

Abstract
A Diels-Alder based route to trans-fused angularly functionalized bicyclic structures has been developed. This transformation features the use of a tetrasubstituted dienophile in the cycloaddition step.
1. Introduction
The development of improved methods for the stereoselective construction of substituted decalin and hydrindane systems remains an important focus of organic synthesis.1 Over the years, our laboratory has been exploring extensions of the Diels-Alder reaction, with a view to gaining access to substructural patterns which are not traditionally filed under Diels-Alder logic. Toward this end, we recently developed a two-step “trans-Diels-Alder” paradigm, which provides access to trans-fused decalin and hydrindane systems from 1-nitrocycloalkene dienophiles (2) and simple dienes (1). As is shown in Figure 1, cycloaddition provides a cis-fused bicyclic adduct, bearing a nitro group in the ring junction (3). Subsequent radical denitration furnishes the target trans-fused system (4) with good selectivity.2 In the case of the hydrindane series, the Diels-Alder step, per se, is even more straightforward; however, denitration results in nearly 1:1 mixtures of diastereomeric products.
Figure 1.

To further expand upon this concept, we considered the possibility of achieving cycloaddition between a tetrasubstituted dienophile of the type 5 and a diene (1).3 It was hoped that the cis-fused nitro-substituted Diels-Alder adduct, 6, following radical-induced denitration, might progress to a trans-fused system bearing substitution (A) at the ring junction (cf. 7). Alternatively, 6 could be converted to a cis-fused angular amine product (cf. 8) through reduction of the nitro functionality. We were not unmindful of the challenges inherent in this proposed sequence. Indeed, tetrasubstituted olefins are known to act as poor dienophiles in the Diels-Alder reaction,4 and relatively few examples of tetrasubstituted cyclic dienophiles bearing nitro functionality have been reported.5 To our knowledge, compound 5 had not been demonstrated to function as a competent Diels-Alder dienophile. We were hopeful that, by installing a second electron-withdrawing group (i.e. CN) as substituent A, we could realize the desired cycloaddition. We describe herein the development of two-step, Diels-Alder-based routes to trans-fused adducts containing angular substitution, such as 7, and to disubstituted, cis-fused adducts of the type 8.6
2. Results and discussion
We first examined the cycloaddition-denitration sequence in the context of dienophile 117 (itself prepared in one step from 9, as shown) and synergistic diene 10 (Scheme 2).8 In the event, 10 and 11 did undergo cycloaddition to afford a 1:1 (exo:endo) mixture of the readily separable adducts 12 and 13. The structure of 13 (endo) was confirmed by X-ray crystallographic analysis. Each adduct was then separately subjected to conditions which favor radical-mediated denitration.9 Following hydrolysis, trans-fused adduct 14 was isolated as the predominant product; its structure was also unambiguously determined by X-ray crystallography. The slightly higher levels of stereocontrol achieved in the denitration of exo cycloadduct 12 (12:1 vs. 10:1) can perhaps be attributed to the axial disposition of the methoxy group, which serves to more effectively shield the “front” face of the transient free radical, leaving the back face exposed to attack by the hydrogen-donor agent.
Scheme 2.

aKey: (a) NaNO2, CAN, CH3CN, RT, 24 h, 56%; (b) toluene, 100 °C, 80 h, 78%; (c) n-Bu3SnH, AIBN, benzene, reflux, 2 h; (d) HF, CH3CN, 1 h.
We next examined the two-step sequence in the context of a cyclopentenyl dienophile. Here, the tetrasubstituted dienophile, 17, was prepared from compound 16, as shown (Scheme 3). Under thermal conditions, 17 underwent cycloaddition with diene 10 to afford the cis-fused adduct, 18, as a mixture of endo and exo isomers, in 95% yield. Upon denitration and subsequent hydrolysis, a 2:1 mixture of trans-fused 19 and cis-fused 20 was isolated. Obviously, this lack of stereocontrol would compromise the value of the method. We also examined the denitration and hydrolysis of hydrindane 22, arising from the Diels-Alder reaction of 21 and 17.10 As shown in Scheme 3, it was observed that, when denitration preceded hydrolysis, the trans-fused adduct (23) was obtained as the predominant product, with only moderate selectivity. By contrast, when hydrolysis was carried out at the stage of compound 22, denitration of the resultant ketone selectively delivered the cis-fused adduct 24 (cis:trans = 7:1).11
Scheme 3.

aKey: (a) NaNO2, CAN, CH3CN, RT, 24 h, 61%; (b) toluene, 100 °C, 10 h, 95%; (c) n-Bu3SnH, AIBN, benzene, reflux, 2 h; (d) HF, CH3CN, 1 h; (e) toluene, 100 C, 36 h, 63%, d.r. = 4:1; (f) HF, CH3CN, RT, 8 h; (g) n-Bu3SnH, AIBN, benzene, reflux, 1.5 h, 48% for two steps.
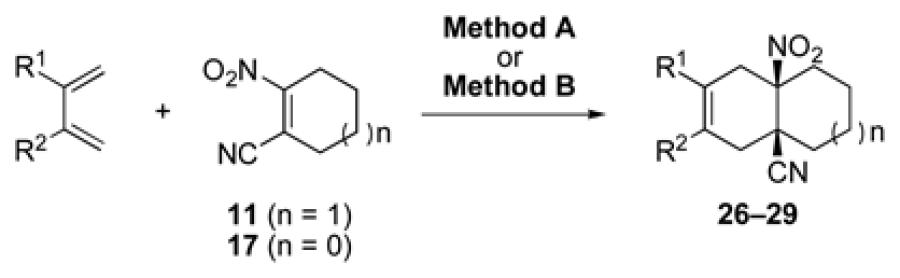
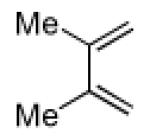
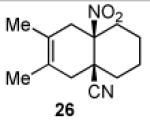
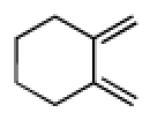
Having explored the cycloaddition-denitration sequence with activated diene coupling partners, we next investigated the use of less reactive hydrocarbon-based dienes in the Diels-Alder step. Our initial efforts to effect Diels-Alder reaction between 2,3-dimethyl-1,3-butadiene and dienophile 11 were unsuccessful. Under moderate thermal conditions (<130 °C), no reaction was observed. At higher temperatures (150 °C), compound 25 was formed in 25-40% yield.12 The structure of adduct 25 was confirmed by X-ray crystallography. It was eventually determined that cycloadduct 26 could be generated in moderate yield at 100 °C, through the addition of 5.0 M LiClO4 in THF,13 which serves to accelerate the Diels-Alder reaction (Table 1, entry 1). In contrast to the moderate Diels-Alder yields obtained with cyclohexenyl dienophile, 11 (Table 1, entries 1-2), the cyclopentenyl dienophile, 17, readily undergoes cycloaddition with hydrocarbon dienes, furnishing cis-fused hydrindane adducts in excellent yields (Table 1, entries 3-4).
Table 1.
 | |||||
|---|---|---|---|---|---|
| Entry | Diene | Dienophile | Method | Adduct | Yield (%) |
| 1 |
|
11 | A |
|
26 |
| 2 |
|
11 | A |
|
35 |
| 3 |
|
17 | B |
|
95 |
| 4 |
|
17 | B |
|
91 |
Key: Method A: 2,6-di-tert-butyl-4-methylphenol, 5.0 M LiClO4 in THF, 100 °C, 60 h; Method B: 2,6-di-tert-butyl-4-methylphenol, toluene, 100 °C, 24 h.
