

Abstract
Ten types of post-translational modifications (PTMs) known to be critical to diverse cellular functions have been described in core histone proteins. However, it remains unclear whether additional PTMs exist in histones, and if so, what roles these undiscovered signals play in epigenetic phenomena. Here, we report a systematic analysis of yeast histone PTMs by mass spectrometry in combination with protein sequence alignment using PTMap, a computer program we recently developed. We have identified, for the first time, multiple sites of lysine propionylation and butyrylation in yeast histones H2B, H3, and H4. We confirmed these modifications by Western blotting using modification-specific antibodies, MS/MS of synthetic peptides, and coelution of synthetic and in vivo-derived peptides from an HPLC column. The presence of multiple modification sites in several yeast histones suggests that these two PTMs are histone marks that are evolutionarily conserved among eukaryotes. In addition, we identified 14 novel mass shifts that do not match any known PTM, suggesting the presence of previously undescribed histone modifications. The chemical natures of these modifications remain to be determined. Our studies therefore expand current knowledge of the “histone code”.
Keywords: protein post-translational modifications, PTMap, histones, lysine propionylation, lysine butyrylation
Introduction
In eukaryotic cells, chromosomal DNA is packaged into a compact structure, chromatin, with the help of four core histones (H2A, H2B, H3, and H4). The fundamental repeating unit of chromatin is the nucleosome, which contains an octamer of core histones, around which ~147 base pairs of DNA are wrapped. Nucleosomes are in turn arranged into progressively higher-order structures. Dynamic chromatin remodeling plays a critical role in regulating diverse DNA-based biological processes, such as transcription of RNA, DNA replication, and DNA repair, as well as chromosome condensation and segregation.1-3 Dysregulation of these processes has been intimately linked with the development of diseases such as cancer.4,5
The core histone proteins are small and highly basic.6 They are predominantly globular except for their N-terminal “tails”, which are unstructured and protrude from the surface of the chromatin. Amino acid sequence analysis shows that histones are highly conserved in eukaryotes from yeast to human, implying that most amino acid residues, if not all, are likely to be important for structure or function. Accordingly, because post-translational modifications (PTMs) alter the properties of the substrate amino acid residue, PTMs affect histone structure and therefore function.
PTMs in histones are generally considered to be a major group of epigenetic marks. Ten types of histone PTMs have been described in the past 4 decades, including phosphorylation, acetylation, methylation, ADP-ribosylation, ubiquitinylation, sumoylation, proline isomerization, citrullination, and butyrylation and propionylation of lysine side chains (the last two being abbreviated KButy and KProp, respectively).1,2,7 Most of these modifications have been carefully analyzed by mass spectrometry,8-15 with KProp and KButy having been recently identified and verified by our group.7 Many histone PTMs contribute to the “histone code”, part of an epigenetic program that dictates diverse DNA-templated biological outputs such as gene expression, DNA replication, and DNA repair.1,2,16,17 While much is known about the currently identified histone PTMs and their functions,1,2,16 it remains unclear whether additional histone PTMs exist in cells, and if so, what roles these undiscovered signals play in epigenetic phenomena. This knowledge gap must be filled to construct a complete atlas of the epigenome, to improve our understanding of chromatin structure and function, and to elucidate the mechanisms by which epigenetic programs in biological processes are regulated.
To test whether lysine propionylation and lysine butyrylation exist in yeast core histones and to identify any novel PTMs present, we performed a systematic analysis of PTMs in yeast histones by mass spectrometry in combination with protein sequence alignment using the software PTMap. The PTMap algorithm can identify all PTMs, whether or not they have been previously described. Our results suggest that the “histone code” is highly complex.
Materials and Methods
Materials
Water and acetonitrile were from Fisher Scientific (Pittsburgh, PA). Trifluoroacetic acid (TFA) was from Sigma-Aldrich (St. Louis, MO). Colloidal Blue Staining Kit was from Invitrogen (Carlsbad, CA). Sequencing-grade trypsin was from Promega (Madison, WI). C18 ZipTips were from Millipore (Bedford, MA).
