

Introduction
Cryopreservation is the process of preserving the viability of cells and tissues by freezing and storing them at the subzero temperatures below which biochemical reactions do not occur [20]. During freezing to such low temperatures cells can be subjected to damage, termed freezing injury. There are two main causes of freezing injury. The first one takes place when cells are cooled slowly. In this case, the formation of ice crystals in the extracellular space causes the osmotic efflux of water from the cell. Osmotic efflux of water leads to the increased concentration of intracellular solutes, causing potential cell damage due to solute toxicity. The other general cause of freezing injury takes place when cells are cooled rapidly. During rapid cooling there is not enough time for water to leave the cell, which leads to the supercooling of cell cytoplasm and the formation of ice crystals inside the cell [18].
To achieve successful cryopreservation of cells and tissues, efforts have typically targeted avoidance of the two aforementioned causes of freezing injury. However, the traditional concept of damage by intracellular ice has recently been challenged; it was shown that intracellular ice formation (IIF) leads to lethal injury of cells in suspension. However, in a confluent cell monolayer – a two dimensional structure formed when cells grow close to each other - IIF can be innocuous [1,5–7]. The phenomenon of innocuous IIF was explained by the propagation of ice between cells through intercellular connections, called gap junctions. This does not cause cell membrane rupture and can protect cells from excessive dehydration during freezing, resulting in improved post-thaw recovery [3,5]. Gap junctions have been proposed to facilitate the exchange of ions and small molecules between adjacent cells [8] and also allow for intercellular ice propagation [3].
Human dental pulp stem cells (DPSCs) are mesenchymal stem cells derived from the dental pulp of extracted third molars. DPSCs have the unique ability to differentiate into various mesodermal tissue types, including bone, muscle, cartilage and dentin. Being a surgical waste, these cells are of interest to many researchers and clinicians due to their easy isolation, ability to reproduce rapidly in culture and also their many potential uses in cell-mediated therapies and tissue engineering applications [21]. To ensure a constant clinical availability of DPSCs, cryopreservation of these cells is needed. Currently DPSC suspensions are preserved using dimethyl sulfoxide (Me2SO) as a cryoprotectant [21]. As DPSCs become sought after for clinical tissue engineering strategies, freezing in suspension may not be ideal, and preservation in more ready-to-use configurations such as two and even three dimensional constructs may be required. Additionally, Me2SO toxicity can reduce the effectiveness of transplantation procedures, an effect which may be exacerbated in scaled-up engineered tissue grafts. Together this creates the need for alternative cryopreservation strategies.
The objective of this study was to examine the effects of IIF on post-thaw viability of both confluent monolayers and suspensions of DPSCs. In this study, it was hypothesized that innocuous IIF in confluent monolayers of DPSCs protects them against freezing injury during cryopreservation. If true, it is anticipated that future efforts will then be able to use these data to scale up the clinical banking of simple engineered tissues without the need for potentially detrimental cryoprotective additives.
Materials and Methods
Cell culture
The primary culture of human DPSCs (General Biotechnology LLC, Indianapolis, IN, USA), derived from dental pulp of extracted third molars, was used as the experimental material. Human DPSCs were cultured using Dulbecco’s modified Eagle’s Medium (DMEM) supplemented with 18% fetal bovine serum (FBS) [15,17,24] in 75 cm2 treated tissue culture flasks in a Steri-Cycle CO2 incubator at 37°C and 5% carbon dioxide level [10,13] (DMEM from GIBCO laboratories, Grand island, NY, USA or HyClone, Logan, UT, USA; FBS from Invitrogen Canada Inc., Burlington, ON, Canada or HyClone, Logan, UT, USA; tissue culture flasks from BD Biosciences, Bedford, MA, USA; Steri-Cycle CO2 incubator from Thermo Electron Corporation, Marietta, OH, USA). DPSCs were passaged every three to four days by exposing them twice to warm 0.25% trypsin-EDTA solution (purchased from Invitrogen, Grand Island, NY, USA or HyClone, Logan, UT, USA) [21], then incubating them at 37°C for five minutes. DPSCs were re-suspended in 5 ml of culture medium and 0.5–1×106 cells were seeded per flask containing 15 ml DMEM.
