

Abstract
Hydrogen bonds stabilize and direct chemistry performed by metalloenzymes. With inspiration from enzymes, we will utilize an approach that incorporates intramolecular hydrogen bond donors to determine their effects on the stability and reactivity of metal complexes. Our premise is that control of secondary coordination sphere interactions will promote new function in synthetic metal complexes. Multidentate ligands have been developed that create rigid organic structures around metal ions. These ligands place hydrogen bond (H-bond) donors proximal to the metal centers, forming specific microenvironments. One distinguishing attribute of these systems is that site-specific modulations in structure can be readily accomplished, in order to evaluate correlations with reactivity. A focus of this research is consideration of dioxygen binding and activation by metal complexes, including developing structure–function relationships in metal-assisted oxidative catalysis.
Introduction
Transition metal-mediated transformations are widespread in chemistry and biology. One only has to give a cursory examination of the vast literature on palladium complexes in organic chemistry to grasp the importance of metal-containing species in the synthesis of new compounds.1 It is no different in biology, in which metal ions have essential functional roles in nearly one-third of all proteins. The diversity of function can be understood within the context of the basic principles of coordination chemistry, as described by Werner over 100 years ago.2 Within this framework, structure–function relationships are developed through the analysis of the primary and secondary coordination spheres. The primary sphere controls basic properties, such as electronic structure and Lewis acidity, whereas the secondary coordination sphere is responsible for, among other things, regulating accessibility and substrate selectivity. Ultimately, it is the integration of these two spheres that produces the functional aspects attributed to metal complexes.
The effects of the primary coordination sphere on metal ion reactivity have been extensively investigated, leading to predictable structure–function relationships. In contrast, the influences of the secondary coordination sphere are less known, and in fact, have often been ignored in the considerations of function. Renewed interest in this area has occurred because cumulative physical evidence suggests a regulatory role for the secondary coordination spheres within the active sites of metalloproteins.3 This has prompted the design of synthetic systems utilizing the secondary coordination sphere to induce new functionality from metal complexes. For instance, sterically bulky ligands have been developed to prevent unwanted intermolecular interactions.4 A prominent example of this effect is the steric tuning of salen ligands to produce metal complexes for the stereoselective oxidation of olefins.5
Hydrogen bonds (H-bonds) in the secondary coordination sphere have also been implicated in the control of metal-mediated processes. The first indication of their importance again arose from biology through the discovery of protein-derived H-bond networks within the active sites of metalloproteins. Their presence are proposed to influence a vast array of functions that span hydrolysis6 to O2 binding and activation—for example, H-bond networks have been linked to O–O bond cleavage and proton transfer in the monooxygenase cytochrome P450 (Fig. 1).7 However, the fundamental aspects of how H-bonds facilitate most of these processes are still largely unknown.
Fig. 1.
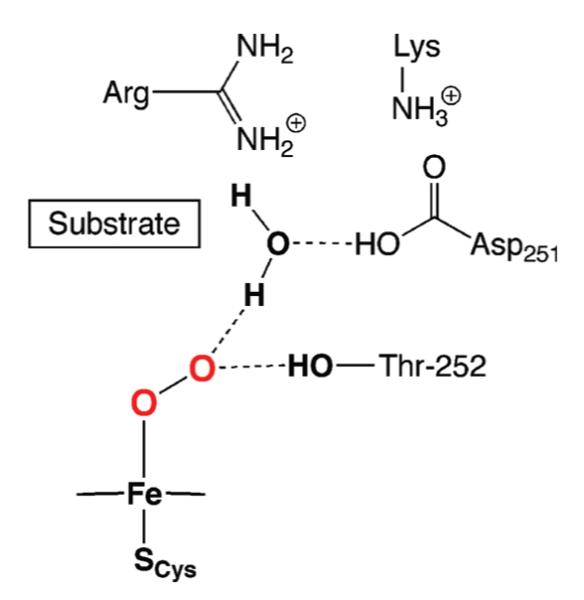
Proposed H-bonding network in cytochrome P450.
The utilization of H-bonds in the design and preparation of synthetic metal complexes is still in the early stages of development. Some notable advances have been reported, which are highlighted in two excellent reviews by Rivas.8 Our group has been investigating the effects of H-bonds on the binding and activation of small molecules by transition metal complexes. A review of our initial findings appeared in 2005 that included the design considerations for the complexes and the reactivity of iron and manganese complexes with dioxygen.9 This Feature Article focuses on results that have occurred since, with an emphasis on systems having varied H-bond networks. Our findings further demonstrate that placement of H-bonds within the secondary coordination sphere can have pronounced effects on the chemical reactivity of metal complexes.
Formation of metal oxo and hydroxo complexes with hydrogen bonds
Monomeric iron and manganese complexes with terminal oxo ligands were our initial targets. These complexes have been implicated in a variety of chemical and biochemical processes but there were few structurally characterized examples when we started this work 10 years ago.10 In fact, there were no examples of mononuclear iron complexes with a terminal oxo ligand prior to our work, presumably because of the strong thermodynamic driving force for the formation of FeIII–O–FeIII units.11 Our premise was that H-bonds involving the terminal oxo ligand would stabilize the M–O unit, allowing for the detection, characterization, and ultimately structural determination of these complexes. The motivation for using H-bonds to stabilize the M–O unit came from metalloproteins, whose active sites are replete with H-bond networks that are essential for oxidative function, as discussed previously.
We were successful in preparing iron and manganese complexes with terminal oxo or hydroxo ligands derived directly from O2 activation.12 This was accomplished with a urea-based tripodal compound, tris[(N’-tert-butylureayl)-N-ethylene]amine (H6buea) that was designed and developed to bind metal ions through the deprotonated α-nitrogen atoms, forming a rigid H-bond donating cavity proximal to the metal ion. As shown in Scheme 1, formation of these complexes was dependent on the amount of base initially added to deprotonate H6buea: 4 equivalents (equiv.) of base were necessary to isolate [MIIIH3buea(O)]2−, whereas 3 equiv. of base afforded [MIIIH3buea(OH)]−. Our studies suggest that the first three equiv. of base deprotonate each of the α-NH groups of the ligand—the fourth equiv. used to make the M(III)–O complexes deprotonates one α’-NH moiety (Fig. 2). [MIIIH3buea(O)]2− was converted to [MIIIH3buea(OH)]− in the presence of 1 equiv. of acid, such as phenols or water, and 1 equiv. of methyl iodide reacts with [FeIIIH3buea(O)]2− to form [FeIIIH3buea(OMe)]−. These results suggest that the coordinated oxo ligand is nucleophilic.
Scheme 1.
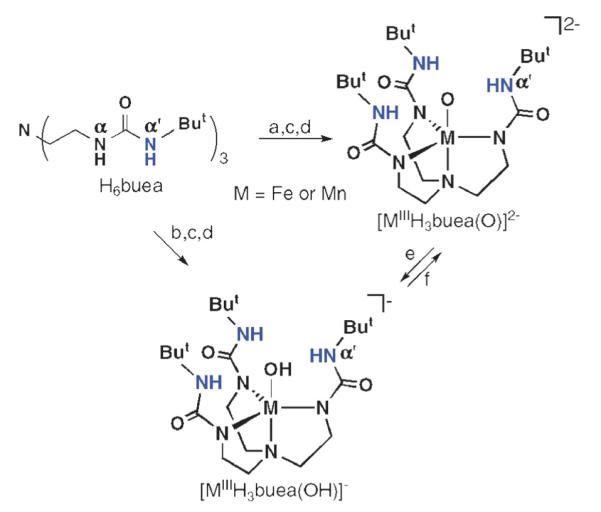
Preparative routes for the formation of the [MIIIH3buea(O)]2− and [MIIIH3buea(OH)]− complexes. Reagents and conditions: (a) 4 equiv. KH, DMA, inert atmosphere (Ar), rt; (b) 3 equiv. KH, DMA, Ar, rt; (c) M(OAc)2 (M = Fe, Mn), DMA, Ar, rt; (d) 0.5 equiv. O2, DMA, rt; (e) H+, DMA rt; (f) base, DMA, rt.
Fig. 2.

Proposed [MIIH3buea]2− intermediate formed during the activation of O2.
The [MIIIH3buea(O)]2− and [MIII H3buea(OH)]− complexes were thoroughly characterized, including with single-crystal X-ray diffraction methods. The complexes all have trigonal bipyramidal structures (Scheme 1) and intramolecular H-bonds to the MIII–O(H) units. Isotopic labeling experiments with 18O2 confirmed that dioxygen is the source of the oxo ligands in the M(III)–O complexes and oxygen atoms in the hydroxo ligands in the M(III)–OH complexes. Key structural and vibrational data are summarized in Table 1. To our knowledge, [MIIIH3buea(O)]2− are the first structural characterized examples of high-spin metal complexes with a terminal oxo ligand and at least four d electrons.
Table 1.
Structural and vibrational results for [MIIIH3buea(O)]2− and [MIIIH3buea(OH)]− complexes
| Complex | M–O/Å | M–N1/Å | α’-N⋯O/Å | ν(M–O)/cm−1 | ν(MO–H)/cm−1 |
|---|---|---|---|---|---|
| [FeIIIH3buea(O)]2− | 1.813(2) | 2.276(4) | 2.711(6) | 671 (645)a | na |
| [FeIIIH3buea(OH)]− | 1.926(1) | 2.180(2) | 2.833(2) | — | 3632 (3621)a |
| [MnIIIH3buea(O)]2− | 1.771(4) | 2.144(5) | 2.731(5) | 700 (672)a | na |
| [MnIIIH3buea(OH)] | 1.872(2) | 2.033(3) | 2.849(3) | — | 3614 (3603)a |
Values for 18O-isotopomer in parentheses.
One of the key intermediates in the formation of the [MIIIH3buea(O)]2− complexes is proposed to be a MII complex with an anionic site within the cavity to assist in scavenging protons (Fig. 2). This concept led us to prepare MII–OH complexes directly from water as shown in Fig. 3A, which were obtained in relatively high yields after recrystallization and have been characterized completely.13 Each complex has the expected trigonal bipyramidal coordination geometry, with the MII–OH unit placed within the cavities. The MnII–OH and FeII–OH complexes could be oxidized to their corresponding MnIII–OH and FeIII–OH complexes. In addition, [FeIIIH3buea(O)]2− and [MnIIIH3buea(O)]2− were synthesized directly from water when two anionic sites were placed within the cavity (Fig. 3B).14 These methodologies permitted the isolation of a unique series of monomeric iron and manganese complexes containing MII–OH, MIII–OH, and MIII–O units, with the same primary and secondary coordination spheres. This is the first instance of such a series of complexes and they have allowed us to investigate fundamental reactivity related to the M–oxo unit.15
Fig. 3.