Preparation of Core Histones
Saccharomyces cerevisiae BY4741 strain was grown to midlog phase in YPDA media, and then was incubated in the presence or absence of inhibitors of histone deacetylases (HDACs) (3 μM trichostatin A (TSA) and 60 mM sodium butyrate (NaB)). After 6 h of continued growth, the cells were harvested by centrifugation at 5000g for 5 min and washed once with distilled water. The cells were washed once with S buffer (1.1 M sorbitol, 50 mM potassium phosphate, pH 6.5, 0.02% β-mercaptoethanol) and resuspended in S buffer. Lyticase (Sigma-Aldrich, St. Louis, MO) at a final concentration of 300 units/mL in S buffer was then added to the cells. Spheroplasting was carried out at 30 °C with gentle shaking. The spheroplasts were harvested by centrifugation at 5000g for 5 min. The pellet was resuspended at a ratio of 3 mL/g of cells in a solution of 18% Ficoll (Sigma-Aldrich, St Louis, MO), 20 mM potassium phosphate, pH 6.8, 1 mM MgCl2, and 0.5 mM EDTA, containing protease inhibitors and HDAC inhibitors. The cells were homogenized with a loose fitting Teflon homogenizer (Kontes Glass Co., Vineland, NJ). Release of nuclei was monitored with a microscope. Cells were removed by centrifugation for 15 min at 2000g. Centrifugation for 30 min at 50 000g pelleted the nuclei, which were then washed with buffer A (10 mM Tris-HCl, pH 8.0, 0.5% NP-40, and 75 mM NaCl) and buffer B (10 mM Tris-HCl, pH 8.0, and 0.4 M NaCl), both of which contained protease inhibitors and HDAC inhibitors. The core histones were extracted with 0.4 N H2SO4 on ice overnight. The extract was centrifuged at 4 °C for 10 min at 16 000g. Histone proteins were precipitated by conventional trichloroacetic acid precipitation. Samples were then washed twice with acetone containing 0.1% HCl (v/v) and twice with pure acetone. The pellet was allowed to dry in the air and resolubilized in water. For immunoblotting, histones were separated on a 15% SDS-PAGE gel, transferred to a 0.2-μM PVDF membrane, and probed with an anti-KProp or -KButy antibody.
Protein In-Gel Digestion
The core histones were resolved in a 15% SDS-PAGE gel and visualized by colloidal Coomassie staining. Gel bands of interest were excised and subjected to in-gel digestion as described previously.7 Briefly, the gel band was sliced into small pieces (~1 mm) and destained with 25 mM ammonium bicarbonate in ethanol/water (50:50, v/v). The destained gel pieces were washed in an acidic buffer (acetic acid/ethanol/water, 10:50:40, v/v/v) three times for 1 h each time, and in water two times for 20 min each time. The gel pieces were dehydrated in acetonitrile and dried in a SpeedVac (ThermoFisher, Waltham, MA). Two hundred nanograms of porcine modified trypsin (Promega, Madison, WI) in 50 mM ammonium bicarbonate was added to the dried gels and incubated overnight at 37 °C. Tryptic peptides were sequentially extracted from the gel pieces with 50% acetonitrile (acetonitrile/water/TFA, 50:45:5, v/v/v) and 75% acetonitrile (acetonitrile/water/TFA, 75:24:1, v/v/v). The peptide extracts were pooled, dried in a SpeedVac, and desalted using a μ-C18 Ziptip before HPLC/MS/MS analysis.
HPLC/MS/MS Analysis
HPLC/MS/MS analysis was carried out by nano-HPLC/LTQ mass spectrometry as described previously.7 Briefly, each tryptic digest was dissolved in 10 μL of HPLC buffer A (0.1% (v/v) formic acid in water), and 2 μL was injected into an Agilent HPLC system (Agilent, Palo Alto, CA). Peptides were separated on a homemade capillary HPLC column (50-mm length × 75-μm inner diameter) containing Jupiter C12 resin (4-μm particle size, 90-Å pore diameter, Phenomenex, St. Torrance, CA) and electrosprayed directly into the mass spectrometer using a nanospray source. The LTQ mass spectrometer was operated in a data-dependent mode, cycling between acquiring one MS spectrum followed by MS/MS spectra of the 10 strongest ions in that MS spectrum.
Protein Sequence Database Search and Manual Verification
All MS/MS spectra were searched against the NCBI-nr protein sequence database using the PTMap software. A PTMap score of 1.0 and SUnmatched scores of 4.0:10.0 (high/low mass ranges) were used to filter false positives.18 Parameters for the PTMap database search included a mass error of ±4.0 Da for parent ions, a mass error of ±0.6 Da for fragment ions, and six allowed missed proteolytic cleavages. All modified peptides identified with PTMap score >1 were manually examined using the rules described previously.19
MS/MS of Synthetic Peptides
Peptides bearing PTMs of interest were synthesized by GL Biochem (Shanghai, P.R. China). MS/MS of synthetic peptides was carried out using the same LTQ settings as those used for the analysis of in vivo peptides.