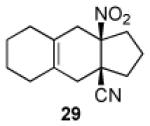
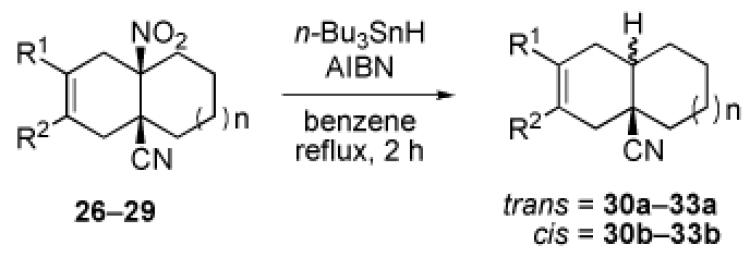
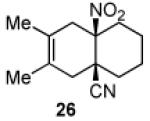
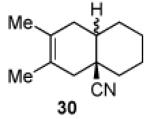
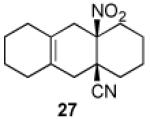
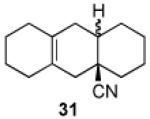
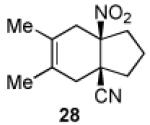
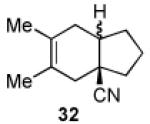
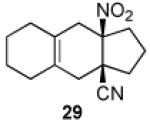
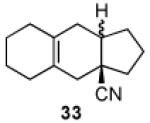
We now turned to an assessment of the feasibility of the denitration step, which we hoped would provide access to substituted, trans-fused hydrindane and decalin systems. As outlined in Table 2, entries 1 and 2, the cis-fused decalin systems (26 and 27) underwent radical-based denitration to provide the corresponding trans-fused adducts in good yield and with excellent levels of diastereocontrol (>20:1 trans: cis).14 However, denitration of hydrindanes 28 and 29 proceeded with quite poor levels of stereoselectivity (Table 2, entries 3 and 4).15
Table 2.
 | ||||
|---|---|---|---|---|
| Entry | Substrate | Product | Yield (%) | trans:cis (a:b) |
| 1 |
|
|
89 | >20:1 |
| 2 |
|
|
85 | >20:1 |
| 3 |
|
|
77 | ~1.5:1 |
| 4 |
|
|
89 | ~1.7:1 |
Finally, we sought to accomplish reduction of the cis-fused Diels-Alder adducts (26 and 28), to provide the corresponding cis-fused amine derivatives. As outlined in Scheme 5, treatment16 of compounds 26 and 28 with zinc dust in the presence of AcOH, followed by Alloc protection of the resultant angular amines, provided compounds 34 and 35 in good yield, and with the cyanide functionality intact. The two-step Diels-Alder/reduction sequence represents a straightforward synthesis of angular amines.
Scheme 5.

aKey: (a) Zn dust, THF/AcOH (2:1), −20 °C, 3 h; (b) AllocCl, THF, sat. NaHCO3, RT, 3 h.
3. Experimental section
3.1. General
Experiments involving moisture- and/or air-sensitive compounds were performed in oven- or flame-dried glassware with rubber septa under a positive pressure of nitrogen or argon using standard Schlenk techniques. Heating was accomplished by heating mantle or silicon oil bath using a temperature controller. Organic solutions were concentrated under reduced pressure using a Büchi rotatory evaporator, unless otherwise noted. Reactions were magnetically stirred and monitored by thin-layer chromatography (TLC) carried out on 0.25 mm E. Merck silica gel plates (60-F254) using UV light as a visualizing agent and a KMnO4 solution, a vanillin solution, an anisaldehyde solution, or a ceric ammonium molybdate (CAM) solution, and heat as developing agent. Flash chromatography was carried out with EM silica gel 60 (230-240 mesh) or Sorbent Technology silica gel 60 (particle size 32-63 μm) according to the method of Still.17 Yields refer to chromatographically and spectroscopically (1H-NMR) homogeneous materials, unless otherwise stated. Unless otherwise noted, all reagents were purchased at the highest commercial quality from commercial suppliers and used without further purification. Lithium perchlorate (battery grade, dry, 99.99%) was purchased from Aldrich and dried under high vacuum at 130 °C for at least 12 h just prior to use. Tetrahydrofuran (THF) was freshly distilled from sodium/benzophenone ketyl under an atmosphere of argon. Benzene, dichloromethane (CH2Cl2) and toluene were freshly distilled over CaH2 or filtered through a column of activated alumina under an atmosphere of argon. Diethyl ether was filtered through a column of activated alumina under an atmosphere of argon. Microwave reactions were performed on a Biotage microwave reactor. 1H and 13C NMR spectra were recorded on a Bruker DRX-400, a Bruker DRX-500 or a Bruker DRX-600 spectrometer at ambient temperature (300 K), unless otherwise stated. Chemical shifts of the 1H NMR (CDCl3: 7.26 ppm) and 13C NMR (CDCl3: 77.0 ppm) spectra were referenced to residual solvent peaks. 1H NMR spectra are reported as follows: chemical shift, multiplicity (br = broad, s = singlet, d = doublet, m = multiplet), coupling constant and integration. 13C NMR spectra were recorded with 1H decoupling. The multiplicities of the carbons of trans-3,4-dimethylbicyclo[4.4.0]dec-3-en-1-carbonitrile (30a) and trans-1,2,3,4,4a,5,6,7,8,9,9a,10-dodecahydroanthracene-4a-carbonitrile (31a) were determined by DEPT (Distortionless Enhancement by Polarization Transfer) experiments. IR spectra were recorded on a Jasco FT/IR-6100 or a Nicolet AVATAR 370 DTGS spectrometer and are reported in terms of frequency of absorption (cm−1). Melting points were measured using MELTEMP® and are uncorrected. Low resolution mass spectra were acquired on a ZQ Micromass spectrometer using the technique of ESI (electrospray ionization). High-resolution mass spectra (HRMS) were obtained from the Columbia University Mass Spectral Core Facility on a JEOL HX 110 mass spectrometer using the technique of EI (electron impact ionization) or FAB (fast atom bombardment).
3.2. General Procedure
3.2.1. General procedure for the nitration of cyloalkenecarbonitriles (9 and 16).7
To a stirred solution of cycloalkenecarbonitrile (20.0 mmol, 1.0 equiv) in anhydrous CH3CN (100 mL) at 0 °C was added NaNO2 (4.14 g, 60.0 mmol, 3.0 equiv) followed by ammonium cerium nitrate (32.8 g, 60.0 mmol, 3.0 equiv). The reaction mixture was stirred at room temperature for 24 h, diluted with H2O, and extracted with EtOAc (20 mL × 3). The organic layer was treated with a saturated solution of NaHCO3 (100 mL) and the resulting mixture was stirred at room temperature for 12 h. The mixture was diluted with H2O and extracted with EtOAc (20 mL × 3). The combined extract was dried over anhydrous MgSO4, filtered, and concentrated on a rotatory evaporator. Purification of the residue by flash column chromatography on silica gel gave the title compound.