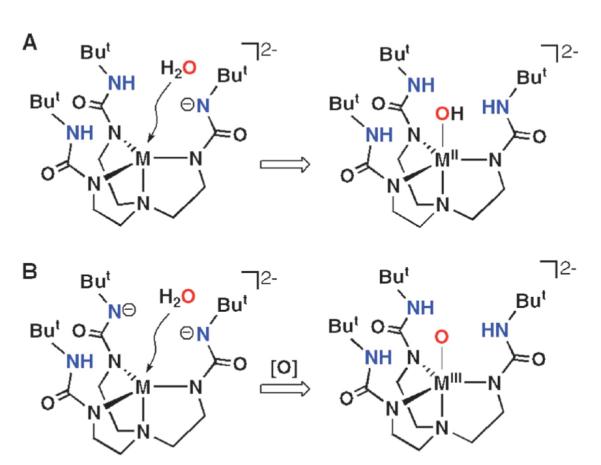
Proposed interactions of H2O with H-bonding cavities.9 Reprinted with permission from reference 9. Copyright 2005, American Chemical Society.
Formation of a MnIV–oxo complex
The discovery of the MIII–O complexes allowed us to probe the formation of higher valent analogs. We examined the oxidation chemistry of [MnIIIH3buea(O)]2− to produce the corresponding Mn(IV)–oxo complex.16 Treating the purple [MnIIIH3buea(O)]2− with [Cp2Fe]BF4 at −45 °C in DMF or in DMSO at room temperature (Scheme 2) produced a new green species (λmax = 635 nm) that is stable for several hours. X-Band EPR measurements at 4 K (Fig. 4) indicated that the green species is a Mn(IV) complex and spin quantification showed that it was the major manganese component of the oxidation (70(10)%).17 The spectrum contained g-values of 5.15, 2.44 and 1.63 that were simulated to show conclusively that this complex has an S = 3/2 ground state with a zero-field splitting constant of D = 3.0 cm−1. The six-line hyperfine pattern centered at the g = 5.15 component had a hyperfine constant of a = 66 G (Fig. 4, inset), a value that is consistent with a monomeric Mn(IV) species. The rhombicity of the EPR signal (E/D = 0.26) is predicted by theory: the DFT geometry-optimized molecular structure of [MnIVH3buea(O)]−, done at the B3LYP/6-311G level, showed large differences in bond distances and angles within the basal plane, producing a distorted ligand field around the Mn(IV) center. These deviations in structure are attributed to a Jahn–Teller distortion arising from the high-spin Mn(IV) ion in C3 symmetry.
Scheme 2.
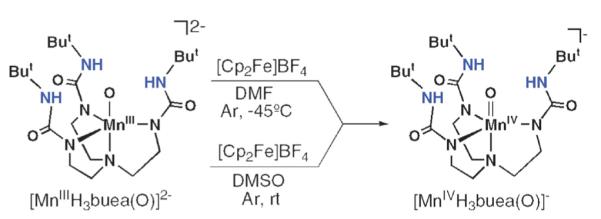
Synthetic routes for the preparation of [MnIVH3buea(O)]−. Reprinted with permission from reference 16. Copyright 2006, American Chemical Society.
Fig. 4.
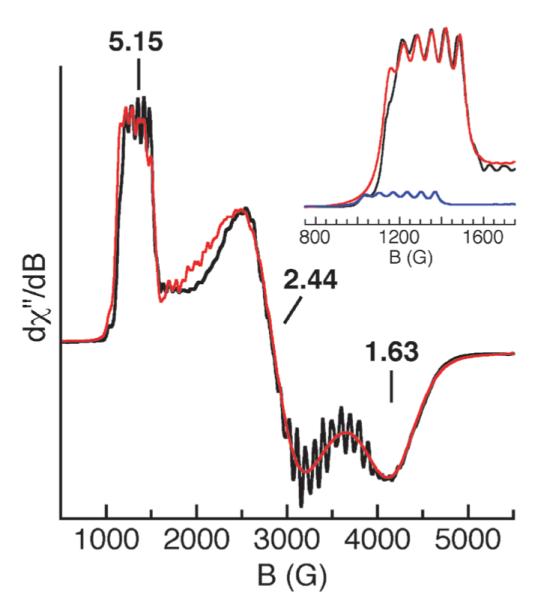
X-Band EPR spectrum (—) of [MnIVH3buea(O)]− measured at 3.6 K and a spectral simulation ( ). Inset: enlargement of spectrum at g = 5 (—) showing the contributions from the ms = 1/2 (
). Inset: enlargement of spectrum at g = 5 (—) showing the contributions from the ms = 1/2 ( ) and ms = 3/2 (
) and ms = 3/2 ( ) doublets.16 Reprinted with permission from reference 16. Copyright 2006, American Chemical Society.
) doublets.16 Reprinted with permission from reference 16. Copyright 2006, American Chemical Society.
The Mn(IV) complex’s moderate stability at room temperature in dry DMSO allowed us to monitor vibrational properties of the complex via solution FTIR methods—these studies confirmed that the Mn–O unit remained intact during oxidation. FTIR spectra collected in DMSO for the green species generated from 16O2 contains a new peak at 737 cm−1 that shifts to 709 cm−1 in the 18O-isotopomer (ν(Mn16O)/ν(Mn18O) = 1.04; calc. 1.05). This value is comparable to the ν(MnO) = 754 cm−1 reported for [MnIVTMP(O)], the only other MnIV=O species whose vibrational properties have been characterized.
These spectroscopic findings are consistent with the product from the oxidation of [MnIIIH3buea(O)]2− being the companion MnIV=O complex, [MnIVH3buea(O)]−. The detection of the oxomanganese complexes gave us the uncommon opportunity to do comparative investigations for two closely related oxometal complexes that are nearly structurally identical and differ electronically by only one electron. Moreover, manganese complexes with terminal oxo ligands have been the subject of many studies because of their purported involvement in various processes that range from the oxidant in olefin conversion to epoxides and a key intermediate in water oxidation in photosynthesis. However, there is still much debate about the chemical properties of oxomanganese complexes, especially those with nearly identical structural features.
In an ongoing study, we are investigating the reactivity of both oxomanganese complexes (the results are summarized in Fig. 5). We have shown so far that [MnIIIH3buea(O)]2− reacts with substrates with C–H bond dissociation energies (BDEC–H)of <80 kcal mol−1. More recently, we found that the Mn(IV)–oxo complex also reacts with similar substrates: for example, [MnIVH3buea(O)]− converts 1,2-diphenylhydrazine (BDEC–H, 69 kcal mol−1) to azobenzene (in >95% yield) and 9,10-dihydroanthracene (BDEC–H, 78 kcal mol−1) to anthracene (~100% yield) at room temperature in DMSO.18 In addition, [MnIVH3buea(O)]− can react with substrates having BDEC–H of >90 kcal mol−1, an example of which is DMSO (BDEC–H, 94 kcal mol−1).19
Fig. 5.
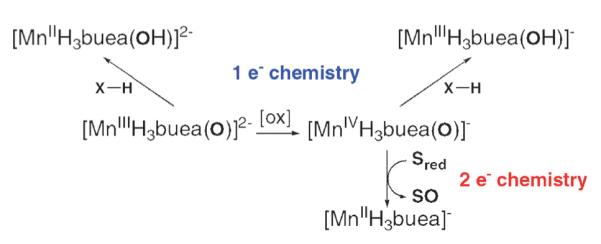
Summary of the reactions involving the Mn–oxo complexes.
Additional reactivity studies showed that O-atom transfer was possible from [MnIVH3buea(O)]−: treating [MnIVH3buea(O)]− with PMePh2 in DMSO at room temperature or at −45 °C in DMF produced O=PMePh2 in ~60% yield. Moreover, 18O=PMePh2 (~50% yield) was produced when the reaction was carried out with [MnIVH3buea(18O)]−, proving that the MnIV–oxo complex was the oxidant. Similarly, O-atom transfer was observed to form O=PMe2Ph when PMe2Ph was employed as the substrate. Monitoring the transfer reactions with EPR spectroscopy showed that a Mn(II) species with axial symmetry is formed when the reaction is completed. This is consistent with a two-electron process that would yield phosphine oxide and the corresponding MnII complex, presumably [MnIIH3buea]−. Note that bulkier phosphines, such as PPh3 and PCy3, do not react with [MnIVH3buea(O)]−. We attributed this selectivity to steric effects arising from the H-bond cavity surrounding the Mn(IV)=O unit.
We have also probed the ability of [MnIIIH3buea(O)]2− to oxidize substrates via O-atom transfer. Similar “two-electron” reactivity is not observed in the Mn(III)–O complex, presumably because of the inaccessibility of the Mn(I) complex.
Formation of terminal sulfido and selenido complexes of iron(III)
We have found over the years that the cavity structure formed by [H3buea]3− is ideally suited for the binding of oxygen-containing ligands. This selectivity has hindered the isolation of complexes with other exogenous ligands with different donor atoms—trace amounts of water appears to be the culprit because it can displace all non-oxygen donating ligands. One area that we have had success was in the formation of iron complexes with terminal sulfido and selenido ligands. Fe–E–M (E = S2−,Se2−; M = metal ion) units are common motifs, especially in protein cofactors,20 yet to our knowledge, iron complexes with terminal sulfido and selenido ligands were unknown prior to our work in 2004.10,21 The lack of these iron complexes has been attributed in part to the propensity of the chalcogenides, especially the heavier congeners, to bridge metal centers.10 Multinuclear complexes with bridging chalcogenido ligands have thus dominated this chemistry.20 However, there are fundamental and practical reasons for preparing and characterizing iron complexes with terminal sulfido and selenido ligands, including obtaining a better understanding of the inherent electronic and bonding properties of Fe–E units and how they relate to chemical reactivity.