Results
Detection of Propionyllysine and Butyryllysine in Yeast Histones H3 and H4 by Western Blotting
Lysine-propionylated and -butyrylated peptides were initially identified in histone H4 from HeLa cells.7 However, it remained unknown whether these two modifications exist in yeast cells and whether they represent evolutionarily conserved histone histone marks. Toward this goal, we performed a systematic analysis of yeast histone PTMs, including propionylation, butyrylation and other novel modifications.
To test whether lysine propionylation and butyrylation exist in yeast, we generated pan propionyllysine- and butyryllysine-specific polyclonal antibodies and used them for Western blotting analysis. These antibodies are specific to propionyllysine and butyryllysine.20 Propionyllysine and butyryllysine were detected in both histone H3 and histone H4 (Figure 1).
Figure 1.
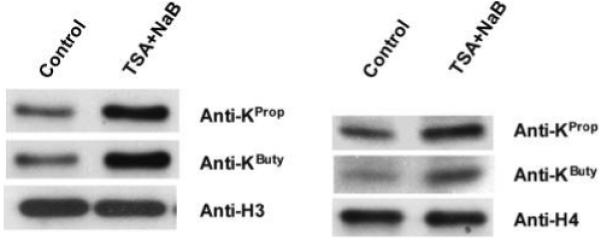
Western blotting analysis of lysine propionylation and butyrylation in yeast histones using anti-KProp and anti-KButy antibodies. Histones were extracted from yeast cells either treated with HDAC inhibitors or left untreated. The preparations were subjected to SDS-PAGE followed by Western blotting using antibodies as indicated. Both propionylation and butyrylation in histones were significantly enhanced in histone H3 (left panel) and histone H4 (right panel) with HDAC inhibitors trichostatin A (TSA) and sodium butyrate (NaB).
Given that histone acetyltransferases can catalyze acetylation, propionylation, and butyrylation of lysine in vitro,7 we hypothesized that HDACs, which remove lysine acetylation, also remove lysine propionylation and lysine butyrylation. To test the effect of HDACs on propionylation and butyrylation in yeast, yeast cells were cultured in buffer with or without 3 μm TSA and 60 mM sodium butyrate, which are inhibitors of HDACs. The core histone proteins were extracted and then analyzed by Western blotting and LC/MS/MS. Both propionylation and butyrylation were significantly enhanced in histones H3 and H4 in the presence of HDAC inhibitors (Figure 1). As we described previously, the two antibodes were specific to the respective modifications (Please see the Supplemental Data in ref 20). Our results therefore confirmed the existence of lysine propionylation and butyrylation in yeast, and that HDAC activity contributes to the regulation of propionyllysine and butyryllysine status in histones.
Identification of Propionyllysine and Butyryllysine Sites in Yeast Histones by Mass Spectrometry and PTMap
To determine the sites of lysine propionylation and butyrylation in yeast histones, we prepared yeast core histones using a procedure previously described.21 To enhance lysine propionylation and lysine butyrylation, the yeast cells were treated with HDAC inhibitors before core histones were prepared. The resulting core histones were resolved by SDS-PAGE and excised from the gel for in-gel digestion and nano-HPLC/MS/MS analysis.
We carried out unrestricted sequence alignment using the PTMap algorithm to analyze MS/MS data sets from the yeast histones.18 PTMap, recently developed in our laboratory, is the first sequence alignment algorithm that emphasizes unmatched peaks for identifying false positives. It incorporates several unique features to improve sensitivity and accuracy of peptide identification, including a unique procedure for peak selection, automatic mass-shift adjustment, and precise PTM localization. The algorithm is able to carry out blind sequence alignment.18
Sequence alignment and subsequent manual inspection using a procedure described previously19 led to identification of 44 sites modified with 26 types of PTM in three histone proteins, after exclusion of frequent in vitro chemical modifications (e.g., oxidation and acrolein addition) (Tables 1 and 2). Among the 26 types of PTM, 14 could not be annotated to known PTMs (http://www.unimod.org) (Table 2), therefore, likely representing novel PTMs. All MS/MS spectra for the modified peptides can be found in Supporting Information S3, S4, and S5.
Table 1.