To obtain cell suspensions, cells were dislodged from a flask, centrifuged at 4°C, 200 g for 10 minutes and the cell pellet re-suspended in fresh DMEM to the required cell concentration. Confluent cell monolayers were obtained by seeding 1×106 or 0.5×106 cells in a 60×15 mm Petri dish, on sterile 12 mm circular glass coverslips, covered with 5 ml of medium (seeding density was approximately 350 cells/mm2) (Petri dishes from Becton Dickinson Labware, Franklin Lakes, NJ, USA; circular glass coverslips from VWR Scientific, West Chester, PA, USA). Petri dishes were incubated for either two days (if 1×106 cells were seeded) or three days (if 0.5×106 cells were seeded), for cells to grow to full confluence. Cells from passage 4 to passage 14 were used in the experiments described in this study.
Connexin-43 expression
The expression of gap junction protein Cx43 was assessed according to the previously described method [22], with modifications. Each coverslip with DPSCs was fixed with 200 μL Phosphate Buffered Saline (PBS) - 1% formaldehyde at 4°C for 30 minutes and permeabilized with 200 μL PBS-0.1% Triton X-100 at 4°C for 30 minutes (PBS from HyClone, Road Logan, Utah; formaldehyde from VWR Scientific, West Chester, PA, USA; Triton X-100 from Sigma-Aldrich, Oakville, ON, Canada). A separate treatment group included samples that were not permeabilized to examine the surface expression of Cx43. Coverslips were stained with 200 μL of rabbit anti-Cx43 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA), diluted 1:200 in PBS-1% FBS, at 4°C for 24 hours. Coverslips were then washed three times with PBS-1% FBS and stained with 200 μL Alexa Fluor - conjugated anti-rabbit immunoglobulin G (IgG) (Molecular Probes, Eugene, OR, USA), diluted 1:100 in PBS-1% FBS, at 4°C for one hour. After three washes with PBS-1% FBS, monolayers were incubated with 20 μL of 12.5 μM SYTO-13 (excitation 490 nm, emission 510 nm) (Molecular Probes, Eugene, OR, USA), diluted in PBS-1% FBS, at room temperature for four minutes. After three washes with PBS-1% FBS, monolayers were examined under an Eclipse TE 2000-4 microscope for red Alexa Fluor fluorescence (excitation 555 nm, emission 565 nm), using NIS-elements imaging software (microscope and imaging software from Nikon, Japan). To exclude the non-specific binding of Alexa Fluor - conjugated anti-rabbit IgG, control coverslips were stained in the fashion described above; however, without addition of rabbit anti-Cx43 antibodies, and examined for Alexa Fluor fluorescence. The filter set used for the SYTO-13 analysis was HQ480/40X EX, Q505LP BS, HQ535/50m EM (41001, Chroma Technology Corp., Bellows Falls, VT), and for Alexa Fluor analysis the filter set was HQ535/50x EX, Q565LP BS, HQ610/75m EM (41007, Chroma Technology Corp.).
