison of the copper binding site in the Cu(II)-bound and Cu(I)-bound CuBD structures. The Cu(II)- and Cu(I)-bound structures are shown in standard atomic colouring and in grey, respectively
More recently we determined the crystal structure of apo CuBD from three different crystal forms at high resolution (Fig. 2a) (PDB ids: 2FKL, 2FJZ, 2FK3; Kong et al. 2007a). These structures revealed eleven different views of the molecule due to multiple copies in the asymmetric unit in some crystal forms. Despite the different crystal lattice forces, these structures superimpose closely amongst each other and with the previously determined NMR structure (Barnham et al. 2003). Major deviations are largely restricted to loops and are attributable to various crystal packing contacts. Backbone atoms of the copper binding residues superimpose closely despite being involved in different types of crystal contacts amongst the crystal forms. The relative positions of those side-chains are also well conserved, although His147 is seen to adopt different side-chain orientations, which may be relevant for copper transport to and from other proteins (see below).
We have just reported the crystal structure of CuBD in metal-free form at ultra-high resolution (0.85 Å) (PDB id: 2FMA; Kong et al. 2007b). The structure shows that the copper binding residues of CuBD (see below) are rather rigid but Met170, thought to be the electron source for Cu(II) reduction, adopts two different side-chain conformations. In one, the thioether is about 7 Å from the copper binding site and points away from the other copper binding residues. In the second conformation, the thioether group is closer to the copper binding site (6 Å) and points towards the copper ion, although Met170 itself is still buried from the surface. The flexibility of the Met170 side-chain may be important in facilitating copper ion binding or the reduction of Cu(II) to Cu(I) after Cu(II) is bound to the CuBD (White et al. 2002; Barnham et al. 2003).
Copper binding to CuBD
The conformation of Cu(II)-binding residues in CuBD could not be visualised by NMR as the paramagnetism of Cu(II) caused severe peak broadening arising from atoms in close proximity to the metal ion and addition of metals such as copper and zinc induced protein aggregation in solution (Barnham et al. 2003). However, this paramagnetic effect implicated His147, His151, Tyr168 and Met170 in Cu(II) binding. Copper ions could not be introduced to CuBD either by co-crystallisation nor by soaking preformed crystals at high concentrations of copper without causing crystal shattering, likely due to numerous copper binding sites on the surface of the protein. However, careful manipulation of the soaking conditions led to crystals in which the high affinity copper binding site was occupied without crystal destruction. Our structure derived from such crystals revealed that the two histidines (His147, His151) and the tyrosine residue (Tyr168) participate in Cu(II) binding but not the methionine residue. In addition, two water ligands were identified: an axial water and an ‘equatorial’ water molecule located ∼1.5 Å above the plane of the other ligands (Fig. 2b) (PDB id: 2FK1; Kong et al. 2007a). The arrangement of ligands about the metal centre is typical of the square pyramidal geometry normally adopted by five coordinate Cu(II) and can be classified as a Type 2 non-blue Cu(II) centre in which the metal is normally bound by two or three nitrogen atoms and oxygen atoms (http://metallo.scripps.edu/PROMISE/CUMAIN.html). The copper binding geometry is consistent with observations suggesting that APP is capable of reducing Cu(II) to Cu(I) (Multhaup et al. 1996) as Type 2 sites are often associated with redox-driven catalysis (Messerschmidt et al. 2001). A similar Type 2 site has been described in nitrosocyanin which is also open to solvent and involves a water ligand (Arciero et al. 2002). The Cu(II) coordination geometry was further confirmed by electron paramagnetic resonance spectroscopy and extended X-ray absorption fine structure spectroscopy of Cu(II)-bound CuBD in solution (Kong et al. 2007a).
We also determined the crystal structure of the Cu(I) form of the protein through the use of a reductant on the Cu(II)-bound CuBD crystals (PDB id: 2FK2; Kong et al. 2007a). The Cu(I) binding geometry is almost identical to that of Cu(II) in CuBD (see above) with such small differences between oxidation states having been noted previously for other copper proteins (Gray et al. 2000).The only significant difference is the absence of the axial water ligand (Fig. 2c). Similar losses of a water ligand upon Cu(II) reduction have been observed in a couple of other copper redox proteins (Murphy et al. 1997; Basumallick et al. 2005). Normally four coordinate Cu(I) adopts a tetrahedral geometry and binds soft ligands (Casella and Gullotti 1993) rather than the distorted square planar arrangement and oxygen ligands seen for CuBD. Thus, the site appears unfavourable for Cu(I) suggesting the Cu(I)-bound state may not be stable, possibly facilitating the transfer of metal to other proteins following CuBD-mediated reduction of Cu(II).