We were successful in preparing [FeIIIH3buea(S)]2− and [FeIIIH3buea(Se)]2− following the procedure outlined for the M–O complexes but used molecular S8 and Se instead of dioxygen.21 X-Band EPR spectra on frozen solutions are consistent with both complexes having nearly axial symmetry around high-spin Fe(III) centers. X-Ray diffraction studies support the monomeric nature of [FeIIIH3buea(S)]2− and [FeIIIH3buea(Se)]2− in the solid state. The molecular structures (Fig. 6) reveal that each complex has three α-N− atoms and the terminal chalcogenido bonded to the iron centers: in [FeIIIH3buea(S)]2−, the Fe1–S1 length is 2.276(4) Å, while the Fe1–Se1 distance is 2.355(1) Å in [FeIIIH3buea(Se)]2−. The apical nitrogen N1 of [H3buea]3− also interacts weakly with the iron centers at Fe⋯N1 distances of 2.666(5) and 2.604(5) Å for [FeIIIH3buea(S)]2− and [FeIIIH3buea(Se)]2−. Their molecular structures differ from that of the related monomeric FeIII–O complex, [FeIIIH3buea(O)]2− (Table 1). Significantly shorter Fe1–O1 and Fe1–N1 bonds are observed in [FeIIIH3buea(O)]2− than Fe1–E and Fe1–N1 bonds in the other two iron–chalcogenido complexes, and the iron(III) center in the oxoiron complex is situated closer to the trigonal plane formed by N2, N4, and N6.
Fig. 6.
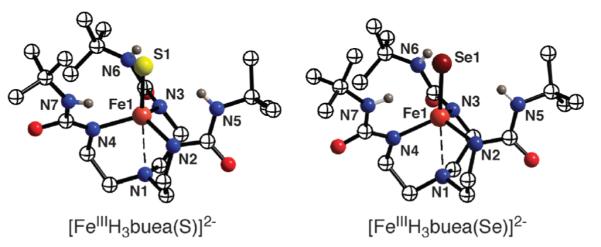
Molecular structures of [FeIIIH3buea(S)]2− and [FeIIIH3-buea(Se)]2−.
The molecular structures of [FeIIIH3buea(S)]2− and [FeIIIH3buea(Se)]2− also indicate the possibility of weak intramolecular H-bonds between the α’-NH’s and the terminal chalcogenido ligands. The average intramolecular Fe–E⋯Hα’-N distances of 3.377(3) Å in [FeIIIH3buea(S)]2− and 3.438(3) Å in [FeIIIH3buea(Se)]2− both suggest the formation of H-bonds.22 In addition, solid-state FTIR spectra of the complexes contain broadened NH signals relative to those of H6buea, which is another indicator of H-bonds.
The [FeIIIH3buea(E)]2− complexes, along with [FeIIIH3buea(O)]2−, represent the first series of iron complexes with terminal chalcogenido ligands, allowing comparative investigations into their properties. For instance, [FeIIIH3buea(S)]2− and [FeIIIH3buea(Se)]2− have limited stabilities compared to [FeIIIH3buea(O)]2−. One possible reason for their relative instabilities is that the Fe(III)–E units are not fully confined within the H-bond cavities, as is the case for the Fe(III)–O unit in [FeIIIH3buea(O)]2−. The urea groups within the Fe(III)–E complexes are tilted relative to the trigonal plane: average angles of > 35° from the normal are observed, resulting in bowl-like structures. The cavities in [FeIIIH3buea(S)]2− and [FeIIIH3buea(Se)]2− are thus relatively open, resulting in greater exposure of the sulfido and selenido ligands (Fig. 7).
Fig. 7.
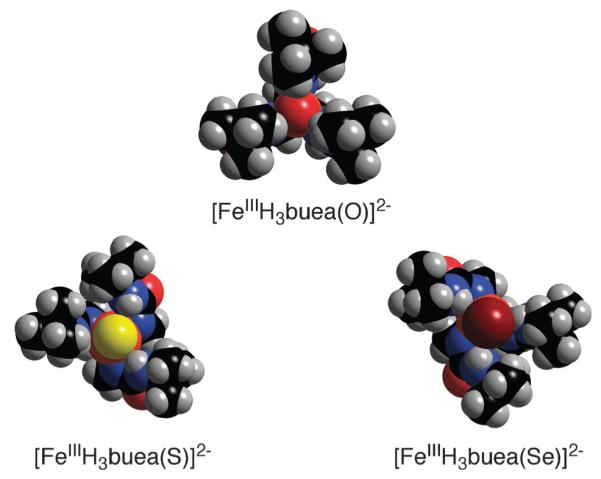
Space-filling representations of the Fe(III)-chalcogenido complexes showing the different cavity architectures.
In a collaborative project with Professor E. Solomon, a series of X-ray absorption experiments were performed to examine the bonding within the FeIII–S unit.23 The sulfur K-edge X-ray absorption spectrum (XAS) of [FeIIIH3buea(S)]2− showed an intense pre-edge transition at 2469.8 eV that was assigned to an envelope of S1s → Fe3d transitions. Because the intensity of the peak is proportional to the %S3p mixing into the five singly-occupied Fe3d orbitals, a fit of the data (2.32 ± 0.02 intensity units) corresponds to 82% S3p orbital character summed over the five half-occupied Fe3d orbitals. For comparison, the sulfur K-edge spectra of Et4N[Fe(SPh)4] and (Et4N)2[Fe2S2Cl4] were also analyzed and significantly lower intensity peaks were observed, corresponding to 68 and 35% S3p orbital character to the Fe–S bond, respectively. These findings indicate that the terminal sulfido ligand in [FeIIIH3buea(S)]2− forms a considerably more covalent FeIII–S bond than those found in complexes with bridging sulfido or terminal thiolate ligands. In fact, to our knowledge [FeIIIH3buea(S)]2− has the most covalent Fe–S bond yet observed.
Theoretical studies on the FeIII–E complexes
There have been several reports on the theoretical aspects of the above iron and manganese oxo complexes.12,24 More recently we have teamed with the Solomon group to examine the bonding in the [FeIIIH3buea(X)]2− complexes (X = O2−, S2−)23—the isolation of these complexes provided the opportunity to examine the bonding within the terminal M–X units and the effects of the H-bonds. A complete XAS study was undertaken (i.e., iron L-edge and K-edge) on [FeIIIH3buea(O)]2− and [FeIIIH3buea(S)]2−; additional sulfur K-edge XAS experiments were done on FeIII–S complex (see above). Experimentally derived electronic structures for the two complexes were determined from the XAS measurements and used to evaluate DFT calculations. Properties of the ground state wave functions obtained from the geometry optimized DFT calculations at the BP86 level are in close agreement with results from XAS measurements. For instance, the average Fe3d character per hole is calculated to be 67 and 72% for [FeIIIH3buea(S)]2− and [FeIIIH3buea(O)]2−, respectively, which are comparable with the 69 and 67% values obtained experimentally. These results indicate that the DFT calculations have acceptably reproduced the experimental ground-state properties, which gave us confidence to pursue further assessments of the bonding within the FeIII–X unit.
Evaluations of the Fe–X bond energies and the H-bond energies in [FeIIIH3buea(X)]3− were complicated by the fact that [H3buea]3− can H-bond to itself. To uncouple this effect, the model of [H3buea]3− (Fig. 8, A) was modified by eliminating the H-bond donor urea arms of the ligand (Fig. 8, B). The H-bond network was simulated by introducing three H2O molecules in approximately C3 arrangement around the Fe(III)–X unit (Fig. 8, C). The geometries for each structure were then fully optimized.
Fig. 8.
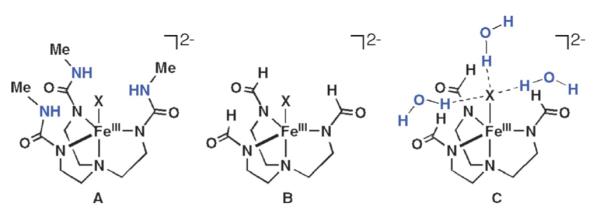
Complexes used in the theoretical studies.
The results show that without H-bonds the Fe–S bond is more covalent (i.e. higher ligand character in the Fe3d manifold) than the Fe–O bond; however, the Fe–O bond energy (EB) is much higher (–102 kcal mol−1 vs. −62 kcal mol−1 for Fe(III)–S). From a simple MO description, the covalent contribution to the bond energy can be approximated by α2Δ, where α2 is the ligand character per orbital and Δ is the energy gap between acceptor FeIII orbitals and the donor X2− orbitals before bonding. Thus the covalency is scaled by the energy of the ligand donor orbitals. We find that even though α2 is higher for the Fe(III)–S complex, the Fe(III)–O complex has a greater value for Δ because of the greater electronegativity of the oxo ligand. The net result is that the Fe(III)–O complex has a larger covalent contribution to the bond energy by approximately 8 kcal mol−1 (determined from the DFT calculated ground states). Note that the FeIII–O unit has a larger ionic contribution (~25 kcal mol−1) because of the higher charge density on the iron and oxo centers, reflecting lower covalency relative to the sulfido and contributes to the greater BEFe–X and shorter Fe(III)–O bond distance.
Inclusion of H-bonds polarizes the Fe–X bonds and localizes charge density on X, weakening the Fe–X bond and results in lowering of the Fe–X bond energies, force constants, and covalencies (Table 2). However, the overall H-bonding energy is favorable even with the weakened Fe–X bonds (Fig. 9). These effects are shown graphically in Fig. 9 for the FeIII–O complex: the overall stabilization results from the compensation of the lowering of the Fe–X bond energy by the large energy gained from H-bonding to the oxo/sulfido group: the energy of the three H-bonds is −25 and −12 kcal mol−1 in the Fe(III)–O and Fe(III)–S complexes, respectively. Therefore, in both complexes, the presence of H-bonds is energetically favorable despite a weakening of the Fe(III)–X bonds. For the Fe(III)–O and Fe(III)–S complexes the H-bonds are mainly dipolar and selectively affect the Fe–X π bonds because of their favorable orientation with respect to the H-bonded dipoles. The Coulombic nature of the interaction leads to a stronger H-bond in the oxo complex because of the shorter O⋯HN distance and high oxo charge, which in turn weakens the Fe–O bond more than the Fe–S bond upon H-bonding.
Table 2.
Effects of H-bonds on the Fe(III)–X bonding23
| X | H-Bond system (Fig. 8) |
Fe–X/Å | Covalency (%Xnp) |
EB(Fe–X)/ kcal mol−1 |
EB(hbs)b/ kcal mol−1 |
|---|---|---|---|---|---|
| O2− | A | 1.81 (1.81)a | 49 | ||
| O2− | B | 1.74 | 73 | −102 | |
| O2− | C | 1.80 | 46 | −83 | −25 |
| S2− | A | 2.23 (2.20)a | 61 | ||
| S2− | B | 2.17 | 81 | −62 | |
| S2− | C | 2.21 | 60 | −55 | −12 |
From XRD measurements.
H-bond stabilization
Fig. 9.