3.2.2. 2-Nitrocyclohexenecarbonitrile (11)
Prepared using cyclohexenecarbonitrile (9, 2.14 g, 20.0 mmol), NaNO2 (4.14 g, 60.0 mmol) and ammonium cerium nitrate (32.8 g, 60.0 mmol) in CH3CN (100 mL), 11 was isolated as a yellow oil (1.70 g, 56%) after column chromatography (hexanes/EtOAc=4:1). 1H NMR (500 MHz, CDCl3) δ 2.75-2.72 (m, 2H), 2.61-2.59 (m, 2H), 1.86-1.82 (m, 2H), 1.77-1.72 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 157.9, 115.1, 114.2, 29.4, 25.7, 20.7, 20.3; IR (neat) 2952, 2869, 2221, 1652, 1528, 1427, 1342, 1331 cm−1; HRMS (EI) exact mass calculated for [M]+ (C7H8N2O2) required m/z 152.0586, found m/z 152.0583.
3.2.3. 2-Nitrocyclopentenecarbonitrile (17)
Prepared using cyclopentenecarbonitrile (16, 3.00 g, 32.2 mmol), NaNO2 (6.67 g, 96.6 mmol) and ammonium cerium nitrate (53.0 g, 96.6 mmol) in CH3CN (200 mL), 17 was isolated as a yellow oil (2.70 g, 61%) after column chromatography (hexanes/EtOAc=4:1). 1H NMR (600 MHz, CDCl3) δ 3.08-3.04 (m, 2H), 2.96-2.92 (m, 2H), 2.24-2.19 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 160.4, 117.2, 112.4, 34.6, 31.2, 20.7; IR (neat) 2964, 2919, 2850, 2229, 1644, 1521, 1435, 1348, 1321, 1177 cm−1. HRMS (EI) exact mass calculated for [M]+ (C6H6N2O2) required m/z 138.0429, found m/z 138.0432.
3.2.4. General procedure for the Diels-Alder reaction of 2-nitrocycloalkenecarbonitriles (11 and 17) with electron-rich dienes (10 and 21)
A sealable vial reactor equipped with a magnetic stirring bar was charged with 2-nitrocycloalkenecarbonitriles (1.0 equiv), an electron-rich diene (2.0 equiv) and anhydrous toluene. The reactor was sealed and the mixture was stirred at 100 °C in a pre-heated oil bath for 10-80 h. At this point, the mixture was cooled to room temperature and the solvent was evaporated. The resulting residue was purified by flash column chromatography on silica gel to afford the desired Diels-Alder adduct as a mixture of endo and exo diastereomers.
3.2.5. A 1:1 mixture of silyl enol ethers 12 and 13
Prepared using 11 (200 mg, 1.31 mmol) and trans-2-(tert-butyldimethylsilyloxy)-4-methoxy-1,3-butadiene (10, 564 mg, 2.63 mmol) in toluene (3.0 mL) at 100 °C for 80 h, an approximate 1:1 mixture of 12 and 13 was isolated as a pale yellow oil (350 mg, 78%) after column chromatography (hexanes/EtOAc=12:1). Diastereomers 12 and 13 were separated by careful column chromatography. Exo isomer 12: 1H NMR (600 MHz, CDCl3) δ 5.14 (d, J = 4.7 Hz, 1H), 4.04 (d, J = 4.9 Hz, 1H), 3.32 (s, 3H), 2.61 (br s, 2H), 2.45-2.44 (m, 1H), 2.37-2.34 (m, 1H), 1.89-1.85 (m, 1H), 1.74-1.71 (m, 2H), 1.65-1.56 (m, 2H), 1.25 (br s, 1H), 0.94 (s, 9H), 0.23 (s, 3H), 0.21 (s, 3H); IR (neat) 2951, 2932, 2884, 2859, 2239, 1752, 1673, 1554, 1454, 1370, 1254, 1230, 1210, 1089 cm−1; MS (ESI) 389.3 [M+Na]+. Endo isomer 13: 1H NMR (600 MHz, CDCl3) δ 4.93 (d, J = 1.7 Hz, 1H), 4.58 (s, 1H), 3.36 (s, 3H), 2.81 (d, J = 17.9 Hz, 1H), 2.66 (d, J = 15.4 Hz, 1H), 2.42 (d, J = 17.9 Hz, 1H), 1.93-1.90 (m, 2H), 1.77-1.75 (m, 1H), 1.67-1.58 (m, 3H), 1.52-1.46 (m, 1H), 0.93 (s, 9H), 0.22 (s, 3H), 0.20 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 141.8, 121.8, 102.3, 92.1, 81.0, 58.5, 39.3, 38.1, 30.6, 29.5, 19.4, 18.5, 17.9, −4.4; IR (neat) 2953, 2932, 2884, 2858, 2237, 1674, 1548, 1455, 1366, 1254, 1226, 1198, 1096, 914 cm−1; HRMS (FAB) exact mass calculated for [M+1]+ (C18H31N2O4Si) required m/z 367.2053, found m/z 367.2063. The structure of 13 was also confirmed by x-ray crystallographic analysis (see supplementary data).
3.2.6. A 1.8:1 mixture of silyl enol ethers 18 and 18'
Prepared using 17 (500 mg, 3.62 mmol) and trans-2-(tert-butyldimethylsilyloxy)-4-methoxy-1,3-butadiene (10, 1.55 g, 7.24 mmol) in toluene (4.0 mL) at 100 °C for 10 h, an approximate 1.8:1 mixture of 18 and 18' was isolated as a pale yellow oil (1.21 g, 95%) after column chromatography (hexanes/EtOAc=10:1). Diastereomers 18 and 18' were separated by careful column chromatography. Compound 18: 1H NMR (600 MHz, CDCl3) δ 4.96-4.93 (m, 1 H), 4.91-4.88 (m, 1 H), 3.32 (s, 3H), 2.73-2.69 (m, 1H), 2.62-2.59 (m, 1H), 2.32-2.28 (m, 1H), 2.14-2.01 (m, 4H), 1.95-1.89 (m, 1H), 0.90 (s, 9H), 0.19 (s, 3H), 0.17 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 147.5, 119.7, 101.0, 100.7, 78.6, 57.7, 45.0, 38.3, 35.8, 28.6, 25.4, 20.0, 17.8, −4.6, −4.7; IR (neat) 2955, 2931, 2887, 2858, 2830, 2241, 1675, 1547, 1462, 1363, 1255, 1218, 1094, 835 cm−1; HRMS (FAB) exact mass calculated for [M+1]+ (C17H29N2O4Si) required m/z 353.1897, found m/z 353.1918. Compound 18': 1H NMR (600 MHz, CDCl3) δ 5.11 (d, J = 4.9 Hz, 1H), 4.26 (d, J = 5.0 Hz, 1H), 3.32 (s, 3H), 2.79 (d of AB pattern, J = 18.3 Hz, 1H), 2.62-2.51 (m, 2H), 2.26 (d of AB pattern, J = 18.3 Hz, 1H), 2.17-2.12 (m, 1H), 2.06-2.01 (m, 1H), 1.93-1.88 (m, 2H), 0.91 (s, 9H), 0.20 (s, 3H), 0.18 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 149.7, 120.8, 99.2, 98.7, 75.6, 57.3, 40.9, 39.8, 35.7, 33.5, 25.4, 18.6, 17.8, −4.6, −4.7; IR (neat) 2955, 2931, 2893, 2858, 2825, 2243, 1671, 1553, 1461, 1369, 1256, 1221, 1087, 835 cm−1; HRMS (FAB) exact mass calculated for [M+1]+ (C17H29N2O4Si) required m/z 353.1897, found m/z 353.1880.