The NMR Cu(II) titration experiments led to evidence of Met170 oxidation and peak broadening of its proton resonances suggesting that it was in the neighbourhood of the metal ion (Barnham et al. 2003). However, in the crystal structures we found that sulfur atom of Met170 is 7 Å from either the Cu(II) or Cu(I) ion (Fig. 2b,c) (Kong et al. 2007a, b). We propose that Met170 may play an important role as an electron donor to Cu(II) either through rapid conformational changes which would bring the sulfur atom in close contact with the metal ion, or through an electron transfer pathway which relays the electron from Met170 to Cu(II) through the copper binding residues. The proximity of Met170 and Tyr168 in CuBD provides a plausible electron transfer pathway; reminiscent of a similar mechanism proposed for Aβ peptide-induced Cu(II) reduction where a methionine residue has been suggested to provide the electron for Cu(II) reduction through a tyrosine ligand (Curtain et al. 2001; Barnham et al. 2004).
The surface location of the copper site in CuBD may facilitate the transfer of metal ions to and from other copper-binding proteins. This is, in a sense, similar to the case of the metal-binding domains of the human Menkes protein (ATP7a), where Cu(I) also binds in an exposed environment after it is delivered from the chaperone HAH1 although the ligands involved are exclusively cysteines (Banci et al. 2005). In the case of the yeast chaperone protein Atx1, its interaction with its downstream target Ccc2 ATPase (Ccc2a) involves bringing the Cu(I) ion from a more shielded environment to the surface where the cysteine ligands of Ccc2a would be able to chelate the Cu(I) from Atx1. The Atx1-Ccc2a interaction, although aided by other forces, is absolutely dependent on Cu(I) binding (Banci et al. 2006). We can similarly envisage the interaction of CuBD and its partners being driven by the presence of copper ions. The water ligands in CuBD could suggest where an endogenous ligand or binding partner of CuBD may dock by displacing these waters (Prigge et al. 2004). The putative ligand could come from a number of sources: (a) another ligand of the CuBD, (b) from an adjacent domain within the same protein molecule, (c) from another APP molecule to form an oligomer, or (d) a binding partner of APP, such as proteins that transport copper ions to or from APP. The third possibility would be consistent with the observation that CuBD may form part of the APP dimerisation interface (Scheuermann et al. 2001; Soba et al. 2005) and the positions of the water molecules may represent where the dimer interactions occur. The crystal packing interactions seen in some of the CuBD crystal forms suggest a propensity of the copper binding residues to take part in protein–protein interactions. However, we have not observed specific CuBD oligomerisation induced by Cu(II) (unpublished results).
Modelling CuBDs from other species
One of the consequences of Cu(II) binding to APP is thought to be neuronal toxicity which has been demonstrated so far in cultured neurones. The hypothesised role of CuBD in neuronal toxicity is that the bound Cu(II) is reduced to Cu(I), which can then lead to lipoprotein peroxidation and free radicals that damage the cell (White et al. 2002). CuBD is one of the best conserved domains within the APP superfamily (Fig. 3). Surprisingly, the effect of copper binding varies between the orthologues and paralogues. For example, it has been shown that the presence of Cu(II) salts in primary murine neuronal cultures causes greater cell death in wild-type neurones compared to APP−/− neurones whereas there is no difference between wild-type and APLP2−/− neurones (White et al.1999a). Lipoprotein peroxidation is enhanced by Cu(II)-treated ectodomain of human APP and APLP2, whereas neither the non-metallated forms of those proteins nor Cu(II)-metallated human APLP1 show increased peroxidation above background (White et al. 2002). Cu(II)-bound CuBD of human APP appears sufficient in the generation of Cu(I) and causing neuronal cell death (White et al. 2002).
Fig. 3.

A sequence alignment of CuBD from selected members of the APP superfamily. The prefixes h, r, m, d and c denote human, rat, mouse, Drosophila melanogaster and Caenorhabditis elegans, respectively. The N-terminal and C-terminal numbering of each CuBD are listed. Conserved residues are indicated by asterisks and coloured in aqua, colons indicate positions with very similar amino acid residues while dots indicate positions with some similarity. The copper binding residues of human APP CuBD and their equivalents in other homologues are coloured in orange
The behaviour of peptides derived from CuBDs of various APP homologues suggests that the toxicity may depend on the presence of the copper binding histidine residues. The replacement of one, and both of those histidines results in an inert and protective effect, respectively, when the peptides are added to cultured neurones incubated with lipoproteins. The peptides studied were derived from human APLP1 (one histidine replaced), Caenorhabditis elegans homologue APL-1 (two histidines replaced) and Drosophila melanogaster homologue APPL (two histidines replaced). However, these fragments do not cover the position corresponding to the copper binding residue Tyr168 and do not preserve the native disulfide bonds. Hence the structure, binding behaviour and Cu(II) reduction mechanism of the peptides are very likely to be different in intact CuBD.
We have produced three-dimensional atomic models of selected CuBDs from various organisms using homology modelling based on the sequence alignments shown in Fig. 3. The sequence alignment shows that the copper binding residues of human APP are not conserved in the APLP-1 family, C. elegans homologue APL-1 or D. melanogaster homologue APPL (Fig. 3). The replacement of the copper binding residues in human APLP-1 (Fig. 4b), C. elegans APL-1 (Fig. 4c) and D. melanogaster APPL (Fig. 4d) likely abolishes copper binding since residues are eliminated that are likely to be involved in copper coordination and the distance between the putative Cu(II) ion binding site to the surrounding residues increases (Fig. 4).