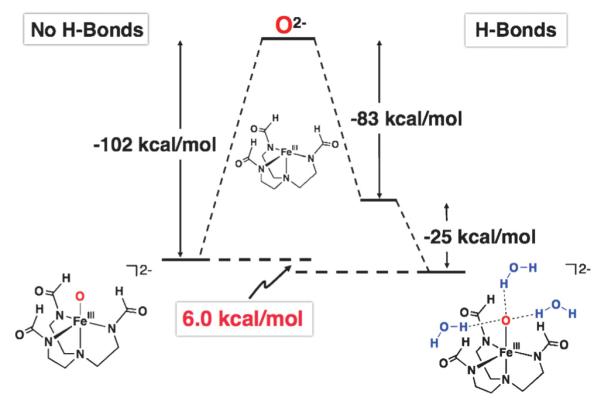
Bonding decomposition scheme for the FeIII–O complex.23
Terminal metal–oxo units are normally stabilized via multiple bonds between the metal ion and the oxo ligand.25 While the degree of π bonding varies, this type of bonding produces low-spin complexes with relatively short M–O bonds. Based on our structural, spectroscopic, and theoretical results on [MIIIH3buea(O)]2− we offer that in some cases H-bond(s) to the terminal oxo ligand can replace π bonds and be used in stabilizing metal–oxo complexes. The H-bonds would decrease the multiple bond character of the M–oxo unit, resulting in high-spin complexes with longer M–O bonds. These predictions are consistent with our results on the [MIIIH3buea(O)]2− complexes. For instance, the properties of the FeIII–O bond in [FeIIIH3buea(O)]2−, such as the reduced covalency and weakened Fe–O bond, are often associated with complex instability; however, in [FeIIIH3buea(O)]2− they are offset by the overall stabilization gained from formation of the H-bonds.
Modular H-bonding networks around metal complexes
The possibility that H-bonds can modulate the properties of metal oxo complexes would have profound implications particularly in metalloproteins, where H-bond networks are prevalent within active sites. Varying the number or type of H-bonds to M–O2 or M–O(H) moieties could modulate reactivity to ensure a desired function. This type of effect has been implicated in some metalloproteins, where both structure and function are known. For example, myoglobin and heme oxygenase both bind dioxygen: myoglobin does it reversibly as it is part of respiration, whereas heme oxygenase cleaves the O–O bond, producing a reactive intermediate for heme catabolism. Comparisons of their structures in the oxygenated states reveal identical primary coordination spheres (Fig. 10) but major differences in the H-bond network that surrounds the Fe–O2 unit.26,27 These differences in the secondary coordination sphere could lead to their divergent functions.
Fig. 10.
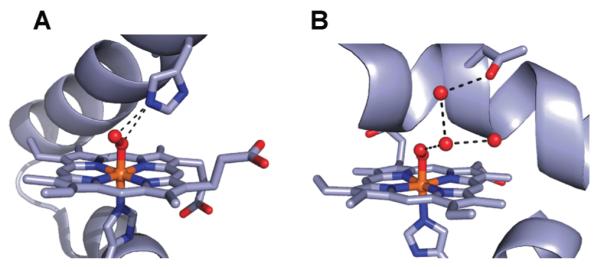
Structures of the active sites in OxyMb (A) (PDB, 1A6M) and the oxy form of heme oxidase (B) (PDB, 1V8X).
To further understand the effects of H-bonds on metal-mediated processes we considered it necessary to know how varying H-bond networks correlated with changes in function. Our initial approach to probe these structure–function relationships required the development of structurally tunable cavities with varied H-bond networks. This can be accomplished through the design and preparation of a series of ligands that enforce similar primary coordination spheres, but create differing architectures proximal to the metal center(s) to render varied H-bonding networks within the secondary coordination spheres. There are only a small number of examples that have utilized this approach; those reports of Masuda, Berreau and Mareque-Rivas are the most notable.28 They have explored the effects of varied H-bonding networks on physical properties such as redox potentials and acidity of coordinated ligands. We have continued the theme of investigating O2 activation with various M(II) complexes and also probed the reactivity of aryl azides with iron(II) complexes.
We designed our initial series of ligands around two tri-anionic ligands, [H3buea]3− and [0]3− (Fig. 11).29 The H-bond network produced for complexes of [H3buea]3− contains three H-bonds within the secondary coordination sphere. Some time ago we introduced the carboxamide-based analog [0]3−,30 which in contrast does not include H-bond donors. We thus sought to develop hybrid ligands of these two parent systems. The hybrid ligands31 [H22R]3− and [H1R]3− (Fig. 11) contain at least one urea arm and, together with [H3buea]3− and [0]3−, constitute a series of ligands whose metal complexes have the desired equivalent primary coordination spheres but different H-bond networks within the secondary coordination sphere. Besides varying the H-bond network around the metal ion, these hybrid ligands can also be used to influence the overall cavity architecture through the choice of R’ groups attached to the carboxamide arms.
Fig. 11.
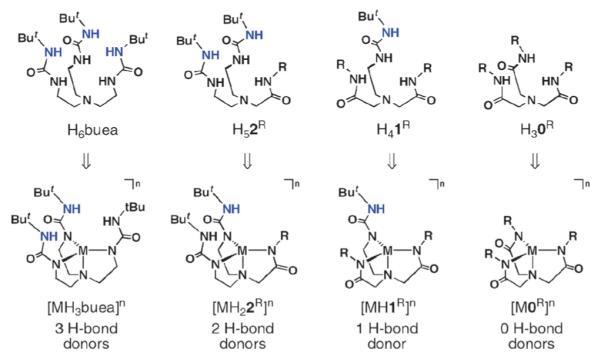
Series of ligands and complexes with varied H-bond networks. Reprinted with permission from reference 9. Copyright 2005, American Chemical Society.
The need for hybrid ligands led us to devise a convergent multi-step synthesis, in which the “arms” of the tripods would be placed on an amine nitrogen atom that ultimately served as the apical nitrogen donor. The preparative routes used the key starting compounds chloroethylurea 3 and N-alkylbromoacetamides 6–8 (Scheme 3). [H22R]3− require two urea arms that were produced from alkylating the secondary amine 4 with 6–8. In a similar fashion, [H1R]3− were prepared by treating primary amine 5 with an N-alkylbromoacetamide. Scheme 1 outlines the experimental conditions employed for the production of the hybrid ligands, which are routinely isolated in multi-gram quantities.
Scheme 3.
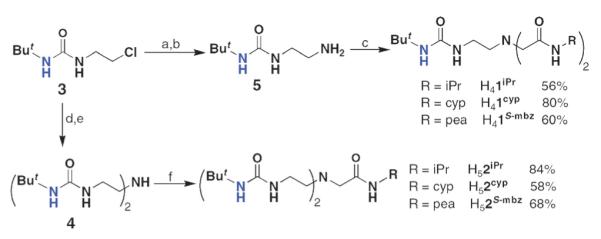
Preparative routes for the hybrid urea/amide ligands. Reagents and conditions: (a) potassium phthalimide, DMF, 80 °C, 73%; (b) H2NNH2·H2O, EtOH, heat, >98%; (c) 6–8, Et3N, heat, NaI (cat.); (d) BnNH2,Et3N, NaI (cat.), THF, heat, 77%; (e) cyclohexene, Pd/MeOH, heat, 86%; (f) 6–8, Et3N, heat.
Formation of CoIII–OH complexes via O2 binding and activation
We first used these ligands to prepare a series of Co(II) complexes and examined their reactivity with dioxygen.29 Our thought was that we could prepare four Co(II) complexes with nearly identical primary coordination spheres but, because of the different number of H-bond donating groups, distinct secondary coordination spheres that could influence reactivity. The study utilized the symmetrical urea-tripod [H3buea]3−, the two hybrid ligands [H22iPr]3− and [H1iPr]3−, and the tri(carboxamide) ligand, [0iPr]3−. The choice of appending isopropyl groups from the carboxamide nitrogen atoms was based on our previous studies that indicated these groups create cavities that are appropriate for external ligand binding. An important structural consequence of our ligand design is that each isopropyl group is positioned such that the methine C–H bond is outside the cavity, nearly eclipsing the carbonyl group of the adjacent amide.32 This conformation of isopropyl groups was fortuitous but advantageous, because the outward placement of these relatively reactive C–H bonds appears to be far enough away from the metal center to prevent ligand oxidation.
The Co(II) complexes were prepared as outlined in Scheme 4 for [CoIIH22iPr]− and isolated in crystalline yields of >75%. Single-crystal X-ray diffraction studies of the series revealed that [CoIIH22iPr]−, [CoIIH1iPr]− and [CoII0iPr]− have nearly identical structural properties. Each complex is four-coordinate, with a trigonal-monopyramidal coordination geometry (Fig. 12). The deprotonated urea/carboxamide nitrogen atoms form the trigonal planes with the amino nitrogen atom N1 as the apical donor. The bond lengths and angles for the complexes are also similar—this is illustrated by the overlay of the primary coordination spheres that is included in Fig. 12. The complexes have the expected differences in their secondary coordination spheres. The appended isopropyl groups from the carboxamide arms have the predicted conformation in which the methyl groups are proximal to the metal center, and thus function as scaffolding for the cavities surrounding the vacant axial coordination sites. In the complexes with the hybrid ligands, the urea groups complete the cavity structures, with two H-bond donors positioned within the cavity for [CoIIH22iPr]− and one for [CoIIH1iPr]−
Scheme 4.
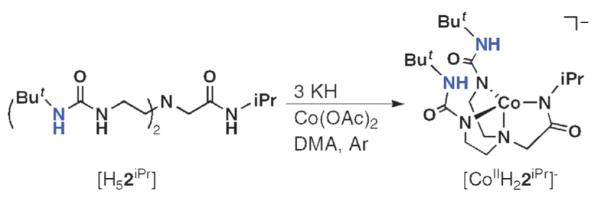
Synthetic procedure for isolation of Co(II) complexes with hybrid ligand.
Fig. 12.
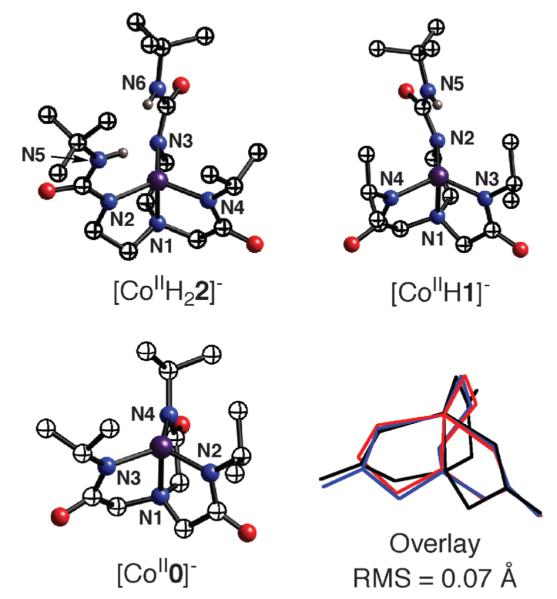
Molecular structures of the Co(II) complexes with differing intramolecular H-bonding networks and an overlay of their primary coordination spheres.