Assessment of the incidence of IIF
On the cryostage, immediately after extracellular ice nucleation, we have assessed the incidence of IIF in DPSC suspensions, based upon the sudden darkening of cell cytoplasm. This darkening is triggered by light scattering across intracellular ice crystals, known as “flashing” (Figure 2) [19]. DPSC monolayers were stained with 12.5 μM SYTO-13 prior to freezing. Immediately after extracellular ice nucleation, the incidence of IIF was evaluated based upon the disruption of fluorescent staining of cytoplasm by ice crystals. This disruption resulted in the formation of the intracellular “honeycomb” pattern (Figure 1) [2]. IIF detection using cryomicroscopy under fluorescent illumination was first described in 1965 by Lozina-Lozinsky, and subsequently in many other publications [7,19], and has proven to bean effective method to assess IIF. Standard “flashing” technique is very hard to use in confluent cell monolayers, because cells are in such close contact with each other that it is difficult to distinguish the borders of each individual cell, especially under bright-field illumination. That is why assessment of IIF based on intracellular “honeycomb” pattern has been used for monolayers. It was shown that at low temperatures, “flashing” technique tends to underestimate the incidence of IIF in monolayers and “honeycomb” method gives more accurate results [2].
Figure 2.
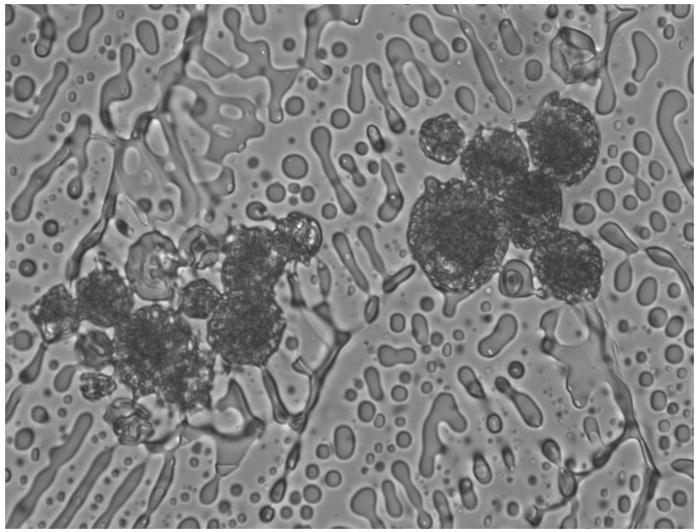
Flashing in DPSC suspension upon intracellular freezing.
Figure 1.
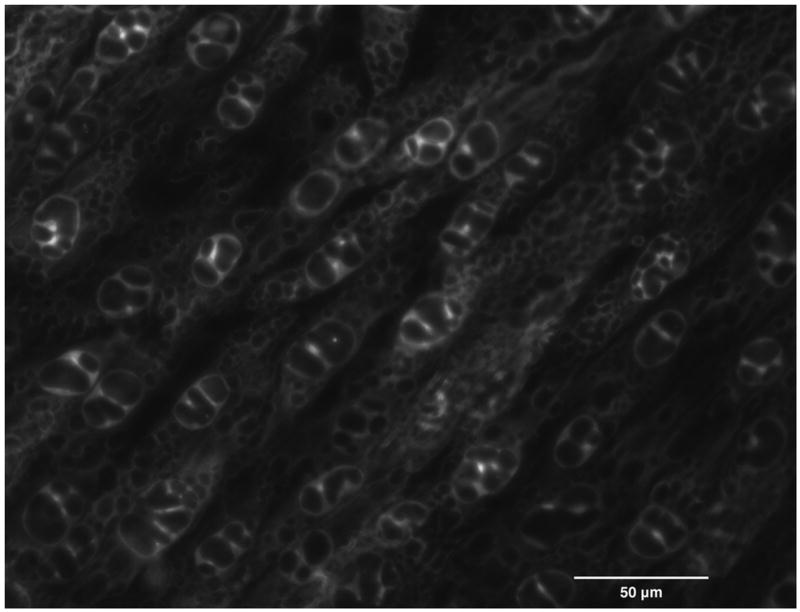
The formation of a honeycomb pattern in the confluent monolayer of DPSCs upon intracellular freezing.