3.2.7. A 4:1 mixture of silyl enol ethers 22 and 22'
Prepared using 17 (75.0 mg, 0.543 mmol) and 2-(tert-butyldimethylsilyloxy)-1,3-butadiene (21, 200 mg, 1.08 mmol) in toluene (1.0 mL) at 100 °C for 36 h, an approximate 4:1 regioisomeric mixture of 22 and 22' was isolated as a pale yellow oil (110 mg, 63%) after column chromatography (hexanes/EtOAc=14:1). Regioisomers 22 and 22' were inseparable and characterization was carried out in the next step. 1H and 13C NMR spectra of the mixture of 22 and 22' are available in the supplementary data.
3.2.8. General procedure for the preparation of bicyclic ketones (14, 15, 19, 20, 23, and 24)
To a stirred solution of a cis-fused silyl enol ether (1.0 equiv) in anhydrous benzene was added tri-n-butyltin hydride (2.0 equiv) followed by azobisisobutyronitrile (0.5 equiv) and the mixture was heated under reflux for 2 h. At this point, the mixture was cooled to room temperature and the solvent was evaporated. In a separate Falcon® tube, the resulting residue was dissolved in CH3CN and treated with HF (48% in H2O) and the mixture was stirred at room temperature for 1 h. The mixture was cooled in an ice-water bath and the reaction was quenched by dropwise addition of saturated NaHCO3 solution. The precipitate thus formed was filtered off and the filtrate was diluted with H2O and extracted with CH2Cl2 (3 × 30 mL). The combined extract was dried over anhydrous Na2SO4, filtered, and concentrated on a rotatory evaporator. The residue was purified by flash column chromatography on silica gel to give the title compound.
3.2.9. A 12:1 mixture of trans-3-oxobicyclo[4.4.0]dec-4-enecarbonitrile (14) and cis-3-oxobicyclo[4.4.0]dec-4-enecarbonitrile (15)
Prepared using an exo isomer 12 (35.0 mg, 95.5 μmol), tri-n-butyltin hydride (55.3 mg, 190 μmol), azobisisobutyronitrile (8.0 mg, 48 μmol) in benzene (4.0 mL), a 12:1 mixture of trans-fused 14 and cis-fused 15 was isolated as a pale yellow oil (13 mg, 78% over two steps) after column chromatography (hexanes/EtOAc=4:1). The trans-fused 14 was isolated in pure form by careful column chromatography. Compound 14: 1H NMR (600 MHz, CDCl3) δ 6.68 (dd, J = 10.1, 1.5 Hz, 1 H), 6.14-6.12 (m, 1H), 2.79 (d of AB pattern, J = 16.7 Hz, 1H), 2.41 (d of AB pattern, J = 16.7 Hz, 1H), 2.38-2.37 (m, 1H), 2.04-2.02 (m, 1H), 1.98-1.94 (m, 2H), 1.83-1.80 (m, 1H), 1.78-1.63 (m, 2H), 1.55-1.50 (m, 1H), 1.48-1.46 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 194.4, 151.4, 130.0, 120.8, 48.3, 43.6, 43.6, 35.9, 28.2, 25.5, 22.0; IR (neat) 2936, 2861, 2232, 1683, 1448, 1384, 1244, 1159, 982 cm− HRMS (EI) exact mass calculated for [M]+ (C11H13NO) required m/z 175.0997, found m/z 175.0998. The structure of 14 was also confirmed by x-ray crystallographic analysis (see supplementary data).
3.2.10. A 10:1 mixture of trans-3-oxobicyclo[4.4.0]dec-4-enecarbonitrile (14) and cis-3-oxobicyclo[4.4.0]dec-4-enecarbonitrile (15)
Prepared using an endo isomer 13 (50.0 mg, 0.136 mmol), tri-n-butyltin hydride (78.6 mg, 0.270 mmol), azobisisobutyronitrile (11.0 mg, 68.0 μmol) in benzene (5.0 mL), a 10:1 mixture of trans-fused 14 and cis-fused 15 was isolated as a pale yellow oil (19 mg, 80% over two steps) after column chromatography (hexanes/EtOAc=4:1).
3.2.11. A 2:1 mixture of trans-3-oxobicyclo[4.3.0]non-4-enecarbonitrile (19) and cis-3-oxobicyclo[4.3.0]non-4-enecarbonitrile (20)
Prepared using a distereomeric mixture of 18 and 18′ (200 mg, 0.567 mmol), tri-n-butyltin hydride (329 mg, 1.13 mmol), azobisisobutyronitrile (47.0 mg, 0.284 mmol) in benzene (6.0 mL), a 2:1 mixture of trans-fused 19 and cis-fused 20 was isolated as a pale yellow oil (47 mg, 51% over two steps) after column chromatography (hexanes/EtOAc=8:1). Diastereomers 19 and 20 were separated by careful column chromatography. Compound 19: 1H NMR (600 MHz, CDCl3) δ 7.04 (dd, J = 10.0, 1.3 Hz, 1H), 6.14 (dd, J = 10.0, 2.8 Hz, 1H), 3.05 (d of AB pattern, J = 16.7 Hz, 1H), 2.63-2.59 (m, 1H), 2.39 (d of AB pattern, J = 16.7 Hz, 1H), 2.30-2.26 (m, 1H), 2.18-2.12 (m, 1H), 2.11-2.05 (m, 1H), 2.01-1.93 (m, 1H), 1.86-1.80 (m, 1H), 1.77-1.71 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 195.3, 147.9, 130.8, 121.6, 48.1, 47.4, 47.4, 35.6, 26.2, 21.0; IR (neat) 2960, 2879, 2231, 1683, 1455, 1381, 1240, 1159 cm−1; HRMS (EI) exact mass calculated for [M]+ (C10H11NO) required m/z 161.0841, found m/z 161.0850. The structure of 19 was also confirmed by x-ray crystallographic analysis (see supplementary data). Compound 20: 1H NMR (600 MHz, CDCl3) δ 6.73 (dd, J = 10.3, 3.7 Hz, 1H), 6.05 (dd, J = 10.3, 1.9 Hz, 1H), 3.17-3.14 (m, 1H), 2.83 (d of AB pattern, J = 16.6 Hz, 1H), 2.73 (d of AB pattern, J = 16.6 Hz, 1H), 2.35-2.23 (m, 2H), 2.00-1.93 (m, 1H), 1.90-1.82 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 193.8, 149.5, 128.5, 123.3, 44.8, 42.0, 41.9, 35.9, 31.0, 23.0; IR (neat) 2923, 2853, 1679, 1446, 1386, 1244, 1151, 1055, 842 cm−1; HRMS (EI) exact mass calculated for [M]+ (C10H11NO) required m/z 161.0841, found m/z 161.0836.