[CoIIH22iPr]−, [CoIIH1iPr]− and [CoII0iPr]− have nearly identical optical properties and EPR spectra that are consistent with S = 3/2 ground states. Electrochemical studies show a linear spread of 0.23 V in anodic potentials (Epa) with [CoIIH22iPr]− being the most negative at −0.385 V vs. [Cp2Fe]+/[Cp2Fe]. The fourth member of the series, [CoIIH3buea]− has similar properties to those of the other complexes based on spectroscopic data. Taken together, these studies show that this series of Co(II) complexes have similar primary coordination spheres, but differ in their cavity structures surrounding the coordinately-unsaturated metal centers.
Differential reactivity with dioxygen
We have investigated the reaction of each complex with dioxygen. [CoIIH3buea]− and [CoIIH22iPr]− react with 0.5 equiv. of O2 at room temperature to afford [CoIIIH3buea(OH)]− and [CoIIIH22iPr(OH)]− as isolatable solids in high yields (>70%, see Scheme 5). Spectroscopic and analytical data support their formulations as monomeric CoIII–OH complexes.33 Isotopic labeling studies confirm that dioxygen is the source of the oxygen atom in the hydroxo ligands: [CoIIIH3buea(16OH)]− has a ν(O–H) band at 3589 cm−1 that shifts to 3579 cm−1 in [CoIIIH3buea(18OH)]−;[CoIIIH22iPr(OH)]− has a ν(16O–H) at 3661 cm−1 and ν(18O–H) at 3650 cm−1.
Scheme 5.
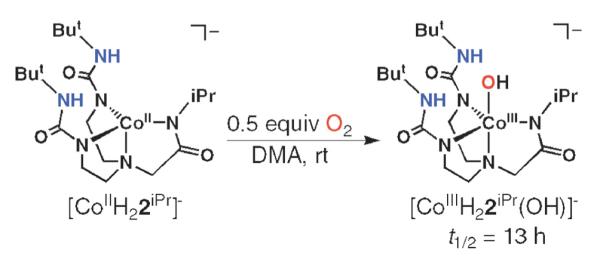
Preparative method for the synthesis of CoIII–OH complex of the hybrid ligand [H22]3− directly from O2 activation.
The remaining two Co(II) complexes with fewer H-bond donors displayed markedly different O2 reactivity. The complex with a single H-bond donor, [CoIIH1iPr]− did not react with 0.5 equiv. of O2 at room temperature—reactivity was only observed when excess dioxygen was introduced into the reaction mixture (Scheme 6). We initially observed a species with an X-band EPR signal at g = 2.0 that could be reverted back to [CoIIH1iPr]− when Ar was bubbled through the system. Based on these observations, we assigned this species to a Co–O2 adduct; however, definitive determination of its formulation was not possible because it was only moderately stable. Our studies suggested that the Co–O2 converted to another compound whose properties were similar to those found for the other CoIII–OH complexes. For example, the FTIR spectrum of a solid sampled obtained via precipitation had a peak at ν = 3633 cm−1, which is consistent with an O–H vibration. This proposed [CoIIIH1iPr(OH)]− also proved to be too unstable for complete characterization, as it slowly decayed to a yet undetermined species.
Scheme 6.
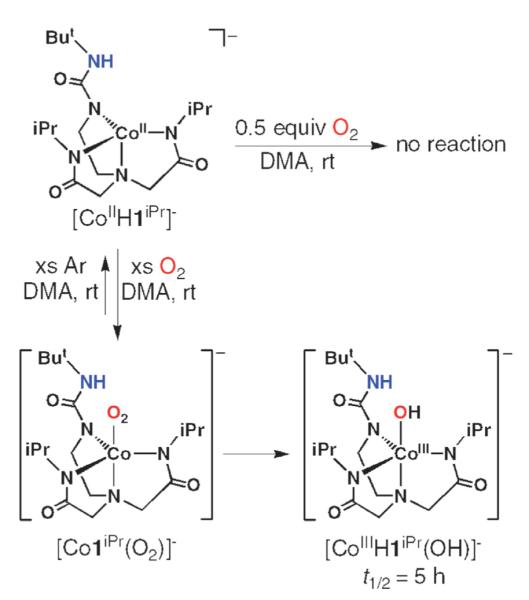
Proposed reaction sequence for the interaction of [CoIIH1iPr]− with dioxygen.
The complex [CoII0iPr]−, which is incapable of forming intramolecular H-bond, did not bind dioxygen under our experimental conditions (Scheme 7). We have ruled-out steric constraints as the cause for the lack of O2 binding. Previous studies in our laboratory found that other complexes of [0iPr]3− readily bind external ligands, including dioxygen. Secondly, we were able to prepare five-coordinate complexes of [CoII0iPr]− using other external ligands. For instance, [CoII0iPr]− binds a cyanide ion to form [CoII0iPr(CN)]2−, whose molecular structure, as determined by X-ray diffraction methods, is shown in Scheme 7. These findings support the premise that the cavity structure formed from the appended isopropyl groups has sufficient flexibility to allow external species access to the cobalt center.
Scheme 7.
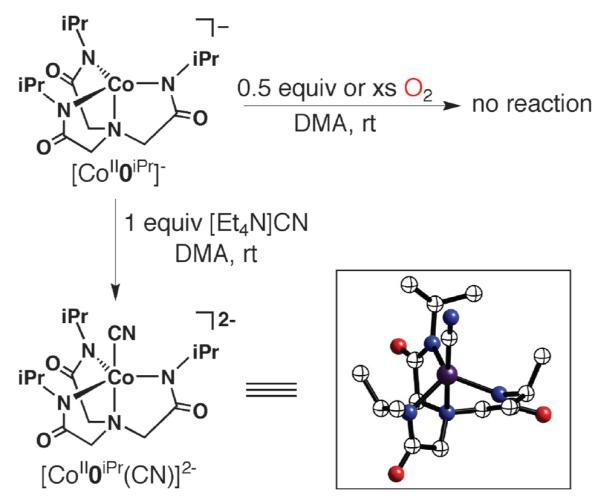
Reactions involving [CoII0iPr]−.
Effects of the intramolecular H-bonding networks
Despite their nearly identical primary coordination sphere, the cobalt complexes in this study have markedly different functional properties toward dioxygen. These differences are correlated, at least in part, to the differing H-bonding networks generated by the tripodal ligands. With two H-bond donors within its cavity, [CoIIH22iPr]− reacts at lower dioxygen concentrations compared to [CoIIH1iPr]−, which only has one H-bond donor. The additional H-bond donor in [CoIIH22iPr]− assists in the initial O2 binding event that is essential in metal-mediated O2 activation. Furthermore, [CoII0iPr]−, which does not have the capability to form intramolecular bonds, does not bind dioxygen. These functional differences observed in our Co(II) complexes are akin to what is found in biology, whereby variation in H-bond networks within secondary coordination spheres alters function, especially O2 binding and activation.
Changes in H-bond networks also correlated with the stabilities of the CoIII–OH complexes that are produced upon O2 activation. [CoIIIH3buea(OH)]− contains a symmetrical H-bond network with up to three H-bond donors that can interact with the coordinated hydroxo ligand—this complex is stable in solution for weeks and can be readily crystallized. On the other hand, the complexes with fewer numbers of H-bond donors have limited stability: [CoIIIH22iPr(OH)]− and [CoIIIH1iPr(OH)]− decay with initial rate constants of 5.9 × 10−8 and 2.5 × 10−7 min−1, respectively. We suggested that the greater number of intramolecular H-bonds involving the OH− ligand increases the stability of the complexes, possibly through a lowering of the nucleophilicity of the CoIII–OH unit. In addition, more intramolecular H-bonds within a complex would reinforce the cavity structure, producing a more protective cavity.
Binucleating H-bonding ligands
As the preceding section described, our initial research goals involved mononuclear metal complexes with H-bond networks—this hinged on our ability to prepare several new tripodal ligands. More recently we have begun a program into the design and preparation of binucleating ligands with H-bond donors. We reasoned that H-bonding binucleating ligands would allow us to prepare complexes with two oxo ligands and thus explore the cooperative influences of two M–O(H) centers within an intramolecular H-bond network. This led to the design concept illustrated in Fig. 13 that began with our pre-existing tripodal urea compound, H6buea. Coupling two of these tripods together by removal of one urea arm and replacing them with a 3,5-disubstituted pyrazole unit results in dinucleating ligands 3,5-bis{bis[(N’-R-ureayl)-N-ethyl]aminomethyl}-1H-pyrazole (H9PRbuam) that have cavities composed of four intramolecular H-bond donors. Using a pyrazole bridge maintains a similar N4 donor set as used in our mononucleating ligands. In addition, there should be flexibility within the H-bond cavity: reports by Okawa and co-workers and Meyer and co-workers.34,35 showed that metal–metal separations range from 3.4 and 4.5 Å when methylene groups are used as linkers to the pyrazole ring. This flexibility would aid in preparing complexes with M–O(H) units and their subsequent chemistry with substrates.
Fig. 13.
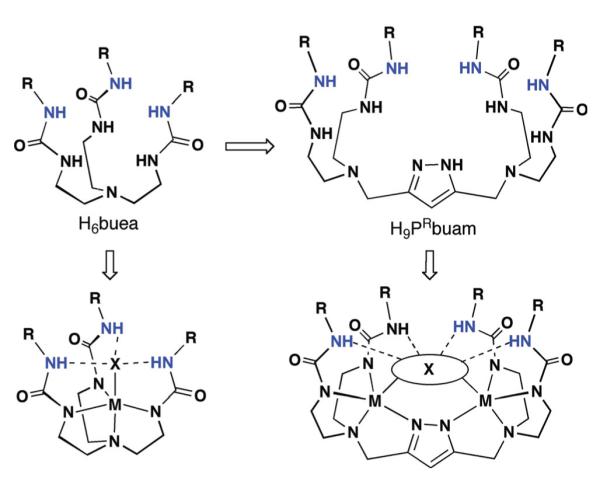
Design evolution for the binucleating H-bonding ligands.