Fig. 4.
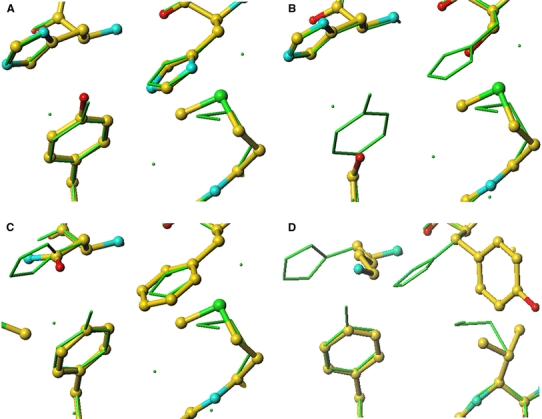
A comparison of the human APP CuBD copper binding site to the equivalent site in orthologues and paralogues. a Human APLP-2, b human APLP-1, c D. melanogaster APPL and d C. elegans APL-1. The human APP residues are shown in greensticks whereas the homology models are in ball-and-stick with the standard atomic colouring
Those APP homologues where the CuBD models are predicted to lack the copper binding site will not be able to bind Cu(II) and thus reduce it to the more toxic Cu(I). This would explain why human APLP-1 is not able to oxidise lipoproteins and cause neuronal death (White et al. 2002). It is also consistent with the protective effect of D. melanogaster APPL and C. elegans APL-1 CuBD peptides against lipoprotein peroxidation by Cu(II) ions. If the hypothesis on Cu(II)-induced toxicity holds, the result of modelling would support the role of His147, His151 and Tyr168 of human APP CuBD in copper binding.
The copper binding site of human APLP-2 CuBD is predicted to be very similar to human APP CuBD based on comparison of primary structures (Fig. 3) and the similarity of the APLP-2 CuBD homology model with the crystal structure (Fig. 4a). In view of the high similarity of the Cu(II)-bound, Cu(I)-bound and apo structures of human APP CuBD (see above), it is expected that the Cu(II)-bound and Cu(I)-bound structures of human APLP-2 CuBD would be very similar as well. The copper binding residues His147, His151 and Tyr168, are also conserved in APLP-2s in other organisms, but the toxicity mediated by APLP-2s appears to be lower than APPs. For example, Cu(II)-bound human APP is more effective than Cu(II)-bound human APLP-2 in oxidising lipoproteins and causing cell death (White et al. 2002). Wild-type murine hippocampal neurones suffer greater cell death compared to mouse APP−/− counterparts while mouse APLP-2−/− offers no protection (White et al. 1999a). The crystal structure of human APP CuBD and the model of human APLP-2 CuBD do not provide an obvious explanation for the different toxicity effects mediated by these two APP homologues.
Modelling CuBD interactions with GFD
The N-terminal GFD is immediately upstream of CuBD (Fig. 1) and together they constitute the cysteine-rich region of the molecule. To determine if these two domains likely interact we used the GRAMM program (Vasker and Aflalo 1994; Vasker 1996) to dock CuBD to the crystal structure of GFD (PDB id: 1MWP; Rossjohn et al. 1999). Visual inspection indicated that the most plausible domain pair structure brought the C-terminus of GFD and the N-terminus of CuBD in close proximity while the arrangement of the domain interface was suggestive of a continuation of the β-sheet from domain 1 into the β-sheet of domain 2 (Fig. 5). The total surface area buried in forming the interface is ∼200 Å2. The residues forming the interface are primarily hydrophobic, including Val86, Leu118, Val119 and Gly120 from GFD and Ala126, Leu127, Leu128, Val129 and Pro130 of CuBD with the ends of this hydrophobic region flanked by polar and charged residues that are exposed to solvent. In total there are nine potential hydrogen bonds and 28 van der Waals interactions in the domain interface. Both the heparin binding site of GFD and the copper binding site of CuBD, are exposed in the model at opposite ends of the cysteine rich region, separated by ∼50 Å.
Fig. 5.