In the methanol bath, the incidence of IIF was assessed, post thaw, using the fixation technique [2]. Prior to freezing, monolayers were stained with 20 μL of 12.5 μM SYTO-13 and left to incubate at room temperature for four minutes. After extracellular ice nucleation was induced, samples were allowed to equilibrate for five minutes at the nucleation temperature. A volume of 800 μL of fixative solution, containing 85 parts of denatured ethanol, 10 parts of 36.5–38.0 % formaldehyde solution and five parts of glacial acetic acid, pre-cooled to the nucleation temperature, was added to the samples to fix the “honeycomb” pattern in place, inside the cells (denatured ethanol from VWR, Brampton, ON, Canada; glacial acetic acid from EM Science, Darmstadt, Germany). Samples were left to equilibrate with the fixative solution at the nucleation temperature for 20 minutes before being thawed in a 37°C water bath. The optimal volume of fixative and equilibration time for fixing the “honeycomb” pattern inside the cells were chosen based upon our previous experiments (data not shown). The presence of fixed “honeycomb” patterns inside the cells was assessed under fluorescent illumination to quantify the incidence of IIF. The filter set used for SYTO-13 analysis was HQ480/40X EX, Q505LP BS, HQ535/50m EM.
In both cases, the incidence of IIF was calculated according to the formula:
| (Eq. 1) |
Assessment of membrane integrity
To assess membrane integrity, DPSC suspensions and confluent DPSC monolayers were stained with vital dyes 12.5 μM SYTO-13 and 2.5 μM ethidium bromide (EB) (Molecular Probes, Eugene, OR, USA) and examined under fluorescent illumination. The filter set used for SYTO-13 analysis was HQ480/40X EX, Q505LP BS, HQ535/50m EM, and for EB analysis the filter set was HQ535/50x EX, Q565LP BS, HQ610/75m EM. On the cryostage, the incidence of membrane damage was calculated as the percentage of cells that underwent membrane damage as a result of freezing compared to the total number of intact cells before freezing. The following formula allowed us to correct for cells with damaged membranes, that are present in the sample before freezing:
| (Eq. 2) |
In the methanol bath, fluorescent images of the samples were not taken before freezing, so there was no opportunity to correct for cells with damaged membranes present in the sample before freezing. The incidence of membrane damage in the methanol bath was calculated as the percentage of cells with damaged membrane post-thaw relative to the total number of cells in the sample:
| (Eq. 3) |
For DPSC suspensions cryopreserved with 10% Me2SO according to the standard protocol, membrane integrity was calculated as the percentage of cells with intact membranes, post-thaw, relative to the total number of cells in a sample:
| (Eq. 4) |
Colony forming assay
DPSC monolayers on coverslips were exposed to 20 μL of warm cell dissociation buffer (GIBCO laboratories, Grand island, NY, USA) at 37°C until cells detached. Cells were then washed from the coverslip using 0.5 ml of fresh medium. An aliquot of the resultant cell suspension was stained with a 0.4% aqueous solution of Trypan Blue (Sigma-Aldrich, Oakville, ON, Canada) at an aliquot to Trypan Blue volumetric ratio of 2:1 for 5–10 minutes and the number of intact cells in a given volume counted using a hemacytometer.
DPSC suspensions cryopreserved with 10% Me2SO, according to the standard protocol, were centrifuged, the supernatant containing Me2SO aspirated, and the resultant cell pellet re-suspended in 1 ml of fresh medium. Cells were counted and the cell suspension was diluted to a final concentration of 0.1×106 cells per ml.