We again used a convergent synthesis to prepare the H9PRbuam ligands that utilized making the bridging unit from 3,5-dimethylpyrazole (Scheme 8).36 The key intermediate 11 was made via a convenient six-step route following procedures first outlined by Bosnich and co-workers37 and Meyer and co-workers.38 The bis[(alkylureayl)-N-ethyl]amine compounds 4R were prepared according to our own methodology. H9PtBubuam and H9PiPrbuam were isolated as white solids in overall yields of 22 and 18% from the coupling of 11 and the diurea compounds 4R. We have optimized the routes to prepare the ligands in multigram quantities without use of chromatography.
Scheme 8.
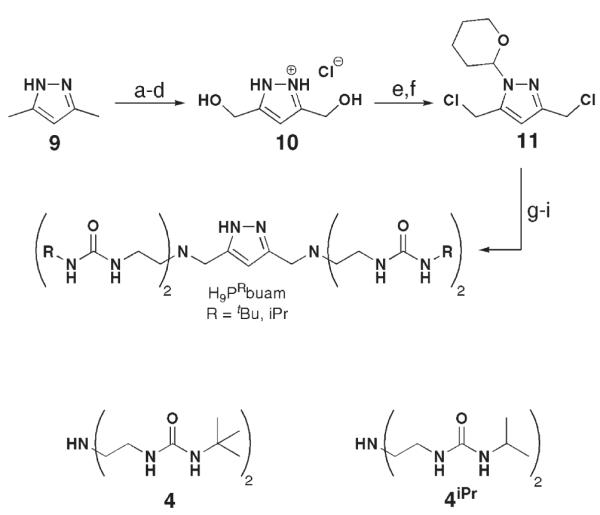
Preparative method for the binucleating ligand, H9PRbuam. Reagents and conditions: (a) KMnO4,H2O, heat, 56%; (b) HCl(g), EtOH, rt, 81%; (c) LiAlH4, ether, heat, N2; (d) ethanolic HCl, 80%; (e) SOCl2, heat, 94%; (f) 1,2-dihydropyran, CH2Cl2, rt, 75%; (g) 4 or 4iPr,Na2CO3,CH3CN, N2, heat, >81%; (h) ethanolic HCl, rt; (i) Na2CO3(aq), >88%. Reprinted with permission from reference 36. Copyright 2006, American Chemical Society.
To evaluate the structural properties of metal complexes with H9PtBubuam and H9PiPrbuam, we prepared the corresponding cobalt dimers following similar procedures described for our monomeric complexes (e.g., Scheme 1) but using CoCl2 (Scheme 9). Single-crystal X-ray diffraction measurements reveal that the cobalt centers are bridged by a single chloride ion with relatively long Co⋯Co for separations: example, in [CoII2H4PiPrbuam(μ-Cl)]2− the Co⋯Co distance is 4.113(1) Å. Each Co(II) ion has a distorted trigonal-bipyramidal geometry containing two urea-α-N− atoms, one pyrazolate nitrogen atom, and a bridging chloride ion with an average CoII–Cl bond length of 2.6279(4) Å for [CoII2H4PiPr-buam(μ-Cl)]2− and 2.570(1) Å for [CoII2H4PtBubuam(μ-Cl)]2−.
Scheme 9.
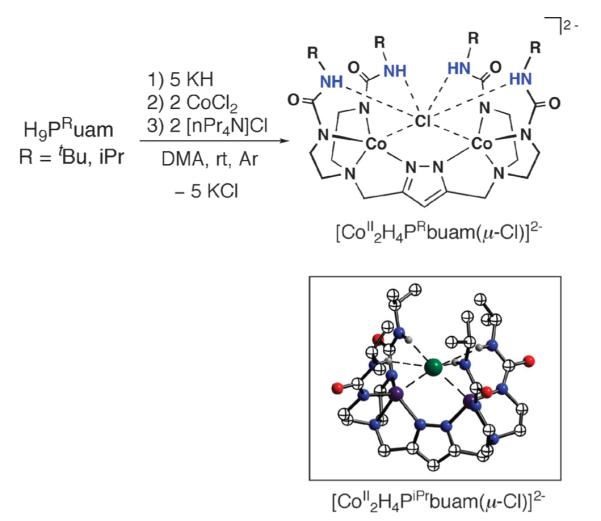
Preparation of [CoII2H4PRbuam(μ-Cl)]2− and the molecular structure of [CoII2H4PiPrbuam(μ-Cl)]2−.
The CoII–Cl bond lengths in the [CoII2H4PRbuam(μ-Cl)]2− complexes differ from the norm; for CoII–μ-Cl–CoII motifs, they are usually <2.30 Å. We proposed that these atypical CoII–Cl bonds result from the H-bonding network surrounding the bridging chloride ion. Intramolecular H-bonds between the binucleating ligands and the chloride ion are evident from their molecular structures: the α’-N⋯Cl distances are <3.5 Å and, along with N–H⋯Cl angles being >140°, are consistent with intramolecular H-bond assignments.39 Further support comes from FTIR measurements that showed a broadening of the peaks associated with the N–H vibrations, which is common for urea groups when strongly H-bonded.
CoII–OH mediated hydration of nitriles to amides
We have also prepared Co(II) dimers with a bridging hydroxo ligand, [CoII2H4PRbuam(μ-OH)]2−.40,41 The procedure (Scheme 10) was similar to those described previously for our mononucleating complexes (Fig. 3), in which an extra equiv. of KH (i.e., 6 equiv. in this case) was initially added to the ligands and water served as the source of the hydroxo ligand. The molecular structure of [CoII2H4PiPrbuam(μ-OH)]2− (Fig. 14A) shows that the Co(II) ions also have trigonal-monopyramidal coordination spheres. However, there are differences in the structure compared to that of [CoII2H4PRbuam(μ-Cl)]2−. The average Co–O(H) bond length is 2.216(2) Å, which is similar to distances found in other Co(II) complexes. The Co⋯Co separation is 3.587(1) Å, a distance that is significantly shorter than the 4.113 Å separation observed in the [CoII2H4PR-buam(μ-Cl)]2− complexes. This coordination appears to position the OH− ligand deeper within the cavity and thus, greatly weakens the H-bond interactions with the urea groups. All the α’-N⋯O distances are > 3.1Å, which are longer than what is normally observed for H-bonds (≤3.0Å). The FTIR spectra of the [CoII2H4PRbuam(μ-OH)]2− complexes support this assertion: the ν(N–H) bands are sharper and at higher energies than we usually find for urea groups involved in H-bonding.
Scheme 10.
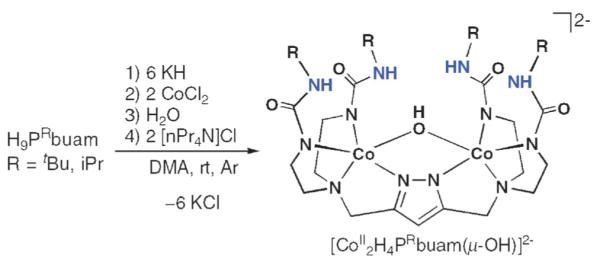
Synthetic route to [CoII2H4PRbuam(μ-OH)]2−. Reprinted with permission from reference 40. Copyright 2007, American Chemical Society.
Fig. 14.
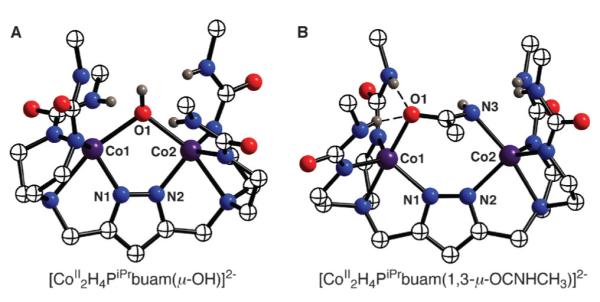
Molecular structures of [CoII2H4PiPrbuam(μ-OH)]2− (A) and [CoII2H4PiPrbuam(1,3-μ-OC(NH)CH3)]2− (B). The methyl groups on the isopropyl groups are omitted for clarity.
The [CoII2–(μ-OH)] complexes are reactive; for example, they can hydrate unfunctionalized nitriles to amides. We discovered this reactivity serendipitously when [CoII2H4PiPrbuam(μ-OH)]2− was dissolved in acetonitrile during purification—a new complex was obtained with a coordinated acetamido ligand. Its formulation was verified by obtaining it in pure form after crystallization. In addition, we have independently prepared this complex from [CoII2H4PiPrbuam(μ-Cl)]2− and [OC(NH)CH3]−. The molecular structure of the complex reveals that the acetamido ligand bridges between the two Co(II) ions in a 1,3-μ manner (Fig. 14B). In [CoII2H4PiPrbuam(1,3-μ-OC(NH)CH3)]2− the Co⋯Co separation is 4.480(1) Å, a lengthening of nearly 0.9 Å compared to that observed in the starting [CoII2(μ-OH)] complex. Furthermore, the acetamido ligand is strongly H-bonded: the acetamido oxygen and nitrogen atoms each have a pair of H-bonds involving the α’-NH groups of the [H4PiPrbuam]5− ligand.42
We propose a mechanism (Fig. 15), whereby displacement of the OH− ligand from one of the cobalt centers occurs upon nitrile coordination. We proposed that this initial step could be facile because of the lack of intra-molecular H-bonding to the hydroxo ligand. Binding of the nitrile places the activated electrophilic carbon atom in position for nucleophilic attack by the adjacent Co(II)–OH unit. A final proton shift produces the 1,3-μ-acetamido bridge, which is further stabilized by four intramolecular H-bonds.43
Fig. 15.
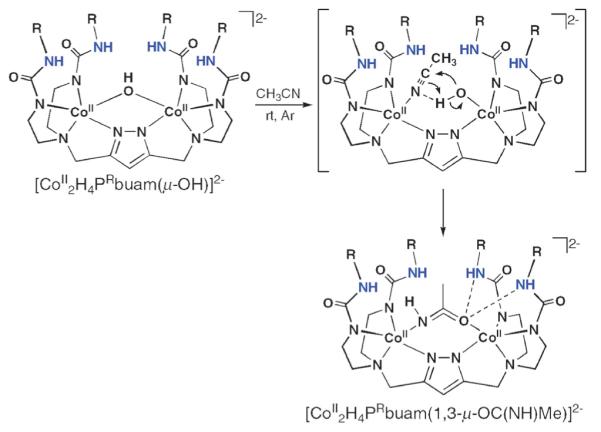
Proposed mechanism for the hydration of acetonitrile by [CoII2H4PRbuam(μ-OH)]2−. Reprinted with permission from reference 40. Copyright 2007, American Chemical Society.