The Modified Peptides Identified from in Vivo Yeast Core Histones
| Histone | GI Number | Position(s) | Residue | Modification | Sequence and mass shift(s) |
|---|---|---|---|---|---|
| H3 | 48428921 | 14 | K | Acetylation | R.KSTGGK(+42)APR.K |
| H3 | 48428921 | 14 | K | Butyrylation | K.STGGK(+70)APR.K |
| H3 | 48428921 | 23 | K | - | R.KQLASK(−19)AAR.K |
| H3 | 48428921 | 23 | K | Propionylation | R.KQLASK(+56)AAR.K |
| H3 | 48428921 | 27 | K | Acetylation | R.K(+42)SAPSTGGVK.K |
| H3 | 48428921 | 36 | K | di-Methylation | K.SAPSTGGVK(+28)KPHR.Y |
| H3 | 48428921 | 36 | K | Methylation | K.SAPSTGGVK(+14)KPHR.Y |
| H3 | 48428921 | 56 | K | Acetylation | R.FQK(+42)STELLIR.K |
| H3 | 48428921 | 56 | K | Propionylation | R.FQK(+56)STELLIR.K |
| H3 | 48428921 | 57 | S | - | K.S(+83)TELLIR.K |
| H3 | 48428921 | 57 | S | Thiophosphorylation | K.S(+96)TELLIR.K |
| H3 | 48428921 | 68 | Q | Deamidation | R.KLPFQ(+l)R.L |
| H3 | 48428921 | 79 | K | Methylation | R.EIAQDFK(+14)TDLR.F |
| H3 | 48428921 | 79 | K | - | R.EIAQDFK(−19)TDLR.F |
| H3 | 48428921 | 79 | K | Methylation | R.EIAQDFK(+14).T |
| H3 | 48428921 | 79 | K | Acetylation | R.EIAQDFK(+42)TDLR.F |
| H3 | 48428921 | 79 | K | - | R.EIAQDFK(+98)TDLR.F |
| H3 | 48428921 | 19, 23 | Q | Deamidation | R.KQ(+1)LASK(+42)AAR.K |
| H3 | 48428921 | 27, 36 | K | Methylation | R.K(+42)SAPSTGGVK(+14).K |
| H3 | 48428921 | 27, 37 | K | Methylation | R.K(+42)SAPSTGGVKK(+14)PHR.Y |
| H3 | 48428921 | 27, 38 | K | di-Methylation | R.K(+42)SAPSTGGVK(+28)KPHR.Y |
| H3 | 48428921 | 27, 36 | K | Butyrylation | R.K(+70)SAPSTGGVK(+23)KPHR.Y |
| H3 | 48428921 | 36, 37 | K | Methylation | K.SAPSTGGVK(+14)K(+14)PHR.Y |
| H3 | 48428921 | 56, 59 | F | Ethylation | R.FQK(+42)STE(+28)LLIR.K |
| H3 | 48428921 | 73, 79 | E | Ethylation | R.E(+28)IAQDFK(+42)TDLR.F |
| H3 | 48428921 | 73, 79 | E | R.E(+42)IAQDFK(+42)TDLR.F | |
| H4 | 122107 | 8 | K | Butyrylation | K.GGK(+70)GLGK.G |
| H4 | 122107 | 25 | D | R.D(+42)NIQGITKPAIR.R | |
| H4 | 122107 | 27 | Q | Deamidation | R.DNIQ(+1)GITKPAIR.R |
| H4 | 122107 | 31 | K | - | R.DNIQGITK(−19)PAIR.R |
| H4 | 122107 | 35 | R | - | R.DNIQGITKPAIR(+74).R |
| H4 | 122107 | 35 | N | - | R.DN(−2)IQGTKPAIR.R |
| H4 | 122107 | 45 | R | - | K.R(+53)ISGLIYEEVR.A |
| H4 | 122107 | 47 | S | - | R.IS(+12)GLIYEEVR.A |
| H4 | 122107 | 47 | S | R.IS(+26)GLIYEEVR.A | |
| H4 | 122107 | 51 | E | - | R.ISGLIYE(+74)EVRA |
| H4 | 122107 | 54 | R | Methylation | R.ISGLIYEEVR(+14).A |
| H4 | 122107 | 58 | K | - | R.AVLK(−19)SFLESVIR.D |
| H4 | 122107 | 59 | S | - | K.S(+81)FLESVIR.D |
| H4 | 122107 | 53 | S | Piperidination | K.S(+68)FLESVIR.D |
| H4 | 122107 | 63 | E | Ethylation | R.DSVTYTE(+28)HAK.R |
| H4 | 122107 | 68 | S | 2-amino-3-oxo-butanoic acid | R.DS(−2)VTYTEHAK.R |
| H4 | 122107 | 76 | K | - | R.DSVTYTEHAKRK(−19).T |