3.2.12. A 2:1 mixture of trans-3-oxobicyclo[4.3.0]nonanecarbonitrile (23) and cis-3-oxobicyclo[4.3.0]nonanecarbonitrile (24)
Prepared using a silyl enol ether 22 (60.0 mg, 0.186 mmol), tri-n-butyltin hydride (81.2 mg, 0.279 mmol), azobisisobutyronitrile (10.0 mg, 56.0 μmol) in benzene (3.0 mL), a 2:1 mixture of trans-fused 23 and cis-fused 24 was isolated as a pale yellow oil (10.3 mg, 34% over two steps) after column chromatography (hexanes/EtOAc=4:1). Diastereomers 23 and 24 were separated by careful column chromatography. Compound 23: 1H NMR (600 MHz, CDCl3) δ 2.93-2.90 (m, 1H), 2.59-2.56 (m, 1H), 2.35-2.26 (m, 3H), 2.19-2.16 (m, 1H), 2.08-1.87 (m, 5H), 1.70-1.61 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 206.3, 120.9, 49.7, 48.3, 47.7, 39.8, 37.0, 27.6, 25.7, 21.9; IR (neat) 2960, 2873, 2229, 1715, 1457, 1423, 1187, 913 cm−1; HRMS (EI) exact mass calculated for [M]+ (C10H13NO) required m/z 163.0997, found m/z 163.0988. Compound 24: 1H NMR (500 MHz, CDCl3) δ 2.71 (d of AB pattern, J = 18.0 Hz, 1H), 2.65-2.60 (m, 1H), 2.53 (d of AB pattern, J = 18.0 Hz, 1H), 2.42-2.33 (m, 2H), 2.28-2.22 (m, 1H), 2.18-2.10 (m, 2H), 1.92-1.83 (m, 2H), 1.77-1.70 (m, 2H), 1.66-1.56 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 207.3, 124.3, 45.2, 43.5, 42.2, 38.2, 37.0, 30.4, 26.0, 23.0; IR (neat) 2957, 2872, 2231, 1719, 1526, 1373, 1350, 1241, 1045 cm−1; HRMS (EI) exact mass calculated for [M]+ (C10H13NO) required m/z 163.0997, found m/z 163.0988.
3.2.13. Stereochemical confirmation of cis-3-oxobicyclo[4.3.0]nonanecarbonitrile (24)
To a solution of 20 (20.0 mg, 0.124 mmol) in degassed CH3OH (4.0 mL) was added Pd(OH)2 (20.0 mg, 20% w/w). A hydrogen-filled balloon (1 atm) was placed over the solution and the mixture was stirred at room temperature for 2 h. At this point, TLC analysis indicated that the reaction was completed. The mixture was filtered through a pad of Celite®, washed thoroughly with EtOAc, and the solvents were evaporated. The crude mixture was purified by flash column chromatography (hexanse/EtOAc=2:1) on silica gel to afford cis-fused 24 (19 mg, 94%), whose spectroscopic properties were in complete accord with the authentic sample.
3.2.14. Preparation of a 1:7 mixture of trans-3-oxobicyclo[4.3.0]nonanecarbonitrile (23) and cis-3-oxobicyclo[4.3.0]nonanecarbonitrile (24)
A Falcon® tube was charged with a silyl enol ether 22 (10.0 mg, 31.0 μmol) and CH3CN (2.0 mL) and HF (48% in H2O, 0.010 mL) was added dropwise. The mixture was stirred at room temperature for 8 h. The mixture was cooled in an ice-water bath and the reaction was quenched by dropwise addition of saturated NaHCO3 solution. The mixture was diluted with H2O and extracted with CH2Cl2 (3 × 4 mL). The combined extract was dried over an hydrous Na2SO4, filtered, and concentrated on a rotatory evaporator. The residue was purified by flash column chromatography (hexanes/EtOAc=4:1) on silica gel to afford cis-6-nitro-3-oxobicyclo[4.3.0]nonanecarbonitrile (4.1 mg, 64%) as a pale yellow oil. cis-6-Nitro-3-oxobicyclo[4.3.0]nonanecarbonitrile: 1H NMR (500 MHz, CDCl3) δ 3.32 (d of AB pattern, J = 18.8 Hz, 1H), 2.84-2.78 (m, 1H), 2.72 (d of AB pattern, J = 18.8 Hz, 1H), 2.70-2.59 (m, 2H), 2.50-2.45 (m, 1H), 2.44-2.38 (m, 1H), 2.35-2.14 (m, 3H), 1.97-1.88 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 203.0, 119.6, 93.5, 47.5, 43.9, 36.0, 35.7, 34.4, 32.3, 18.1; IR (neat) 2962, 2919, 2889, 2850, 2239, 1724, 1675, 1539, 1419 cm−1; HRMS (EI) exact mass calculated for [M]+ (C10H12N2O3) required m/z 208.0848, found m/z 208.0855. To a stirred solution of cis-6-nitro-3-oxobicyclo[4.3.0]nonanecarbonitrile (27.0 mg, 0.130 mmol, 1.0 equiv) in anhydrous benzene (2.0 mL) was added tri-n-butyltin hydride (75.7 mg, 0.260 mmol, 2.0 equiv) followed by azobisisobutyronitrile (11.0 mg, 70.0 μmol, 0.5 equiv) and the mixture was heated under reflux for 1.5 h. At this point, the mixture was cooled to room temperature and the solvent was evaporated. The residue was purified by flash column chromatography (hexanes/EtOAc=6:1) on silica gel to give a 1:7 mixture of trans-fused 23 and cis-fused 24 (16 mg, 75%).
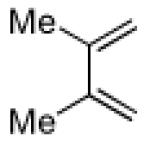
3.2.15. Preparation of tricyclic compound 25
A sealable vial reactor equipped with a magnetic stirring bar was charged with 2-nitrocyclohexenecarbonitrile (11, 70.0 mg, 0.460 mmol, 1.0 equiv), 2,3-dimethyl-1,3-butadiene (379 mg, 4.60 mmol, 10 equiv), 2,6-di-tert-butyl-4-methylphenol (10.1 mg, 46.0 μmol, 0.1 equiv) and anhydrous toluene (2.0 mL). The reactor was sealed and the mixture was stirred at 150 °C in a pre-heated oil bath for 24 h. At this p oint, the mixture was cooled to room temperature and the solvent was evaporated. The resulting residue was purified by flash column chromatography (hexanes/EtOAc=14:1) on silica gel to afford the title compound (32.0 mg, 33%) as a tan/orange sticky solid. 1H NMR (500 MHz, CDCl3) δ 2.81-2.74 (m, 1H), 2.67-2.64 (m, 1H), 2.41-2.29 (m, 2H), 2.12-1.88 (m, 5H), 1.70 (s, 3H), 1.65 (s, 3H), 1.56-1.54 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 180.0, 172.2, 124.2, 117.1, 47.4, 38.3, 32.4, 30.5, 27.7, 26.3, 24.2, 19.4, 18.5; IR (neat) 2931, 2863, 1785, 1715, 1452, 1386, 1086 cm−1; mp 80-86 °C; HRMS (EI) exact mass calculated for [M]+ (C13H17NO2) required m/z 219.1259, found m/z 219.1250. The structure of 25 was also confirmed by x-ray crystallographic analysis (see supplementary data).
3.2.16. General procedure for the Diels-Alder reaction of 2-nitrocyclohexenecarbonitriles (11) with simple dienes
A sealable tube equipped with a magnetic stirring bar was charged with 2-nitrocyclohexenecarbonitrile (11, 76.1 mg, 0.500 mmol, 1.0 equiv), a simple diene (5-10 equiv), 2,6-di-tert-butyl-4-methylphenol (5.5 mg, 25 mmol, 5.0 mol%) and anhydrous lithium perchlorate (532 mg, 5.00 mmol, 10 equiv). Under an atmosphere of argon, the mixture was cooled at 0 °C and anhydrous THF (1.0 mL) was added slowly. After stirring of the mixture for 10 min, the cooling bath was removed and the tube was sealed. The mixture was then stirred at 100 °C in a pre-heated oil bath for 60 h. At this p oint, the mixture was cooled to room temperature, diluted with H2O, and extracted with CH2Cl2 (3 × 15 mL). The combined extract was washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated on a rotatory evaporator. The resulting residue was purified by gradient column chromatography (hexanes/EtOAc=9:1→8:1) on silica gel to afford the title compound.