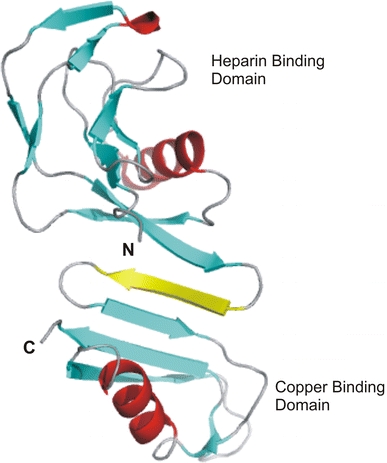
Model of the APP growth factor domain docked onto CuBD. Note that an extra β-strand (coloured yellow), suggested by the modelling, has been inserted between the two domains to form an extended sheet
We have determined the crystal structure of a longer length fragment of CuBD124–189 to 2.5 Å resolution (Kong et al. 2007a). This crystal from was found to have two CuBD molecules in the asymmetric unit. An interesting feature of this structure is that the two molecules interact head-to-head via new β-strand (β0) involving residues 127–129 of one molecule hydrogen bonding onto strand β2 of a neighbouring molecule to form an extended β-sheet (Fig. 6). The interaction buries about 1,550 Å2 of surface area between the molecules. However, there are a few contacts between the two chains. The new strand β0 of chain B provides some of those interactions through hydrogen bonds from the backbone amides of Ala126 (B) and Leu127 (B) to the backbone carbonyl of Met170 (A), and that from the backbone carbonyl of Tyr168 (A) to backbone amide of Val129 (B). There are van der Waals contacts between the rings of His147 (A) and Phe135 (B), between the backbone of Gly175 (A) and side-chain of Leu171 (B), between the side-chains of Asp177 (A) and His137 (B), and a hydrogen bond between the backbone amide of Asp177 (A) and carboxylate group of Glu183 (B). Strand β0 is also within van der Waals distance of the copper binding site of the latter molecule. However, it is unlikely that the dimer seen in this crystal form has a significant existence in solution because there is no evidence of dimerisation of CuBD124–189 in the NMR experiment (Barnham et al. 2003). Nevertheless, the structure shows that N-terminal residues 124–132 of CuBD have an inherent propensity to form a β-strand and thus lends support to the suggestion discussed above that this region of CuBD could be induced to form a strand leading to the extended β-sheet with GFD in intact APP (Fig. 5).
Fig. 6.
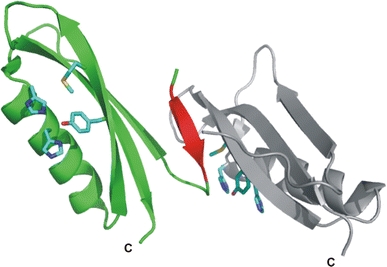
A ribbon diagram of CuBD124–189. Chains A and B are coloured in grey and green, respectively. Strand β0 of chain B is highlighted in red. The side-chains of the copper binding residues and Met170 are shown as blue sticks
In a recent collaboration an ab initio reconstruction of a molecular model of the complete extracellular region of APP (sAPP) was presented based on data from high-resolution solution X-ray scattering (SAXS) data together with the fitting of domains from published crystal structures (Gralle et al. 2006). In this model the N-terminal growth-factor domain and CuBD would lie at a distance of ∼100 Å from the membrane. The results from SAXS, analytical ultracentrifugation (AUC) and size-exclusion chromatography (SEC) analysis indicated that sAPP is monomeric in solution. On the other hand, SEC, fluorescence, AUC and SAXS measurements showed that sAPP forms a 2:1 complex with heparin. A model for the sAPP:heparin complex was also derived from the SAXS data.
Copper binding to APP: implications for signalling
Our structures do not directly explain how copper binding leads to decreasing Aβ production since we see no evidence of significant conformational changes in the structure of CuBD upon metal binding or reduction. However, it must be mediated by some sort of signalling mechanism, for example, modulation of proteins that cleave APP (Angeletti et al. 2005), a change in conformation of APP, recruitment of other proteins or a change in the oligomerisation state of the APP. APP can associate with Go as a G-protein coupled receptor (Brouillet et al. 1999) and signalling can be initiated upon binding of an antibody, 22C11, that binds to the GFD and is thought to act as a ligand mimetic (Okamoto et al. 1995). Scheuermann et al. (2001) were able to isolate endogenous membrane-bound APP as dimers and showed that Aβ production increased when APP was cross-linked to form dimers. However, it cannot be excluded that the effect on Aβ production could have been as a consequence of modifying APP near its α-secretase cleavage site. Putative dimerisation domains have been proposed. Wang and Ha (2004) suggested the E2 domain (Fig. 1) as a candidate. On the other hand, Scheuermann et al. (2001) demonstrated that a fragment encompassing the GFD, CuBD and the acidic-rich region existed in dimeric form by gel filtration, while Soba et al. (2005) showed that the GFD and CuBD might be a major site of dimerisation of APP in the cellular context. If the CuBD does contribute to APP dimerisation in the absence of Cu ions, we propose that the effect of Cu(II) in lowering Aβ production may perhaps be elicited through either a shift of monomer-dimer equilibrium to favour monomers or a re-orientation of the APP dimers to a form that disfavours Aβ production (Fig. 7). If the copper site is exposed in the monomeric or re-orientated dimer form, it could also aid the interaction of APP with other copper binding proteins in the transfer of copper ions. The copper binding pocket, perhaps together with other parts of CuBD or APP, will provide a site for the docking of interacting partners. Based on this hypothesis, new approaches to Alzheimer’s therapeutics could be devised, as compounds that can disrupt the formation or organisation of pre-formed APP dimers may be beneficial in reducing Aβ production from APP.
Fig. 7.