| H4 | 122107 | 90 | K | - | R.KTVTSLDVVYAL(−19)R.Q |
| H4 | 122107 | 5,8, 12,16 | K | Acetylation | R.GK(+42)GGK(+42)GLGK(+42)GGAK(+42)R.H |
| H4 | 122107 | 8, 12,16 | K | Acetylation | K.GGK(+42)GLGK(+42)GGAK(+42)R.H |
| H2B | 403314 | 51 | S | K.S(+198)MSILNSFVNDIFER.I | |
| H2B | 403314 | 21,22 | K | Acetylation | K.KPAAK(+42)K(+42)TSTSVDGK.K |
| H2B | 403314 | 51 | S | K.S(+174)MSILNSFVNDIFER.I | |
| H2B | 403314 | 70 | N | Deamidation | K.SMSILNSFVN(+1)DIFER.I |
| H2B | 403314 | 37 | K | di-Methylation | R.K(+23)ETYSSYIYK.V |
| H2B | 403314 | 16, 21 | K | Acetylation | K.APAEK(+42)KPAAK(+42)K.T |
| H2B | 403314 | 38 | E | Ethylation | R.KE(+28)TYSSYIYK.V |
| H2B | 403314 | 74 | E | Ethylation | K.SMSILNSFVNDIFE(+28)R.I |
| H2B | 403314 | 45 | K | K.ETYSSYIYK(−19)VLK.Q | |
| H2B | 403314 | 111 | K | di-Methylation | R.LILPGELAK(+28)HAVSEGTR.A |
| H2B | 403314 | 21 | K | Butyrylation, Methylation | K.APAEK(+42)KPAAK(+70)K(+14)TSTSVDGK.K |
Table 2.
Known and Previously Undescribed Mass Shifts Identified in Yeast Core Histones
| residue | histone H3 | histone H4 | histone H2B |
|---|---|---|---|
| Known Mass Shifts | |||
| Lys | +14, +28, +42, 56, +70 |
+14, +42, +70 | +14, +28, +42, +70 |
| Arg | +14 | ||
| Undescribed Mass Shifts | |||
| Lys | +98, −19 | −19 | −19 |
| Arg | +53, +74 | ||
| Asn | −2 | +1a | |
| Ser | +83, +96a | +12, +26, +68a, +81, −2a |
+174, +198 |
| Glu | +28a, +42 | +28a, +74 | +28a |
| Asp | +42 | ||
| Gln | +1a | +1a | |
This mass shift may represent a previously documented PTM (http://www.unimod.org).
PTMs Identified in Yeast Histones
We identified 30 sites in yeast histones bearing known PTMs, including lysine acetylation, lysine methylation (mono- and dimethylation), lysine propionylation, lysine butyrylation, and arginine methylation (Table 1). Lysine propionylation and lysine butyrylation are PTMs recently discovered by our laboratory in HeLa cells.7 Our initial work confirmed these two PTMs by observing identical MS/MS peak patterns with in vivo-derived and synthetic peptides, and by identifying enzymes that catalyzed the modification reaction in vitro. Interestingly, we found 2 lysine propionylation sites and 2 lysine butyrylation sites in histone H3, 1 lysine butyrylation site in histone H4, and 1 lysine butyrylation site in histone H2B (Figure 2). These results demonstrate the existence of these PTMs in a simple eukaryotic organism and suggest that the PTMs are evolutionarily conserved among eukaryotes.
Figure 2.
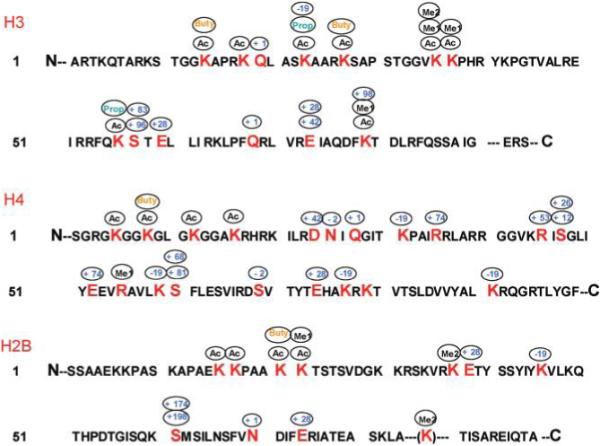
Identified modification sites within histones H3, H4 and H2B from yeast. The known modifications include Ac, acetyl; Me1, methyl; Me2, dimethyl; Buty, butyryl; Prop, propionyl. Unknown modifications are indicated by the sizes of the mass shifts, in daltons.