These findings illustrate that tunable H-bonding cavity structures are possible in complexes of [H4PRbuam]5−. Our preliminary work suggests that structural changes are achieved because the pyrazolate unit serves as a flexible linker that allows the system to accommodate varied metal⋯metal separations. The urea arms that form the H-bond cavities are relatively rigid, but their positions are correlated to changes in the metal⋯metal separation. This tunability may subtle allow for adjustments in structure that are necessary for the development of functional metal complexes. In the hydration of nitriles, the [CoII2(μ-OH)] motif is not H-bonded, presumably enabling the complex to bind substrate and react. After hydration, the acetamido product is positioned within the cavity such that four intramolecular H-bonds are formed adding to the overall stability of the product. These properties are reminiscent of structural effects attributed to active sites in metalloproteins, in which secondary sphere H-bond networks are often proposed to influence transition states and stability of the species formed during turnover.
Conclusions
We are just beginning to realize the effects that non-covalent interactions have on metal-mediated transformations. Their importance has been denoted within the active sites of metalloproteins and some structure–function relationships have been established. These influences in biomolecules, as exemplified by H-bonding networks, appear to be mostly centered within the secondary coordination spheres of the metal complex. Therefore the function and dysfunction of metalloproteins can be understood within the context of changes in their secondary coordination spheres.
Our premise is that similar relationships need to be established in synthetic systems through the rational design of complexes with intramolecular H-bond networks. Our goals are to obtain fundamental understanding into the effects of H-bonds on the properties of metal complexes and to produce new systems with new or enhanced functions. We have described a set of complexes containing a variety of different H-bonding networks and discussed their reactivities toward small molecules, especially dioxygen. The monomeric iron and manganese complexes we produced have multiple intramolecular H-bonds involving terminal oxo or hydroxo ligands. In addition, related FeIII–S and FeIII–Se complexes have also been isolated. Our studies have shown that H-bonds directly affect the strength of the Fe–O(S) bonds, at the expense of π-bonds. These findings led us to the idea that variation in the number of H-bonds involving oxometal species should have functional consequences, an idea that was tested with a series of four-coordinate Co(II) complexes with varied numbers of H-bond donors. A clear dependence was observed: only the systems with H-bond donors were able to bind and activate dioxygen, albeit with different affinities. Moreover, the stability of the CoIII–OH products were dependent on the number of intramolecular H-bonds. Finally, our efforts in developing binucleating systems that form intramolecular H-bonding network about two metal ions were reviewed.
Future work in this area will undoubtedly seek to establish new correlations between H-bonds and function in both biological and synthetic systems. One possible scenario where this may be pertinent is in the oxidation of organic substrates. Multiple H-bonds to a metal oxo center could contribute to cleaving C–H bonds, whereas a lower number could promote O-atom transfer processes. Similarly, numerous hydrolytic metalloenzymes have H-bond networks surrounding M–OH units that could produce metal hydroxo centers with enhanced nucleophilicity. Whether these ideas are viable must await further studies but they do point to some intriguing possibilities for the role of intramolecular H-bonds in metal complexes.
Acknowledgements
We are exceedingly grateful to the numerous co-workers who have contributed to the work presented in this article, including C. MacBeth, R. Gupta, R. Lucas, P. Zinn, J. Mukherjee, T. Parsell, P. Larsen, M. Zart, M. Hendrich and E. Solomon. This worked was funded by the National Institutes of Health, USA (GM 050781).
Biography
 Ryan L. Shook was raised in Muscatine, Iowa. He received his BSc in Chemistry from the University of Northern Iowa in 2003, then joined the research group of A. S. Borovik at the University of Kansas to pursue his PhD. In 2006, the group moved to the University of California–Irvine where Ryan is expecting his PhD in the Spring of 2009. His research investigates the activation of dioxygen by non-heme metal complexes containing hydrogen bond donors.
Ryan L. Shook was raised in Muscatine, Iowa. He received his BSc in Chemistry from the University of Northern Iowa in 2003, then joined the research group of A. S. Borovik at the University of Kansas to pursue his PhD. In 2006, the group moved to the University of California–Irvine where Ryan is expecting his PhD in the Spring of 2009. His research investigates the activation of dioxygen by non-heme metal complexes containing hydrogen bond donors.
 A. S. Borovik was raised in Chicago, Illinois. He received his BS degree in Chemistry from Humboldt State University in 1981, his PhD in Chemistry at the University of North Carolina–Chapel Hill in 1986 under Tom Sorrell, and held postdoctoral positions with Larry Que at the University of Minnesota (1986–1988) and Ken Raymond at the University of California–Berkeley (1990–1992). He has been on the faculty at Ithaca College, Kansas State University, the University of Kansas, and the University of California–Irvine, where he is currently a professor.
A. S. Borovik was raised in Chicago, Illinois. He received his BS degree in Chemistry from Humboldt State University in 1981, his PhD in Chemistry at the University of North Carolina–Chapel Hill in 1986 under Tom Sorrell, and held postdoctoral positions with Larry Que at the University of Minnesota (1986–1988) and Ken Raymond at the University of California–Berkeley (1990–1992). He has been on the faculty at Ithaca College, Kansas State University, the University of Kansas, and the University of California–Irvine, where he is currently a professor.
Notes and references
- 1.Forum On Palladium Chemistry For Organic Synthesis Inorg. Chem. 2007;46:1865–1947. [Google Scholar]; Weck M, Jones CW. Inorg. Chem. 2007;46:1865–1875. doi: 10.1021/ic061898h. [DOI] [PubMed] [Google Scholar]; Köohler K, Kleist W, Pröckl SS. Inorg. Chem. 2007;46:1876–1883. doi: 10.1021/ic061907m. [DOI] [PubMed] [Google Scholar]; Astruc D. Inorg. Chem. 2007;46:1884–1894. doi: 10.1021/ic062183h. [DOI] [PubMed] [Google Scholar]; Tsuji Y, Fujihara T. Inorg. Chem. 2007;46:1895–1902. doi: 10.1021/ic061872q. [DOI] [PubMed] [Google Scholar]; Cornell CN, Sigman MS. Inorg. Chem. 2007;46:1903–1909. doi: 10.1021/ic061858d. [DOI] [PubMed] [Google Scholar]; Kotov V, Scarborough CC, Stahl SS. Inorg. Chem. 2007;46:1910–1923. doi: 10.1021/ic061997v. [DOI] [PubMed] [Google Scholar]; Deprez NR, Sanford MS. Inorg. Chem. 2007;46:1924–1935. doi: 10.1021/ic0620337. [DOI] [PubMed] [Google Scholar]; Hartwig JF. Inorg. Chem. 2007;46:136–1947. doi: 10.1021/ic061926w. [DOI] [PubMed] [Google Scholar]
- 2.Colquhoun HM, Stoddart JF, Williams DJ. Angew. Chem., Int. Ed. Engl. 1986;25:487–507. [Google Scholar]
- 3.Dawson JH. Science. 1988;240:433–439. doi: 10.1126/science.3358128. [DOI] [PubMed] [Google Scholar]; de Montellano P. R. Ortiz. Acc. Chem. Res. 1987;20:289–294. [Google Scholar]; Lu Y, Valentine JS. Curr. Opin. Struct. Biol. 1997;7:495–500. doi: 10.1016/s0959-440x(97)80112-1. [DOI] [PubMed] [Google Scholar]
- 4.Kitajima N, Tolman WB. Prog. Inorg. Chem. 1995;43:419–531. [Google Scholar]
- 5.Zhang W, Loebach JL, Wilson SR, Jacobsen EN. J. Am. Chem. Soc. 1990;112:2801–2803. [Google Scholar]; Jacobsen EN, Zhang W, Muci AR, Ecker JR, Deng L. J. Am. Chem. Soc. 1991;113:7063–7064. [Google Scholar]
- 6.Christianson DW, Cox JD. Annu. Rev. Biochem. 1999;68:33–57. doi: 10.1146/annurev.biochem.68.1.33. [DOI] [PubMed] [Google Scholar]
- 7.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Pestko GA, Sligar SG. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]; Nagano S, Poulos TL. J. Biol. Chem. 2005;280:31659–31663. doi: 10.1074/jbc.M505261200. [DOI] [PubMed] [Google Scholar]; Gerber NC, Sligar SG. J. Am. Chem. Soc. 1992;114:8742–8743. [Google Scholar]
- 8.Natale D, Mareque-Rivas JC. Chem. Commun. 2008:425–437. doi: 10.1039/b709650j. [DOI] [PubMed] [Google Scholar]; Mareque-Rivas JC. Curr. Org. Chem. 2007;11:1434–1449. [Google Scholar]