Either 150 cells or 750 cells (for monolayers or for suspensions cryopreserved with Me2SO according to the standard protocol, respectively) were seeded into 25 cm2 tissue culture flasks (Becton Dickinson, Oxford, England) containing 5 ml of medium, and incubated at 37°C, 5% carbon dioxide for five days. On day 5, the medium was removed and cells were fixed with 1 ml of 70% isopropanol (Ricca Chemical Company, Arlington, TX, USA) and incubated at room temperature for two minutes. Isopropanol was removed, cells were stained with 0.25 ml of 0.4% Trypan Blue and rinsed with distilled water [6]. The number of colonies was quantified using light microscopy, and the survival percentage was calculated as follows:
| (Eq. 5) |
Overall study design
Two human DPSC model systems were used: single cells in suspension and confluent cell monolayers. DPSCs (0.5×106 cells) were seeded into 60×15 mm Petri dishes on sterile 12 mm circular glass coverslips and cultured for five days. The expression of gap junction protein Cx43, both within the cytoplasm and on the cell surface, was monitored daily, for five days, as previously described (see Connexin-43 expression section). The DPSCs were then cooled either on a cryostage or in a methanol bath using different freezing profiles. Specifically, for the low temperature microscope stage, DPSC suspensions and confluent monolayers were cooled to different subzero temperatures, where extracellular ice formation was induced by touching the samples with a cold metal probe (isothermal nucleation). Alternately, in a methanol bath, DPSC monolayers were either subjected to isothermal nucleation at various subzero temperatures or cooled continuously, to examine the effects of the kinetics of intracellular freezing. The incidence of IIF and corresponding post-thaw cell viability (membrane integrity for cryostage experiments; membrane integrity and colony forming ability for methanol bath experiments) were assessed for each freezing profile. In addition, cell loss was calculated during cryopreservation.
For comparison, a separate experiment was performed, where DPSC suspensions were cryopreserved according to the standard method using 10% Me2SO [21] and their viability (membrane integrity and colony forming ability) was measured post thaw.
Standard cryopreservation procedure for DPSC suspensions
For this procedure, 0.7 ml of Me2SO (Cryoserv, Edwards Lifesciences Research Medical Inc., Bedford, OH, USA, purity ≥ 99%)was added dropwise to 6×106 DPSCs suspended in 6 ml of DMEM to form a suspension of cells in 10% (v/v) Me2SO in DMEM. After 20 minutes of incubation on wet ice, 1 ml of cell suspension was transferred into a 1.8 ml sterile cryogenic vial (NUNC, Roskilde, Denmark) and subjected to cooling at approximately 1–3°C/min [16] in a −80°C freezer. Samples were left in the freezer for 48 hours before thawing. Vials were subsequently thawed in a 37°C circulating water bath until the first liquid phase appeared. To remove Me2SO, samples were centrifuged, supernatant containing Me2SO aspirated, and the cell pellet re-suspended in 1 ml of fresh medium. Immediately post-thaw, and one hour after Me2SO was removed, samples were stained with vital dyes 12.5 μM SYTO-13 and 2.5 μM EB to assess membrane integrity (see Assessment of membrane integrity section). Membrane integrity was compared to that of fresh control samples and also to that of unfrozen samples incubated with Me2SO for 20 minutes. A colony forming assay was set up immediately after Me2SO was removed, as described previously (see Colony forming assay section) and compared to that of fresh control suspensions.
Freezing procedure on the cryostage
Freezing profiles were applied using a cryomicroscope system. The system consisted of an ECLIPSE 80i microscope, an ORCA-ER digital camera, an FDCS 196 cryostage and a TMS 94 programmer and was operated using Linksys 32 software (ECLIPSE 80i microscope from Nikon, Japan; ORCA-ER digital camera from Hamamatsu, Japan; FDCS 196 cryostage and a TMS 94 programmer both from Linkam Scientific Instruments Ltd., England; Linksys 32 software from Linkam Scientific Instruments Ltd., Tadworth, Surrey, United Kingdom). No cryoprotectant was added to DPSCs in these freezing experiments. Prior to freezing, DPSC monolayers and suspensions were stained with 12.5 μM SYTO-13 and 2.5 μM EB. For monolayers, coverslips were placed on the cryostage crucible monolayer cell-side down. Alternatively, for suspensions, 2 μL of cell suspension at a concentration of 6–12×106 cells/ml were placed on the cryostage crucible and covered with a 12 mm circular glass coverslip.