We also identified a series of mass shifts that may represent previously documented PTMs (http://www.unimod.org), including glutamine and asparagine deamidation (+1 Da), serine thiophosphorylation (+96 Da), serine piperidination (+68 Da), serine dehydrogenation (−2 Da), and addition of 28 Da to glutamic acid (Table 2, Figure 2). These results suggest the existence of a variety of novel PTMs in yeast histones.
In addition to the known PTMs, we identified 14 residues bearing 12 different mass shifts that could not be matched to the shifts of known PTMs. Since we identified both the modified and unmodified versions of these peptides, the mass shifts are unlikely to be caused by mutations in the gene, and therefore are probably novel PTMs.
Verification of Lysine Propionylation and Lysine Butyrylation Sites in Yeast Histones
To further confirm the lysine propionylation and lysine butyrylation sites in histones, we compared MS/MS spectra of the in vivo-derived peptides bearing lysine modifications and their synthetic counterparts. The MS/MS spectrum of a histone H3 peptide bearing a propionylation at residue K56 (FQKPropSTELLIR) almost completely overlapped with that of its synthetic peptide counterpart (Figure 3A,B). In addition, the MS/MS spectrum derived from a mixture of the histone H3 tryptic digest and synthetic FQKPropSTELLIR (Figure 3C) was almost same as the spectra from either the in vivo-derived peptide or the synthetic peptide. That the lysine propionylated peptide has been correctly identified is further evidenced by coelution from an HPLC column of the in vivo-derived peptide, the synthetic peptide, and a mixture of the two (Figure 3D–F). In addition, we also compared the MS/MS spectrum and HPLC retention time of the lysine propionylated peptide with the corresponding unmodified and acetylated peptide (Supporting Information S2). The identified lysine propionylated peptide could be paired with MS/MS spectra of their corresponding unmodified and acetylated peptides. In addition, the HPLC-retention times were determined to be 39.18, 48.59 and 50.37 min for the unmodified, lysine acetylated, and lysine propionylated, respectively, in a 100-min HPLC MS/MS run. The propionylation on the side chain of lysine cause the increase of the retention time significantly, due to an increase of hydrophobicity.
Figure 3.
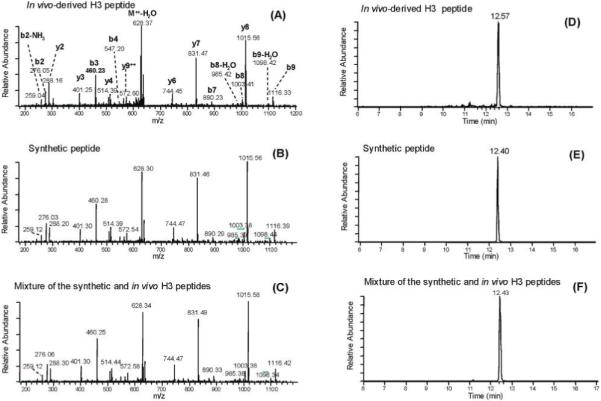
Identification and verification of a lysine-propionylated histone H3 peptide. (A) MS/MS of a tryptic peptide ion (FQKPropSTELLIR) from histone H3 isolated from yeast. (B) MS/MS of a synthetic peptide corresponding to the sequence identified in (A), showing a similar fragmentation pattern. (C) MS/MS of a mixture of the in vivo-derived histone H3 peptide identified in (A) and its synthetic counterpart as indicated in (B). (D) Selected reaction monitoring (SRM) chromatogram of the in vivo-derived histone H3 peptide. (E) SRM chromatogram of the synthetic peptide corresponding to the sequence identified in (A), showing a similar retention time. (F) SRM chromatogram of a mixture of the peptide from the in vivo-derived histone H3 digest and its synthetic counterpart, showing coelution of the two peptides from an HPLC column.
Likewise, we confirmed the lysine butyrylation at histone H3K14 (STGGKButyAPR) by performing MS/MS of its corresponding synthetic peptide, observing coelution in HPLC/MS/MS analysis (Figure 4). We also observed the differential HPLC-retention times of its corresponding lysine acetylation peptide (Supporting Information S2).
Figure 4.