3.2.17. cis-3,4-Dimethyl-6-nitrobicyclo[4.4.0]dec-3-en-1-carbonitrile (26)
Prepared using 11 (76.1 mg, 0.500 mmol) and 2,3-dimethyl-1,3-butadiene (410 mg, 5.00 mmol), 26 was isolated as an off-white solid (30.5 mg, 26%) after careful column chromatography. 1H NMR (CDCl3, 400 MHz, 333.15 K) δ 2.85 (d, J = 18.0 Hz, 1H), 2.76 (d of AB pattern, J = 18.8 Hz, 1H), 2.60 (d of AB pattern, J = 18.8 Hz, 1H), 2.32-2.24 (m, 2H), 2.10-2.02 (m, 2H), 1.86-1.55 (m, 5H), 1.64 (s, 6H); 13C NMR (CDCl3, 100 MHz, 333.15 K) δ 121.8, 121.4, 120.8, 90.4, 40.5, 39.7, 37.1, 33.9, 32.7, 22.0, 20.9, 18.4, 18.2; IR (KBr) 2933, 2865, 2232, 1789, 1544, 1455, 851 cm−1; HRMS (EI) exact mass calculated for [M]+ (C13H18NO2) required m/z 234.1368, found m/z 234.1383.
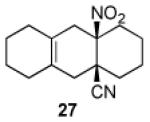
3.2.18. cis-9a-Nitro-1,2,3,4,4a,5,6,7,8,9,9a,10-dodecahydroanthracene-4a-carbonitrile (27)
Prepared using 11 (76.1 mg, 0.500 mmol) and 1,2-dimethylenecyclohexane18 (270 mg, 2.50 mmol), 27 was isolated as an off-white solid (45.6 mg, 35%) after careful column chromatography. 1H NMR (CDCl3, 400 MHz, 333.15 K) δ 2.79 (d, J = 17.6 Hz, 1H), 2.68 (d of AB pattern, J = 18.8 Hz, 1H), 2.54 (d of AB pattern, J = 18.8 Hz, 1H), 2.31-2.24 (m, 1H), 2.20 (d, J = 18.0 Hz, 1H), 2.10-2.02 (m, 2H), 1.92-1.56 (m, 13H); 13C NMR (CDCl3, 100 MHz, 333.15 K) δ 124.2, 123.1, 121.5, 90.4, 39.7, 39.4, 36.1, 34.0, 32.8, 29.7, 29.5, 29.3, 22.6, 22.1, 21.0; IR (KBr) 2930, 2236, 1789, 1546, 1439, 851 cm−1; HRMS (EI) exact mass calculated for [M+1]+ (C15H21N2O2) required m/z 261.1603, found m/z 261.1607.
3.2.19. General procedure for the Diels-Alder reaction of 2-nitrocyclopentenecarbonitriles (17) with simple dienes
A sealable vial reactor equipped with a magnetic stirring bar was charged with 2-nitrocyclopentenecarbonitriles (17, 34.5 mg, 0.250 mmol, 1.0 equiv), a simple diene (0.750 mmol, 3.0 equiv), 2,6-di-tert-butyl-4-methylphenol (2.8 mg, 13 μmol, 5.0 mol%) and anhydrous toluene (0.5 mL). The reactor was sealed and the mixture was stirred at 100 °C in a pre-heated oil bath for 24 h. At this point, the mixture was cooled to room temperature and the solvent was evaporated. The resulting residue was purified by column chromatography (hexanes/EtOAc=9:1) on silica gel to afford the title compound.
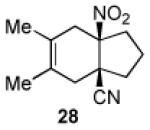
3.2.20. cis-3,4-Dimethyl-6-nitrobicyclo[4.3.0]non-3-en-1-carbonitrile (28)
Prepared using 17 (34.5 mg, 0.250 mmol) and 2,3-dimethyl-1,3-butadiene (61.6 mg, 0.750 mmol), 28 was isolated as a pale yellow oil (52.3 mg, 95%) after column chromatography. 1H NMR (600 MHz, CDCl3) δ 2.80 (d of AB pattern, J = 18.4 Hz, 1H), 2.72 (d of AB pattern, J = 18.0 Hz, 1H), 2.64-2.60 (m, 1H), 2.49 (d of AB pattern, J = 18.4 Hz, 1H), 2.35-2.31 (m, 1H), 2.28 (d of AB pattern, J = 18.0 Hz, 1H), 2.15-2.10 (m, 1H), 2.09-1.02 (m, 1H), 1.99-1.94 (m, 1H), 1.91-1.85 (m, 1H), 1.66 (s, 6H); 13C NMR (150 MHz, CDCl3) δ 121.4, 121.3, 121.2, 95.0, 43.4, 38.8, 37.6, 35.0, 34.5, 18.5, 18.3; IR (neat) 2986, 2964, 2915, 2891, 2861, 2237, 1537, 1457, 1431, 1354, 1133 cm−1; HRMS (EI) exact mass calculated for [M]+ (C12H16N2O2) required m/z 220.1212, found m/z 220.1214.
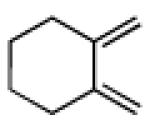
3.2.21. cis-9a-Nitro-2,3,3a,4,5,6,7,8,9,9a-decahydro-1H-cyclopenta[b]naphthalene-3a-carbonitrile (29)
Prepared using 17 (34.5 mg, 0.250 mmol) and 1,2-dimethylenecyclohexane18 (81.1 mg, 0.750 mmol), 29 was isolated as a pale yellow oil (56.0 mg, 91%) after column chromatography. 1H NMR (CDCl3, 400 MHz) δ 2.75 (d of AB pattern, J = 18.0 Hz, 1H), 2.70-2.60 (m, 2H), 2.44 (d of AB pattern, J = 18.0 Hz, 1H), 2.38-2.31 (m, 1H), 2.23 (d, J = 17.6 Hz, 1H), 2.19-1.82 (m, 8H), 1.67-1.57 (m, 4H); 13C NMR (CDCl3, 100 MHz) δ 123.6, 123.5, 121.3, 94.8, 43.2, 37.8, 36.6, 35.0, 34.5, 29.3, 29.2, 22.5, 22.5, 18.4; IR (neat) 2924, 2239, 1726, 1549, 1439, 1361, 845 cm−1; HRMS (EI) exact mass calculated for [M]+ (C14H18N2O2) required m/z 246.1368, found m/z 246.1383.
3.2.22. General procedure for the radical denitration of cis-fused decalin (26 and 27) and hydrindane (28 and 29) systems
A 15-mL Schlenk-type flask equipped with a magnetic stirring bar and a reflux condenser was charged with a cis-fused decalin or hydrindane derivative (1.0 equiv), tri-n-butyltin hydride (2.0 equiv), azobisisobutyronitrile (0.3-0.5 equiv) and anhydrous benzene. The mixture was heated under reflux for 2 h. At this point, the mixture was cooled to room temperature and the solvent was evaporated. The resulting residue was purified by gradient column chromatography (hexanes/EtOAc=1:0→97:3→19:1) on silica gel to afford the title compound.