Samples were cooled at 25°C/min to a final isothermal nucleation temperature (from −2°C to −13°C), which resulted in various degrees of supercooling of cells (from 1.4°C to 12.4°C) [23]. To induce extracellular ice formation, the crucible with coverslip was touched with a copper probe pre-cooled in liquid nitrogen. Samples were held at the nucleation temperature for several minutes, then warmed at 25°C/min to 22°C and held at 22°C for one minute to allow EB penetration into those cells possessing damaged membranes. Observations were made at three points in time: before ice nucleation, immediately after ice nucleation, and after thawing. Bright-field and fluorescent images were taken using NIS-elements imaging software. The incidence of IIF was assessed immediately after extracellular ice nucleation, as described previously (see Assessment of the incidence of IIF section). The extracellular ice nucleation temperature, at which 100% IIF was observed, was used in subsequent experiments to examine the effect of 100% IIF on post-thaw cell viability. The incidence of membrane damage was assessed immediately after thawing as described earlier (see Assessment of membrane integrity section).
Freezing procedure in a methanol bath
DPSC monolayers on glass coverslips were placed at the bottom of 15×45 mm glass vials (Kimble Glass Inc., Vineland, NJ, USA), covered with 20 μL of 12.5 μM SYTO-13, and left to incubate at room temperature for four minutes. Samples were then transferred into a methanol bath (FTS Systems, Stone Ridge, NY, USA). The temperature during cooling was monitored by a thermocouple placed in an adjacent fluid control (glass vial with 20 μL of medium), and recorded using Daq View software (Omega, Laval, Quebec, Canada).
In order to assess the incidence of IIF as a function of extracellular ice nucleation temperature, confluent monolayers were cooled at 0.75°C/min to different subzero temperatures (−4°C to −8°C) and allowed to equilibrate at these temperatures for five minutes. Extracellular ice nucleation was induced by touching a vial with forceps cooled in liquid nitrogen. After that the intracellular “honeycomb” pattern was fixed and the incidence of IIF was assessed, post-thaw, according to the previously described procedure (see Assessment of the incidence of IIF section). As with cryostage freezing, the extracellular ice nucleation temperature at which 100% IIF was observed was used in subsequent experiments to examine the effect of 100% IIF on cell viability, post-thaw.
A different experiment was performed, in the absence of fixative solution, to assess cell viability after 100% IIF. Here no pre-freeze staining with SYTO-13 was performed, and monolayers were covered with 20 μL of medium instead. During the assessment of the incidence of IIF in a methanol bath, we determined that 100% IIF occurs upon extracellular ice nucleation at −7°C (see Results, The incidence of IIF). Based on this data, confluent monolayers were divided into four treatment groups and cooled according to different freezing profiles. Group 1 was placed in a methanol bath at −7°C, extracellular ice was nucleated at −7°C and samples were subsequently thawed at 37°C water bath to examine the effect of 100% IIF on cell viability. Group 2 was placed in a methanol bath at −7°C, extracellular ice was nucleated at −7°C and samples were cooled at approximately 90°C/min to −78.5°C (plunged into a mixture of dry ice and methanol). Group 3 was placed in a methanol bath at −7°C, extracellular ice was nucleated at −7°C, samples were cooled at 0.75°C/min to −30°C and then at approximately 90°C/min to −78.5°C. Groups 2 and 3 were designed to investigate the effect of cooling rate, after IIF, on cell viability. Group 4 was placed in a methanol bath at −7°C, cooled at 0.75°C/min to −30°C and then at approximately 90°C/min to −78.5°C. Group 4 was designed to investigate the effect of continuous cooling, without direct induction of extracellular ice, on cell viability. Samples of Groups 2, 3 and 4 were transferred into a −80°C freezer (Ultima II, Kendro Laboratory Products, Asheville, NC, USA) for 48 hours and then thawed in a 37°C water bath. Either immediately post-thaw, or one hour post-thaw, coverslips were stained with vital dyes 12.5 μM SYTO-13 and 2.5 μM EB to assess membrane integrity (see Assessment of membrane integrity section), which was compared to the membrane integrity of fresh control samples. The colony forming assay was performed immediately post-thaw, as previously described (see Colony forming assay section), and monolayers incubated at room temperature during the course of freeze-thaw procedure served as the control.