Identification and verification of lysine-butyrylated histone H3 peptides. (A) MS/MS of a lysine butyrylated peptide (STGGKButyAPR) from histone H3 isolated from yeast. (B) MS/MS of a synthetic peptide corresponding to the sequence identified in (A), showing a similar fragmentation pattern. (C) MS/MS of a mixture of the in vivo-derived histone H3 peptide identified in (A) and its synthetic counterpart as indicated in (B). (D) SRM chromatogram of the in vivo-derived histone H3 peptide. (E) SRM chromatogram of the synthetic peptide corresponding to the sequence identified in (A), showing a similar retention time. (F) SRM chromatogram of a mixture of the peptide from the in vivo-derived histone H3 tryptic digest and synthetic counterpart, demonstrating coelution of the two peptides from an HPLC column.
Novel PTMs in Yeast Histones
PTMap identified a series of novel mass shifts in yeast histones that have not been described previously (http://www.unimod.org), at lysine, arginine, asparagine, aspartic acid, glutamic acid, and serine (Tables 1 and 2, Figure 2). We manually inspected the MS/MS spectra of each putatively modified peptide and its unmodified counterpart to ensure the quality of analysis. The MS/MS spectra of all the modified peptides are provided in Supporting Information S2, S3, S4, and S5. To learn more about these novel mass shifts in yeast histones, we compared the spectral counts and retention times for each pair of modified and unmodified peptides (Supporting Information S1). The unmodified peptides were usually much more abundant than their corresponding modified counterparts, suggesting that the novel mass shifts are caused by protein modification instead of gene mutations. In addition, each PTM causes an apparent shift in HPLC retention time, which excludes the possibility that the precursor ions of the modified peptide came from in-source fragmentation of the corresponding unmodified peptide.
Discussion
Our systematic studies of yeast core histones demonstrated that lysine propionylation and lysine butyrylation are present in yeast proteins, suggesting the evolutionary conservation of these modifications in eukaryotic cells. These two modifications in yeast histones were further confirmed by Western blotting using pan anti-KProp and anti-KButy antibodies, MS/MS of synthetic peptides, and coelution in HPLC/MS/MS analysis. Induction of lysine propionylation and lysine butyrylation in yeast core histones, demonstrated by Western blot analysis, suggest that some enzymes that remove lysine propionylation and lysine butyrylation are likely to be shared with lysine deacetylation enzymes. This study, in combination with our initial report on propionylation and butyrylation,7 conclusively establishes that KProp and KButy are present in eukaryotic cells.
In addition to KProp and KButy, we identified 14 novel mass shifts which, because they do not match any known PTMs, are likely to represent novel protein modifications. The limited mass accuracy of our low-resolution mass spectrometer makes it impossible to determine elemental compositions for the modifications. Future studies using high-resolution mass spectrometers will aid structural elucidation of these novel PTMs in histones.
KProp and KButy have lower stoichometry that KAc. For example, MS signal intensities of H3K56 Ac is 85 times higher than H3K56Prop. It is unclear at this time if this difference is caused by differential enzymatic activities of transferases or demodification enzymes, or the concentration differences among the cofactors (acetyl CoA, propionyl CoA, or butyryl CoA). In Western blotting analysis, weak signals were detected for both H2A and H2B from S. cerevisiae. However, neither KProp or KButy was identified in H2A. We believe that this is largely caused by lower stoichometry of the two modifications in H2A as Western blot is usually more sensitive than mass spectrometric analysis. It is also unclear what are the functional differences among lysine acetylation, lysine propionylation, and lysine butyrylaiton. Each one of these three modified lysines may create a docking site for different bromo-domain proteins. Alternatively, status of lysine propionylation and lysine butyrylation might be regulated by the concentration of substrate CoA compounds, which can be modulated by physiological conditions and nutrition sources (e.g., glucose vs lipids). Demonstration of KProp and KButy in S. cerevisiae will allow future use of this model organism for genetics and biochemistry studies of these two PTM pathways.
Supplementary Material
Acknowledgment
This work was supported by The Robert A. Welch Foundation (I-1550 to Y.Z.), NIH (CA107943 to Y.Z.), and NSFC (20845004 to K.Z.).