3.2.23. trans-3,4-Dimethylbicyclo[4.4.0]dec-3-en-1-carbonitrile (30a)
Prepared using 26 (41.0 mg, 0.175 mmol), tri-n-butyltin hydride (102 mg, 0.350 mmol) and azobisisobutyronitrile (8.6 mg, 52 μmol) in benzene (1.8 mL), trans-fused 30a was isolated as a pale yellow oil (29.5 mg, 89%) after column chromatography. The minor isomer, cis-fused 30b was not detected within the limits of 1H and 13C NMR. 1H NMR (CDCl3, 400 MHz) δ 2.21 (d, J = 16.8 Hz, 1H), 2.05-1.89 (m, 3H), 1.80-1.75 (m, 1H), 1.72-1.58 (m, 3H), 1.62 (s, 6H), 1.49-1.41 (m, 1H), 1.37-1.23 (m, 4H); 13C NMR (CDCl3, 100 MHz) δ 125.8, 122.6, 121.9, 43.2 (CH2), 39.9 (CH), 39.6, 36.9 (CH2), 36.6 (CH2), 29.9 (CH2), 25.5 (CH2), 23.1 (CH2), 18.7 (CH3), 18.6 (CH3); IR (neat) 2926, 2230, 1738, 1448, 1382 cm−1; HRMS (EI) exact mass calculated for [M]+ (C13H19N) required m/z 189.1517, found m/z 189.1516.
3.2.24. trans-1,2,3,4,4a,5,6,7,8,9,9a,10-Dodecahydroanthracene-4a-carbonitrile (31a)
Prepared using 27 (21.0 mg, 80.7 μmol), tri-n-butyltin hydride (47 mg, 161 μmol) and azobisisobutyronitrile (6.6 mg, 40 μmol) in benzene (0.8 mL), trans-fused 31a was isolated as an off-white solid (14.8 mg, 85%) after column chromatography. The minor isomer, cis-fused 31b was not detected within the limits of 1H and 13C NMR. 1H NMR (CDCl3, 400 MHz) δ 2.15 (d, J = 16.8 Hz, 1H), 2.05-1.95 (m, 2H), 1.88-1.45 (m, 14H), 1.37-1.24 (m, 4H); 13C NMR (CDCl3, 100 MHz) δ 128.0, 124.3, 122.7, 42.1 (CH2), 39.8 (CH), 39.4, 36.8 (CH2), 35.7 (CH2), 30.0 (CH2), 29.7 (CH2), 29.6 (CH2), 25.5 (CH2), 23.2 (CH2), 22.8 (CH2), 22.7 (CH2); IR (KBr) 2925, 2830, 2228, 1738, 1449, 1320 cm−1; HRMS (EI) exact mass calculated for [M]+ (C15H21N) required m/z 215.1674, found m/z 215.1658.
3.2.25. A 1.5:1 mixture of trans-3,4-dimethylbicyclo[4.3.0]non-3-enecarbonitrile (32a) and cis-3,4-dimethylbicyclo[4.3.0]non-3-enecarbonitrile (32b)
Prepared using 28 (30.8 mg, 0.140 mmol), tri-n-butyltin hydride (81.5 mg, 0.280 mmol) and azobisisobutyronitrile (11.5 mg, 70.0 μmol) in benzene (1.4 mL), an inseparable 1.5:1 mixture of trans-fused 32a and cis-fused 32b was isolated as an pale yellow oil (18.9 mg, 77%) after column chromatography. The relative stereochemistry of the major isomers was assigned according to Casadevall's observation and our previous study.2 Diastereomeric ratios were estimated by the integration of the methine carbon signals. 1H and 13C NMR spectra of the mixture of 32a and 32b are available in the supplementary data. IR (neat) 2959, 2914, 2836, 2231, 1727, 1452, 1275, 1124, 1073 cm−1; HRMS (EI) exact mass calculated for [M]+ (C12H17N) required m/z 175.1361, found m/z 175.1358.
3.2.26. A 1.7:1 mixture of trans-2,3,3a,4,5,6,7,8,9,9a-decahydro-1H-cyclopenta[b]naphthalene-3a-carbonitrile (33a) and cis-2,3,3a,4,5,6,7,8,9,9a-decahydro-1H-cyclopenta[b]naphthalene-3a-carbonitrile (33b)
Prepared using 29 (34.5 mg, 0.140 mmol), tri-n-butyltin hydride (81.5 mg, 0.280 mmol) and azobisisobutyronitrile (11.5 mg, 70.0 μmol) in benzene (1.4 mL), an inseparable 1.7:1 mixture of trans-fused 33a and cis-fused 33b was isolated as an pale yellow oil (25.2 mg, 89%) after column chromatography. The relative stereochemistry of the major isomers was assigned according to Casadevall's observation and our previous study.2 Diastereomeric ratios were estimated by the integration of the methine carbon signals. 1H and 13C NMR spectra of the mixture of 32a and 32b are available in the supplementary data. IR (neat) 2927, 2833, 2230, 1729, 1440, 1312 cm−1; HRMS (EI) exact mass calculated for [M]+ (C14H19N) required m/z 201.1517, found m/z 201.1523.
3.2.27. General procedure for the preparation of cis-fused angular amine derivatives
To a stirred solution of a cis-fused decalin or hydrindane (1.0 equiv) in a mixture of glacial acetic acid and THF (1:2, v/v) at −20 °C was added Zn dust (10 equiv). After stirring for 3 h, the mixture was filtered through a pad of Celite® and the filtrate was concentrated on a rotatory evaporator. The resulting residue was dissolved in THF and the mixture was treated successively with a saturated solution of NaHCO3 and allyl chloroformate (2.5 equiv). The mixture was stirred for 3 h, diluted with H2O, and extracted with EtOAc (3 × 10 mL). The combined extract was washed with brine, dried over anhydrous MgSO4, filtered, and concentrated on a rotatory evaporator. The resulting residue was purified by gradient column chromatography on silica gel to afford the title compound.
3.2.28. cis-Allyl 8a-cyano-6,7-dimethyl-1,2,3,4,4a,5,8,8a-octahydronaphthalen-4a-ylcarbamate (34)
Prepared using 26 (20.0 mg, 85.4 μmol), Zn dust (55.8 mg, 854 μmol) and allyl chloroformate (25.7 mg, 214 μmol), 34 was isolated as a pale yellow oil (10 mg, 40%) after column chromatography. 1H NMR (600 MHz, CDCl3) δ 5.95-5.88 (m, 1H), 5.32-5.28 (m, 1H), 5.23-5.21 (m, 1H), 4.92 (s, 1H), 4.54-4.48 (m, 2H), 2.67 (d of AB pattern, J = 17.9 Hz, 1H), 2.50-2.42 (m, 2H), 2.25 (d of AB pattern, J = 17.9 Hz, 1H), 2.12-1.98 (m, 2H), 1.81-1.77 (m, 1H), 1.72-1.62 (m, 4H), 1.63 (s, 3H), 1.62 (s, 3H), 1.52-1.47 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 154.4, 132.8, 123.2, 126.7, 119.3, 117.8, 65.3, 55.0, 41.5, 38.5, 36.2, 31.3, 30.2, 29.7, 21.2, 18.7, 18.3; IR (neat) 3346, 2933, 2865, 2232, 1724, 1509, 1450, 1294, 1231, 1111, 1046, 985 cm−1; HRMS (FAB) exact mass calculated for [M+1]+ (C17H25N2O2) required m/z 289.1916, found m/z 289.1919.