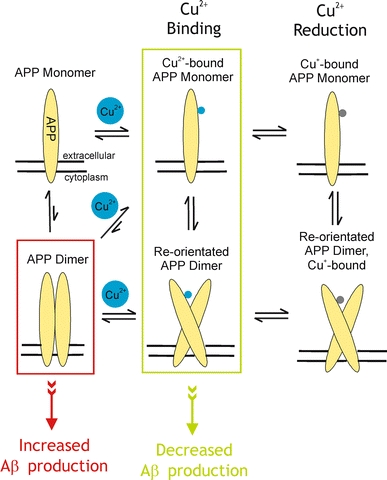
A model of the putative effects of Cu(II) binding on APP dimerisation and Aβ production
Concluding remarks
Copper binding to APP can yield paradoxical outcomes. On the one hand copper binding to the CuBD of APP results in Aβ depletion whereas copper binding to the Aβ peptide generates toxic species. From a therapeutic point-of-view these observations suggest that a molecule that mimics the effect of copper binding on CuBD could be of significant clinical benefit.
Acknowledgements
M. W. P. would like to thank all present and past members of his laboratory and his collaborators who have made major contributions to the structural studies of APP discussed in this paper. This work was supported in part by grants from the Australian Research Council (ARC) and National Health and Medical Research Council of Australia (NHMRC). G. K.-W. K. was an Australian Postgraduate Award Scholar and a recipient of a Ludo Frevel Scholarship from the International Centre for Diffraction Data, W. J. M. was a NHMRC Industry Fellow, R. C is a NHMRC Senior Research Fellow and M. W. P. is an ARC Federation Fellow and NHMRC Honorary Fellow.
Footnotes
Australian Society for Biophysics Special Issue: Metals and Membranes in Neuroscience.
References
- Anastassopoulou I, Banci L, Bertini I, Cantini F, Katsari E, Rosato A. Solution structure of the apo and copper(I)-loaded human metallochaperone HAH1. Biochemistry. 2004;43:13046–13053. doi: 10.1021/bi0487591. [DOI] [PubMed] [Google Scholar]
- Angeletti B, Waldron KJ, Freeman KB, Bawagan H, Hussain I, Miller CCJ, Lau K-F, Tennant ME, Dennison C, Robinson NJ, Dingwall C. BACE1 cytoplasmic domain interacts with the copper chaperone for superoxide dismutase-1 and binds copper. J Biol Chem. 2005;280:17930–17937. doi: 10.1074/jbc.M412034200. [DOI] [PubMed] [Google Scholar]
- Arciero DM, Pierce BS, Hendrich MP, Hooper AB. Nitrosocyanin, a red cupredoxin-like protein from Nitrosomonas europaea. Biochemistry. 2002;41:1703–1709. doi: 10.1021/bi015908w. [DOI] [PubMed] [Google Scholar]
- Arnesano F, Banci L, Bertini I, Huffman DL, O’Halloran TV. Solution structure of the Cu(I) and apo forms of the yeast metallochaperone Atx1. Biochemistry. 2001;40:1528–1539. doi: 10.1021/bi0014711. [DOI] [PubMed] [Google Scholar]
- Banci L, Bertini I, Conte R, D’Onofrio M, Rosato A. Solution structure and backbone dynamics of the Cu(I) and apo forms of the second metal-binding domain of the Menkes protein ATP7A. Biochemistry. 2004;43:3396–3403. doi: 10.1021/bi036042s. [DOI] [PubMed] [Google Scholar]
- Banci L, Bertini I, Ciofi-Baffono S, Chasapis CT, Hadjiliadis N, Rosato A. An NMR study of the interaction between the human copper(I) chaperone and the second and the fifth metal-binding domains of the Menkes protein. FEBS J. 2005;272:865–871. doi: 10.1111/j.1742-4658.2004.04526.x. [DOI] [PubMed] [Google Scholar]
- Banci L, Bertini I, Cantini F, Felli IC, Gonnelli L, Hadjiliadis N, Pierattelli R, Rosato A, Voulgaris P. The Atx1-Ccc2 complex is a metal-mediated protein-protein interaction. Nature Chem Biol. 2006;2:367–368. doi: 10.1038/nchembio797. [DOI] [PubMed] [Google Scholar]
- Barnham KJ, McKinstry WJ, Multhaup G, Galatis D, Morton CJ, Curtain CC, Williamson NA, White AR, Hinds MG, Norton RS, Beyreuther K, Masters CL, Parker MW, Cappai R. Structure of the Alzheimer’s disease amyloid precursor protein copper binding domain. J Biol Chem. 2003;278:17401–17407. doi: 10.1074/jbc.M300629200. [DOI] [PubMed] [Google Scholar]
- Barnham KJ, Haeffner F, Ciccotosto GD, Curtain CC, Tew D, Mavros C, Beyreuther K, Carrington D, Masters CL, Cherny RA, Cappai R, Bush AI. Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer’s disease β-amyloid. FASEB J. 2004;18:1427–1429. doi: 10.1096/fj.04-1890fje. [DOI] [PubMed] [Google Scholar]
- Basumallick L, Sarangi R, DeBeerGeorge S, Elmore B, Hooper AB, Hedman B, Hodgson KO, Solomon EI. Spectroscopic and density functional studies of the red copper site in nitrosocyanin: role of the protein in determining active site geometric and electronic structure. J Am Chem Soc. 2005;127:3531–3544. doi: 10.1021/ja044412+. [DOI] [PubMed] [Google Scholar]