- 9.Borovik AS. Acc. Chem. Res. 2005;38:54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]
- 10.Parkin G. Prog. Inorg. Chem. 1998;47:1–165. [Google Scholar]; Trnka TM, Parkin G. Polyhedron. 1997;16:1031–1045. [Google Scholar]
- 11.Kurtz DM., Jr Chem. Rev. 1990;90:585–606. [Google Scholar]; Feig AL, Lippard SJ. Chem. Rev. 1994;94:759–805. [Google Scholar]; Wieghardt K. Angew. Chem., Int. Ed. Engl. 1989;28:1153. [Google Scholar]
- 12.Shirin Z, Hammes BS, Young VG, Jr, Borovik AS. J. Am. Chem. Soc. 2000;122:1836–1837. [Google Scholar]; MacBeth CE, Golombek AP, Young VG, Jr, Yang C, Kuczera K, Hendrich MP, Borovik AS. Science. 2000;289:938–941. doi: 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]; Gupta R, Borovik AS. J. Am. Chem. Soc. 2003;125:13234–13242. doi: 10.1021/ja030149l. [DOI] [PubMed] [Google Scholar]; MacBeth CE, Gupta R, Mitchell-Koch KR, Young VG, Jr, Lushington GH, Thompson WH, Hendrich MP, Borovik AS. J. Am. Chem. Soc. 2004;126:2556–2567. doi: 10.1021/ja0305151. [DOI] [PubMed] [Google Scholar]
- 13.MacBeth CE, Hammes BS, Young VG, Jr, Borovik AS. Inorg. Chem. 2001;40:4733–4741. [PubMed] [Google Scholar]
- 14.Gupta R, MacBeth CE, Young VG, Jr, Borovik AS. J. Am. Chem. Soc. 2002;124:1136–1137. doi: 10.1021/ja016741x. [DOI] [PubMed] [Google Scholar]
- 15.Gupta R, Borovik AS. J. Am. Chem. Soc. 2003;125:13234–13242. doi: 10.1021/ja030149l. [DOI] [PubMed] [Google Scholar]
- 16.Parsell TH, Miller M, Hendrich MP, Green MT, Borovik AS. J. Am. Chem. Soc. 2006;128:5728–5729. doi: 10.1021/ja062332v. [DOI] [PubMed] [Google Scholar]
- 17.The multi-line hyperfine signal near g = 2 is most likely a minor impurity (<10%) from a mixed-valent Mn2 species
- 18.Bordwell FG. J. Am. Chem. Soc. 1991;113:9750–9795. [Google Scholar]; Zhao Y, Bordwell FG, Cheng J-P, Wang D. J. Am. Chem. Soc. 1997;119:9125–9129. [Google Scholar]
- 19.Bordwell FG, Liu W-Z. J. Phys. Org. Chem. 1998;11:397–406. [Google Scholar]
- 20.Holm RH. Acc. Chem. Res. 1977;10:427–434. [Google Scholar]; Beinert H, Holm RH, Münck E. Science. 1997;277:653–659. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 21.Larsen PL, Gupta R, Powell DR, Borovik AS. J. Am. Chem. Soc. 2004;126:6622–6623. doi: 10.1021/ja049118w. [DOI] [PubMed] [Google Scholar]
- 22.Huang J, Ostrander RL, Rheingold AL, Leung Y, Walters MA. J. Am. Chem. Soc. 1994;116:6769–6776. [Google Scholar]; Desiraju GR, Steiner T. The Weak Hydrogen Bond In Structural Chemistry and Biology. Oxford University Press; Oxford: 1999. [Google Scholar]
- 23.Dey A, Hocking RK, Larsen PL, Borovik AS, Hedman B, Hodgson KO, Solomon EI. J. Am. Chem. Soc. 2006;128:9825–9833. doi: 10.1021/ja061618x. [DOI] [PubMed] [Google Scholar]
- 24.Budria JG, Raugei S, Cavallo L. Inorg. Chem. 2006;45:1732–1738. doi: 10.1021/ic051372h. [DOI] [PubMed] [Google Scholar]; Conradie J, Tangen E, Ghosh A. J. Inorg. Biochem. 2006;100:707–715. doi: 10.1016/j.jinorgbio.2006.01.029. [DOI] [PubMed] [Google Scholar]
- 25.Nugent WA, Mayer JM. Metal–Ligand Multiple Bonds. Wiley; New York: 1988. [Google Scholar]
- 26.Chu K, Berendzen J, Sweet RM, Schlichting I. Biophys. J. 1999;77:2153–2174. doi: 10.1016/S0006-3495(99)77056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Unno M, Matsui T, Chu GC, Couture M, Yoshida T, Rousseau DL, Olson JS, Ideda-Saito M. J. Biol. Chem. 2004;279:21055–21061. doi: 10.1074/jbc.M400491200. [DOI] [PubMed] [Google Scholar]
- 28.Wada A, Honda Y, Yamaguchi S, Nagatomo S, Kitagawa T, Jitsukawa K, Masuda H. Inorg. Chem. 2004;43:5725–5735. doi: 10.1021/ic0496572. [DOI] [PubMed] [Google Scholar]; Makowska-Grzyska MM, Jeppson PC, Allred RA, Arif AM, Berreau LM. Inorg. Chem. 2002;41:4872–4887. doi: 10.1021/ic0255609. [DOI] [PubMed] [Google Scholar]; Mareque-Rivas JC, Prabaharan R, Parsons S. Dalton Trans. 2004:1648–1655. doi: 10.1039/b402084g. [DOI] [PubMed] [Google Scholar]
- 29.Lucas RL, Muhkerjee J, Zart MK, Sorrell TN, Powell DR, Borovik AS. J. Am. Chem. Soc. 2006;128:15476–15489. doi: 10.1021/ja063935+. [DOI] [PubMed] [Google Scholar]
- 30.Ray M, Yap GPA, Rheingold AL, Borovik AS. J. Chem. Soc., Chem. Commun. 1995:1777–1778. [Google Scholar]; Ray M, Golombek AP, Hendrich MP, Yap GPA, Liable-Sands LM, Rheingold AL, Borovik AS. Inorg. Chem. 1999;38:3110–3115. [Google Scholar]; Ray M, Hammes BS, Yap GPA, Rheingold AL, Borovik AS. Inorg. Chem. 1998;37:1527–1532. [Google Scholar]; Shirin Z, Young VG, Jr, Borovik AS. Chem. Commun. 1997;4:1967–1968. [Google Scholar]; Hammes BS, Maldonado-Ramos D, Yap GAP, Liable-Sands L, Rheingold AL, Borovik AS. Inorg. Chem. 1997;36:3210–3211. doi: 10.1021/ic970124l. [DOI] [PubMed] [Google Scholar]
- 31.The bold numbers in the abbreviations for the hybrid ligands denote the number of H-bond donating groups—H41R and H52R have one and two H-bond donors, respectively. The ligands containing only amide groups, H30R has no H-bond donors and is thus indicated by the zero. The ligand with three urea groups, H6buea has been reported previously and its name is unchanged for consistency.
- 32.Ray M, Golombek AP, Hendrich MP, Young VG, Jr, Borovik AS. J. Am. Chem. Soc. 1996;118:6084–6085. [Google Scholar]
- 33.Note: [CoIIIH3buea(OH)]− and [CoIIIH22iPr(OH)]− have been independently prepared from water. The properties of these complexes are identical to those derived from dioxygen.
- 34.Kamiusuki T, Okawa H, Matsumoto N, Kida S. J. Chem. Soc., Dalton Trans. 1990:195–198. [Google Scholar]; Kamiusuki T, Okawa H, Inoue K, Matsumoto N, Kodera M, Kida S. J. Coord. Chem. 1991;23:201–211. [Google Scholar]; Itoh M, Motoda K-I, Shindo K, Kamiusuki T, Sakiyama H, Matsumoto N, Okawa H. J. Chem. Soc., Dalton Trans. 1995:3635–3641. [Google Scholar]
- 35.Meyer F, Beyreuther S, Heinze K, Zsolnai L. Chem. Ber./Recl. 1997;130:605–613. [Google Scholar]; Meyer F, Heinze K, Nuber B, Zsolnai L. J. Chem. Soc., Dalton Trans. 1998:207–213. [Google Scholar]; Buchler S, Meyer F, Kaifer E, Pritzkow H. Inorg. Chim. Acta. 2002;337:371–386. [Google Scholar]
- 36.Zinn PJ, Powell DR, Hendrich MP, Sorrell TN, Borovik AS. Inorg. Chem. 2006;45:3484–3486. doi: 10.1021/ic060009n. [DOI] [PubMed] [Google Scholar]
- 37.Schenck TG, Downes JM, Milne CRC, Mackenzie PB, Boucher H, Whelan J, Bosnich B. Inorg. Chem. 1985;24:2334–2337. [Google Scholar]
- 38.Röder JC, Meyer F, Pritzkow H. Organometallics. 2001;20:811–817. [Google Scholar]
- 39.Steiner T. Acta Crystallogr., Sect. B. 1998;54:456–463. doi: 10.1107/s090744499701500x. [DOI] [PubMed] [Google Scholar]; Aullón G, Bellamy D, Brammer L, Bruton EA, Orpen AG. Chem. Commun. 1998:653–654. [Google Scholar]
- 40.Zinn PJ, Sorrell TN, Powell DR, Day VW, Borovik AS. V, W, Day. 2007;46:10120–10132. doi: 10.1021/ic700685g. [DOI] [PubMed] [Google Scholar]
- 41.Representative examples of metal-mediated hydration: Parkins AW. Platinum Met. Rev. 1996;40:169–174., and references therein: Ghaffar T, Parkins AW. J. Mol. Catal. A: Chem. 2000;160:249–261. Murahashi S-I, Takaya H. Acc. Chem. Res. 2000;33:225–233. doi: 10.1021/ar980085x. Kukushkin VY, Pombeiro AJL. Inorg. Chim. Acta. 2005;358:1–21., and references therein; Allred RA, Doyle K, Arif AM, Berreau LM. Inorg. Chem. 2006;45:4097–4108. doi: 10.1021/ic051603k. Feng G, Mareque-Rivas JC, Williams NH. Chem. Commun. 2006:1845–1847. doi: 10.1039/b514328d.
- 42.Examples of synthetic coordination complexes of Co(III) related to the hydration of nitriles: Ellison JJ, Nienstedt A, Shoner SC, Barnhart D, Cowen JA, Kovacs JA. J. Am. Chem. Soc. 1998;120:5691–5700. Shearer J, Jackson HL, Schweitzer D, Rittenberg DK, Leavy TM, Kaminsky W, Scarrow RC, Kovacs JA. J. Am. Chem. Soc. 2002;124:11417–11428. doi: 10.1021/ja012555f. Shearer J, Kung IY, Lovell S, Kaminsky W, Kovacs JA. J. Am. Chem. Soc. 2001;123:463–468. doi: 10.1021/ja002642s. Jackson HL, Shoner SC, Rittenberg D, Cowen JA, Lovell S, Barnhart D, Kovacs JA. Inorg. Chem. 2001;40:1646–1653. doi: 10.1021/ic001271d. Tyler LA, Noveron JC, Olmstead MM, Mascharak PK. Inorg. Chem. 2003;42:5751–5761. doi: 10.1021/ic030088s. Harrop TC, Mascharak PK. Acc. Chem. Res. 2004;37:253–260. doi: 10.1021/ar0301532., and references therein; Harrop TC, Olmstead MM, Mascharak PK. Inorg. Chem. 2005;44:9527–9533. doi: 10.1021/ic051183z.
- 43.Similar mechanisms, but ones without intramolecular H-bonds, have been proposed in other dinuclear systems. For example see: Curtis NJ, Hagen KS, Sargeson AM. J. Chem. Soc., Chem. Commun. 1984:1571–1573. Alcock NW, Creaser II, Curtis NJ, Roecker L, Sargeson AM, Willis AC. Aust. J. Chem. 1990;43:643–654. Frey ST, Murthy NN, Weintraub ST, Thompson LK, Karlin KD. Inorg. Chem. 1997;36:956–957. doi: 10.1021/ic961439o. Meyer F, Kaifer E, Kircher P, Heinze K, Pritzkow H. Chem.–Eur. J. 1999;5:1617–1630.