3.2.29. cis-Allyl 7a-cyano-5,6-dimethyl-2,3,3a,4,7,7a-hexahydro-1H-inden-3a-ylcarbamate (35)
Prepared using 28 (20.0 mg, 90.8 μmol), Zn dust (59.4 mg, 908 μmol) and allyl chloroformate (27.4 mg, 227 μmol), 35 was isolated as a pale yellow oil (13 mg, 52%) after column chromatography. 1H NMR (600 MHz, CDCl3) δ 5.95-5.89 (m, 1H), 5.33-5.30 (m, 1H), 5.23 (dd, J = 10.4, 1.3 Hz, 1H), 5.01 (s, 1H), 4.54 (d, J = 5.5 Hz, 2H), 2.50-2.39 (m, 4H), 2.25-2.16 (m, 2H), 2.05-1.91 (m, 2H), 1.88-1.75 (m, 2H), 1.63 (s, 3H), 1.62 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 155.0, 132.6, 123.9, 122.7, 119.8, 118.0, 65.5, 62.5, 45.2, 37.7, 37.6, 34.7, 29.7, 19.3, 18.8, 18.4; IR (neat) 3332, 2916, 2886, 2849, 2233, 1724, 1510, 1445, 1274, 1238, 1086, 916 cm−1; HRMS (FAB) exact mass calculated for [M+1]+ (C16H23N2O2) required m/z 275.1760, found m/z 275.1772.
Supplementary Material
Scheme 4.

aKey: (a) 2,6-di-tert-butyl-4-methylphenol, toluene, 130 °C, 24 h. (b) 2,6-di-tert-butyl-4-methylphenol, toluene, 150 °C, 24 h.
Acknowledgments
Support was provided by the NIH (HL25848 to SJD). W.H.K. is grateful for a Korea Research Foundation Grant funded by the Korean government (KRF-2007-357-c00060). We thank Rebecca Wilson for assistance with the preparation of the manuscript and Dr. Peter K. Park (Columbia University), Dr. Bernhard Fasching (MSKCC), Dr. Jennifer Stockdill (MSKCC) and Dr. Pavel Nagorny (MSKCC) for helpful discussions. We also thank Dr. George Sukenick, Ms. Hui Fang, Sylvi Rusli (NMR Core Facility, Sloan-Kettering Institute) and Dr. Yasuhiro Itagaki (Mass Spectral Core Facility, Columbia University) for mass spectral and NMR.
We dedicate this paper to Professor Steven Ley on the occasion of his receipt of the Tetrahedron Prize for Creativity in Organic Chemistry, honoring his pioneering accomplishments in organic synthesis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
1H and 13C NMR spectra of all new compounds are available. Crystallographic data for the structures reported in this article have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC-763857 (13), CCDC-763854 (14), CCDC-763855 (19), and CCDC-763856 (25). Copies of the data can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK [Tel: (+44)1223 336 408, Fax: (+44)1223 336 033 or deposit@ccdc.cam.ac.uk].
References and notes
- 1.For recent reviews, see: Trost BM, Jiang C. Synthesis. 2006:369–396. Douglas C, Overman LE. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5363–5367. doi: 10.1073/pnas.0307113101. Corey EJ, Guzman-Perez A. Angew. Chem. Int. Ed. 1998;37:388–401. doi: 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V.
- 2.Kim WH, Lee JH, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:12576–12578. doi: 10.1021/ja9058926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For examples of Diels-Alder reactions of cyclohexenecarboxaldehyde or its corresponding acetal to obtain cis-fused bicyclic ring system with all-carbon quaternary center on the bridgehead, see: Szmuszkowicz J, Bergmann ED. Bull. Res. Counc. Israel. 1953;3:93–95. Bergmann ED, Becker A. J. Am. Chem. Soc. 1959;81:221–225. Lee JH, Kim WH, Danishefsky SJ. Tetrahedron Lett. 2010;51:1252–1253. doi: 10.1016/j.tetlet.2009.12.127.
- 4.Most examples of tetrasubstituted cyclic dienophiles are limited to quinone and maleic anhydride derivatives. For examples, see: Kwon O, Park SB, Schreiber SL. J. Am. Chem. Soc. 2002;124:13402–13404. doi: 10.1021/ja028086e. Nicolaou KC, Vassilikogiannakis G, Mägerlein W, Kranich R. Angew. Chem. Int. Ed. 2001;40:2482–2486. doi: 10.1002/1521-3773(20010702)40:13<2482::AID-ANIE2482>3.0.CO;2-A.
- 5.(a) Fuji K, Tanaka K, Abe H, Matsumoto K, Taga T, Miwa Y. Tetrahedron: Asymmetry. 1992;3:609–612. [Google Scholar]; (b) Piaz VD, Giovannoni MP, Ciciani G, Giomi D, Nesi R. Tetrahedron Lett. 1993;34:161–162. [Google Scholar]; (c) Nesi R, Giomi D, Papaleo S, Corti M. J. Org. Chem. 1990;55:1227–1230. [Google Scholar]
- 6.Our efforts to accomplish the Diels-Alder reaction of methyl 2-nitrocyclohexenecarboxylate (i.e. 5, A = CO2Me) with a range of dienes were unsucessful.
- 7.Jayakanthan K, Madhusudanan KP, Vankar YD. Tetrahedron. 2004;60:397–403. [Google Scholar]
- 8.Danishefsky S, Kitahara T. J. Am. Chem. Soc. 1974;96:7807–7808. [Google Scholar]
- 9.Ono N, Miyake H, Kamimura A, Hamamoto I, Tamura R, Kaji A. Tetrahedron. 1985;41:4013. [Google Scholar]
- 10.Jung ME, McComb CA. Tetrahedron Lett. 1976;17:2935–2938. [Google Scholar]
- 11.The relative stereochemistry of 24 was confirmed by its preparation through the catalytic hydrogenation of cis-fused 20 (H2, Pd(OH)2, CH3OH, room temperature, 2 h, 94%). For details, see the experimental section.
-
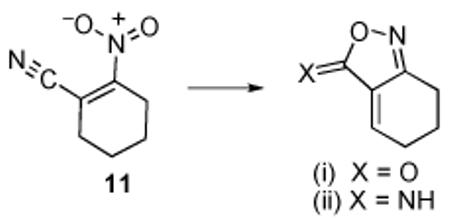
12.The operative dienophile formally corresponds to either (i) or (ii), in principle derivable from the thermolysis of 11 via cyclization, tautomerization, and dehydration in a sequence which has not been defined.
- 13.(a) Koprowski M, Skowrońska A, Glówka ML, Fruzi ński A. Tetrahedron. 2007;63:1211–1228. [Google Scholar]; (b) Grieco PA, Nunes JJ, Gaul MD. J. Am. Chem. Soc. 1990;112:4595–4596. [Google Scholar]
- 14.Comparison with authentic sample of the cis-isomers indicates the presence of only the trans-isomers within the limits of 1H and 13C NMR. For the analytical data of the cis-isomers, see reference iiic. In accord with Casadevall's observation, the ring junction carbons of trans-fused 30a and 31a are more deshielded than those of cis-fused 30b and 31b: Metzger P, Casadevall E, Pouet MJ. Org. Magn. Reson. 1982;19:229–234.
- 15.Hydrindanes 30 and 32 were obtained as inseparable mixtures of the cis- and trans-isomers. The relative stereochemistry of the major isomers was assigned according to Casadevall's observation and our previous study.2 Diastereomeric ratios were estimated by the integration of the methine carbon signals.
- 16.Corey EJ, Estreicher HJ. J. Am. Chem. Soc. 1978;100:6294–6295. [Google Scholar]
- 17.Still WC, Kahn M, Mitra A. J. Org. Chem. 1978;43:2923–2925. [Google Scholar]
- 18.(a) Arenz T, Vostell M, Frauenrath H. Synlett. 1991:23–24. [Google Scholar]; (b) Block E, Aslam M. Org. Synth. 1987;65:90–97. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.