- Bayer TA, Schäfer S, Simons A, Kemmling A, Kamer T, Tepest R, Eckert A, Schüssel K, Eikenberg O, Sturchler-Pierrat C, Abramowski D, Staufenbiel M, Multhaup G.Dietary Cu stabilizes brain superoxide dismutase-1 activity and reduces amyloid Aβ production in APP23 transgenic mice Proc Natl Acad Sci USA 200310014187–14192. 10.1073/pnas.23328181002003PNAS..10014187B [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellingham S, Ciccotosto GD, Needham BE, Fodero LR, White AR, Masters CL, Cappai R, Camakaris J. Gene knockout of amyloid precursor protein and amyloid precursor-like protein-2 increases cellular copper levels in primary mouse cortical neurons and embryonic fibroblasts. J Neurochem. 2004;91:423–428. doi: 10.1111/j.1471-4159.2004.02731.x. [DOI] [PubMed] [Google Scholar]
- Borchardt T, Camakaris J, Cappai R, Masters CL, Beyreuther K, Multhaup G. Copper inhibits β-amyloid production and stimulates the non-amyloidogenic pathway of amyloid precursor protein secretion. Biochem J. 1999;344:461–467. doi: 10.1042/0264-6021:3440461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchardt T, Schmidt C, Camakaris J, Cappai R, Masters CL, Beyreuther K, Multhaup G. Differential effects of zinc on amyloid precursor protein (APP) processing in copper-resistant variant of cultured Chinese hamster ovary cells. Cell Mol Biol. 2000;46:785–795. [PubMed] [Google Scholar]
- Brouillet E, Trembleau A, Galanaud D, Volovitch M, Bouillot C, Valenza C, Prochiantz A, Allinquant B. The amyloid precursor protein interacts with Go heterotrimeric protein within a cell compartment specialised in signal transduction. J Neurosci. 1999;19:1717–1727. doi: 10.1523/JNEUROSCI.19-05-01717.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush AI. Metals and neuroscience. Curr Opin Chem Biol. 2000;4:184–191. doi: 10.1016/S1367-5931(99)00073-3. [DOI] [PubMed] [Google Scholar]
- Cao X, Südhof T. A transcriptively active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- Cappai R, Cheng F, Ciccotosto GD, Needham BE, Masters CL, Multhaup G, Fransson LA, Mani K. The amyloid precursor protein (APP) of Alzheimer disease and its paralog, APLP2, modulate the Cu/Zn-nitric oxide catalyzed degradation of glypican-1 heparin sulfate in vivo. J Biol Chem. 2005;280:13913–13920. doi: 10.1074/jbc.M409179200. [DOI] [PubMed] [Google Scholar]
- Casella L, Gullotti M. Dioxygen activation of biomimetic dinuclear complexes. In: Karlin KD, Teyklar Z, editors. Bioinorganic chemistry of copper. New York: Chapmann & Hall; 1993. pp. 292–305. [Google Scholar]
- Coulson EJ, Paliga K, Beyreuther K, Masters CL. What the evolution of the amyloid protein precursor supergene family tells us about its function. Neurochem Intl. 2000;36:175–184. doi: 10.1016/S0197-0186(99)00125-4. [DOI] [PubMed] [Google Scholar]
- Curtain CC, Ali F, Volitakis I, Cherny RA, Norton RS, Beyreuther K, Barrow CJ, Masters CL, Bush AI, Barnham KJ. Alzheimer’s disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J Biol Chem. 2001;276:20466–20473. doi: 10.1074/jbc.M100175200. [DOI] [PubMed] [Google Scholar]
- Strooper B, Annaert W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci. 2000;113:1857–1870. doi: 10.1242/jcs.113.11.1857. [DOI] [PubMed] [Google Scholar]
- Gralle M, Oliveira CLP, Guerreiro LH, McKinstry WJ, Cappai R, Parker MW, Ramos CHI, Torriani I, Ferreira ST. Solution conformation and heparin-induced dimerization of the full-length extracellular domain of the human amyloid precursor protein. J Mol Biol. 2006;357:493–508. doi: 10.1016/j.jmb.2005.12.053. [DOI] [PubMed] [Google Scholar]
- Gray HB, Malmström BG, Williams RJP. Copper coordination in blue proteins. J Biol Inorg Chem. 2000;5:551–559. doi: 10.1007/s007750000146. [DOI] [PubMed] [Google Scholar]
- Guenette S, Chang Y, Hiesberger T, Richardson JA, Eckman CB, Hammer RE, Herz J. Essential roles for the FE65 amyloid precursor protein-interacting proteins in brain development. EMBO J. 2006;25:420–431. doi: 10.1038/sj.emboj.7600926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Hung AY, Selkoe DJ. Processing of beta-amyloid precursor protein in microglia and astrocytes favors an internal localization over constitutive secretion. J Neurosci. 1991;11:3783–3793. doi: 10.1523/JNEUROSCI.11-12-03783.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Haass C, Steiner H. Alzheimer disease γ-secretase: a complex story of GxGD-type presenilin proteases. Trends Cell Biol. 2002;12:556–562. doi: 10.1016/S0962-8924(02)02394-2. [DOI] [PubMed] [Google Scholar]