Footnotes
Supporting Information Available: Supplemental Figure S1, spectral counts and retention time of the unmodified and modified peptides with novel mass shifts. Supporting Information S2, MS/MS spectra of the peptides containing KProp and KButy. Supporting Information S3, MS/MS spectra of the modified peptides from yeast histone H3. Supporting Information S4, MS/MS spectra of the modified peptides from yeast histone H4. Supporting Information S5, MS/MS spectra of the modified peptides from yeast histone H2B. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- (2).Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143):407–12. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- (3).Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- (4).Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447(7143):433–40. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- (5).Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Garcia BA, Mollah S, Ueberheide BM, Busby SA, Muratore TL, Shabanowitz J, Hunt DF. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2007;2(4):933–8. doi: 10.1038/nprot.2007.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, Falck JR, Peng J, Gu W, Zhao Y. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell. Proteomics. 2007;6(5):812–9. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Garcia BA, Pesavento JJ, Mizzen CA, Kelleher NL. Pervasive combinatorial modification of histone H3 in human cells. Nat. Methods. 2007;4(6):487–9. doi: 10.1038/nmeth1052. [DOI] [PubMed] [Google Scholar]
- (9).Taverna SD, Ueberheide BM, Liu Y, Tackett AJ, Diaz RL, Shabanowitz J, Chait BT, Hunt DF, Allis CD. Long-distance combinatorial linkage between methylation and acetylation on histone H3 N termini. Proc. Natl. Acad. Sci. U.S.A. 2007;104(7):2086–91. doi: 10.1073/pnas.0610993104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Jiang L, Smith JN, Anderson SL, Ma P, Mizzen CA, Kelleher NL. Global assessment of combinatorial post-translational modification of core histones in yeast using contemporary mass spectrometry. LYS4 trimethylation correlates with degree of acetylation on the same H3 tail. J. Biol. Chem. 2007;282(38):27923–34. doi: 10.1074/jbc.M704194200. [DOI] [PubMed] [Google Scholar]
- (11).Chu F, Nusinow DA, Chalkley RJ, Plath K, Panning B, Burlingame AL. Mapping post-translational modifications of the histone variant MacroH2A1 using tandem mass spectrometry. Mol. Cell. Proteomics. 2006;5(1):194–203. doi: 10.1074/mcp.M500285-MCP200. [DOI] [PubMed] [Google Scholar]
- (12).Wisniewski JR, Zougman A, Kruger S, Mann M. Mass spectrometric mapping of linker histone H1 variants reveals multiple acetylations, methylations, and phosphorylation as well as differences between cell culture and tissue. Mol. Cell. Proteomics. 2007;6(1):72–87. doi: 10.1074/mcp.M600255-MCP200. [DOI] [PubMed] [Google Scholar]
- (13).Beck HC, Nielsen EC, Matthiesen R, Jensen LH, Sehested M, Finn P, Grauslund M, Hansen AM, Jensen ON. Quantitative proteomic analysis of post-translational modifications of human histones. Mol. Cell. Proteomics. 2006;5(7):1314–25. doi: 10.1074/mcp.M600007-MCP200. [DOI] [PubMed] [Google Scholar]
- (14).Zhang L, Su X, Liu S, Knapp AR, Parthun MR, Marcucci G, Freitas MA. Histone H4 N-terminal acetylation in Kasumi-1 cells treated with depsipeptide determined by acetic acid-urea polyacrylamide gel electrophoresis, amino acid coded mass tagging, and mass spectrometry. J. Proteome Res. 2007;6(1):81–8. doi: 10.1021/pr060139u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Galasinski SC, Resing KA, Ahn NG. Protein mass analysis of histones. Methods. 2003;31(1):3–11. doi: 10.1016/s1046-2023(03)00082-3. [DOI] [PubMed] [Google Scholar]
- (16).Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell Biol. 2007;8(12):983–94. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hake SB, Xiao A, Allis CD. Linking the epigenetic ‘language’ of covalent histone modifications to cancer. Br. J. Cancer. 2007;96:R31–9. [PubMed] [Google Scholar]
- (18).Chen Y, Chen W, Cobb M, Zhao Y. PTMap, a novel algorithm for systematic identification of all the possible post-translational protein modifications in a protein. PNAS. 2008 in press. [Google Scholar]
- (19).Chen Y, Kwon SW, Kim SC, Zhao Y. Integrated approach for manual evaluation of peptides identified by searching protein sequence databases with tandem mass spectra. J. Proteome Res. 2005;4(3):998–1005. doi: 10.1021/pr049754t. [DOI] [PubMed] [Google Scholar]
- (20).Cheng Z, Tang Y, Chen Y, Kim S, Liu H, Li SS, Gu W, Zhao Y. Molecular characterization of propionyllysines in non-histone proteins. Mol. Cell. Proteomics. 2008 doi: 10.1074/mcp.M800224-MCP200. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Fischle W. In nucleo enzymatic assays for the identification and characterization of histone modifying activities. Methods. 2005;36(4):362–7. doi: 10.1016/j.ymeth.2005.03.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.