- Hesse L, Beher D, Masters CL, Multhaup G. The βA4 amyloid precursor protein binding to copper. FEBS Lett. 1994;349:109–116. doi: 10.1016/0014-5793(94)00658-X. [DOI] [PubMed] [Google Scholar]
- Kang J, Lemaire H-G, Unterbeck A, Salbaum JM, Masters CL, Grzeschik K-H, Multhaup G, Beyreuther K, Müller U.The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor Nature 1987325733–736. 10.1038/325733a01987Natur.325..733K [DOI] [PubMed] [Google Scholar]
- Kong GK-W, Adams JJ, Harris HH, Boas JF, Curtain CC, Galatis D, Masters CL, Barnham KJ, McKinstry WJ, Cappai R, Parker MW. Structural studies of the Alzheimer’s amyloid precursor protein copper-binding domain reveals how it binds copper ions. J Mol Biol. 2007;367:148–161. doi: 10.1016/j.jmb.2006.12.041. [DOI] [PubMed] [Google Scholar]
- Kong GK-W, Adams JJ, Cappai R, Parker MW (2007b) Structure of Alzheimer’s disease amyloid precursor protein copper-binding domain at atomic resolution. Acta Crystallogr Sect F (in press) [DOI] [PMC free article] [PubMed]
- Maynard CJ, Cappai R, Volitakis I, Cherny R, White AR, Beyreuther K, Masters CL, Bush AI, Li Q-X. Overexpression of Alzheimer’s disease amyloid-β opposes the age-dependent elevations of brain copper and iron. J Biol Chem. 2002;277:44670–44676. doi: 10.1074/jbc.M204379200. [DOI] [PubMed] [Google Scholar]
- Messerschmidt A, Huber R, Poulos T, Wieghardt K. Handbook of Metalloproteins, vol 2. Chichester, England: Wiley; 2001. [Google Scholar]
- Multhaup G, Ruppert T, Schlicksupp A, Hesse L, Beher D, Masters CL, Beyreuther K. Reactive oxygen species and Alzheimer’s disease. Biochem Pharmacol. 1997;54:533–539. doi: 10.1016/S0006-2952(97)00062-2. [DOI] [PubMed] [Google Scholar]
- Multhaup G, Schlicksupp A, Hesse L, Beher D, Ruppert T, Masters CL, Beyreuther K.The amyloid precursor protein of Alzheimer’s disease in the reduction of copper(II) to copper(I) Science 19962711406–1409. 10.1126/science.271.5254.14061996Sci...271.1406M [DOI] [PubMed] [Google Scholar]
- Murphy MEP, Turley S, Adman ET. Structure of nitrite bound to copper-containing nitrite reductase from Alcaligenes faecalis. J Biol Chem. 1997;272:28455–28460. doi: 10.1074/jbc.272.45.28455. [DOI] [PubMed] [Google Scholar]
- Ohsawa I, Takamura C, Kohsaka S. The amino-terminal region of amyloid precursor protein is responsible for neurite outgrowth in rat neocortical explant culture. Biochem Biophys Res Commun. 1997;236:59–65. doi: 10.1006/bbrc.1997.6903. [DOI] [PubMed] [Google Scholar]
- Okamoto T, Takeda S, Murayama Y, Ogata E, Nishimoto I. Ligand-dependent G protein coupling function of amyloid transmembrane precursor. J Biol Chem. 1995;270:4205–4208. doi: 10.1074/jbc.270.9.4205. [DOI] [PubMed] [Google Scholar]
- Pearl F, Todd A, Sillitoe I, Dibley M, Redfern O, Lewis T, Bennett C, Marsden R, Grant A, Lee D, Akpor A, Maibaum M, Harrison A, Dallman T, Reeves G, Diboun I, Addou S, Lise, Johnston C, Sillero A, Thornton J, Orengo C. The CATH domain structure database and related resources Gene3D and DHS provide comprehensive domain family information for genome analysis. Nucl Acids Res. 2005;33:D247–D251. doi: 10.1093/nar/gki024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phinney AL, Drisaldi B, Schmidt SD, Lugowski S, Coronado V, Liang Y, Horne P, Yang J, Sekoulidis J, Coomaraswamy J, Chishti MA, Cox DW, Mathews PM, Nixon RA, Carlson GA, St George-Hyslop P, Westaway D.In vivo reduction of amyloid-β by a mutant copper transporter Proc Natl Acad Sci USA 200310014193–14198. 10.1073/pnas.23328511002003PNAS..10014193P [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigge ST, Eipper BA, Mains RE, Amzel LM.Dioxygen binds end-on to mononuclear copper in a precatalytic enzyme complex Science 2004304864–867. 10.1126/science.10945832004Sci...304..864P [DOI] [PubMed] [Google Scholar]
- Rossjohn J, Cappai R, Feil SC, Henry A, McKinstry WJ, Galatis D, Hesse L, Multhaup G, Beyreuther K, Masters CL, Parker MW. Crystal structure of the N-terminal growth factor-like domain of Alzheimer’s amyloid precursor protein. Nat Struct Biol. 1999;6:327–331. doi: 10.1038/7562. [DOI] [PubMed] [Google Scholar]
- Scheuermann S, Hambsch B, Hesse L, Stumm J, Schmidt C, Beher D, Bayer TA, Beyreuther K, Multhaup G. Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer’s disease. J Biol Chem. 2001;276:33923–33929. doi: 10.1074/jbc.M105410200. [DOI] [PubMed] [Google Scholar]
- Schubert W, Prior R, Weidemann A, Dircksen H, Multhaup G, Masters CL, Beyreuther K. Localization of Alzheimer beta A4 amyloid precursor protein at central and peripheral synaptic sites. Brain Res. 1991;563:184–194. doi: 10.1016/0006-8993(91)91532-6. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ.Alzheimer’s disease is a synaptic failure Science 2002298789–791. 10.1126/science.10740692002Sci...298..789S [DOI] [PubMed] [Google Scholar]
- Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Kelm P, Knops J, Lieberberg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, Seubert P, Suomensaari SM, Wang S, Walker D, Zhao J, McConlogue L, John V.Purification and cloning of amyloid precursor protein β-secretase from human brain Nature 1999402537–540. 10.1038/9901141999Natur.402..537S [DOI] [PubMed] [Google Scholar]
- Smith DG, Cappai R, Barnham KJ. The redox chemistry of the Alzheimer’s disease amyloid beta peptide. Biochim Biophys Acta. 2007;1768:1976–1990. doi: 10.1016/j.bbamem.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Soba P, Eggert S, Wagner K, Zentgraf H, Siehl K, Kreger S, Löwer A, Langer A, Merdes G, Paro R, Masters CL, Müller U, Kins S, Beyreuther K. Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 2005;24:3624–3634. doi: 10.1038/sj.emboj.7600824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondag CM, Combs CK. Amyloid precursor protein mediates proinflammatory activation of monocytic lineage cells. J Biol Chem. 2004;279:14458–14463. doi: 10.1074/jbc.M313747200. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Nakamura S, Ueda K, Kameyama M, Shiojiri A, Takahashi Y, Kitaguchi N, Ito H. Three types of amyloid protein precursor mRNA in human brain: their differential expression in Alzheimer’s disease. Biochem Biophys Res Commun. 1988;157:472–479. doi: 10.1016/S0006-291X(88)80273-0. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, McClatchey AI, Lamperti ED, Villa-Komaroff L, Gusella JF, Neve RL.Protease inhibitor domain encoded by an amyloid protein precursor mRNA associated with Alzheimer’s disease Nature 1988331528–530. 10.1038/331528a01988Natur.331..528T [DOI] [PubMed] [Google Scholar]
- Treiber C, Simons A, Strauss M, Hafner M, Cappai R, Bayer TA, Multhaup G. Clioquinol mediates copper uptake and counteracts Cu efflux activities of the amyloid precursor protein of Alzheimer’s disease. J Biol Chem. 2004;279:51958–51964. doi: 10.1074/jbc.M407410200. [DOI] [PubMed] [Google Scholar]
- Vasker IA, Aflalo C. Hydrophobic docking: a proposed enhancement to molecular recognition techniques. Proteins. 1994;20:320–329. doi: 10.1002/prot.340200405. [DOI] [PubMed] [Google Scholar]
- Vasker IA. Long-distance potentials: an approach to the mulitple-minima problem in ligand-receptor interaction. Protein Eng. 1996;9:37–41. doi: 10.1093/protein/9.1.37. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ha Y. The X-ray structure of an antiparallel dimer of the human amyloid precursr protein E2 domain. Mol Cell. 2004;15:343–353. doi: 10.1016/j.molcel.2004.06.037. [DOI] [PubMed] [Google Scholar]
- White AR, Multhaup G, Maher F, Bellingham S, Camakaris J, Zheng H, Bush AI, Beyreuther K, Masters CL, Cappai R. The Alzheimer’s disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. J Neurosci. 1999;19:9170–9179. doi: 10.1523/JNEUROSCI.19-21-09170.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AR, Reyes AE, Mercer JFB, Camakaris J, Zheng H, Bush AI, Multhaup G, Beyreuther K, Masters CL, Cappai R. Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knockout mice. Brain Res. 1999;842:439–444. doi: 10.1016/S0006-8993(99)01861-2. [DOI] [PubMed] [Google Scholar]
- Weidemann A, Konig G, Bunke D, Fischer P, Salbaum JM, Masters CL, Beyreuther K. Identification, biogenesis, and localization of precursors of Alzheimer’s disease A4 amyloid protein. Cell. 1989;57:115–126. doi: 10.1016/0092-8674(89)90177-3. [DOI] [PubMed] [Google Scholar]
- Yan R, Blenkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashler JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME.Membrane-anchored aspartyl protease with Alzheimer’s disease β-secretase activity Nature 1999402533–536. 10.1038/9901071999Natur.402..533Y [DOI] [PubMed] [Google Scholar]