Cell loss during cryopreservation processing
Cell loss can occur at different stages during the pre- and post-cryopreservation processing of DPSC monolayers and was characterized at three different points. We calculated the number of DPSCs attached to a coverslip in untreated control samples, after control incubation at room temperature, and also after nucleation at −7°C and thawing. We also calculated the number of intact DPSCs that were harvested from coverslips both after control incubation at room temperature and after nucleation at −7°C and thawing, as well as their colony forming ability at both stages. Each coverslip was stained with 12.5 μM SYTO-13 and the images of 5 random fields of view were taken to determine the mean number of cells per unit area. The filter set used for the SYTO-13 analysis was HQ480/40X EX, Q505LP BS, HQ535/50m EM. The surface areas of captured fields of view were measured, and, knowing the surface area of the coverslip, we calculated the number of cells present on each coverslip. The number of intact DPSCs harvested from each coverslip was counted and their colony forming ability was determined as previously described (see Colony forming assay section).
Statistical analysis
Experimental data are expressed as mean±SEM. Linear regression analysis was performed by the method of least squares, using Excel software. The differences between experimental groups were determined using the Mann-Whitney test. The level of significance was set at 5%.
Results
Viability of DPSC suspensions after standard cryopreservation procedure
After cryopreservation by the standard procedure, where DPSC suspenions were incubated with 10% Me2SO and cooled slowly by being placed in −80°C freezer, membrane integrity was 95.0±1.9% immediately post-thaw, compared to 98.5±1.2% membrane integrity of unfrozen control samples, incubated with 10% Me2SO, on ice, for 20 minutes (p=0.644). After Me2SO was removed and samples were incubated at 37°C for one hour, membrane integrity was reduced to 93.0±2.1% (p=0.027 vs unfrozen Me2SO control, Figure 3). An alternative measure of viability, the colony forming ability of cryopreserved DPSC suspensions was 85.3±10.2%.
Figure 3.
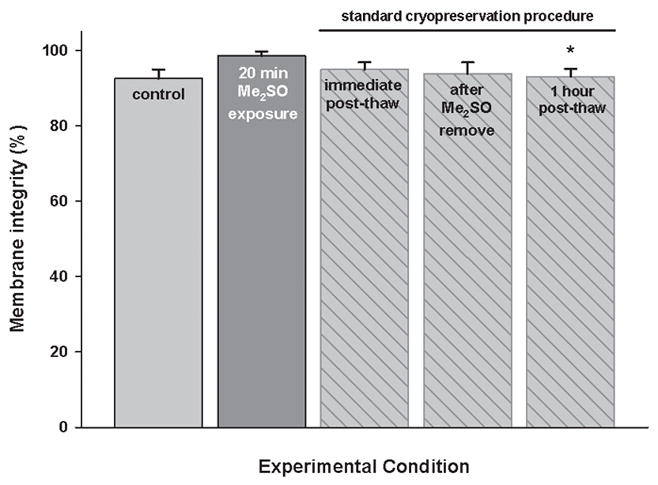
Membrane integrity of DPSC suspensions after the standard cryopreservation procedure, according to membrane integrity assay. Controls are untreated suspensions or suspensions incubated with Me2SO for 20 minutes. Mean ± SEM (n = 3). * p < 0.05 vs. unfrozen control samples, incubated with 10% Me2SO, on ice, for 20 minutes (Mann-Whitney test).
Connexin-43 expression
On the first day of culture, random sites of Cx43 fluorescence were seen in the cytoplasm of single attached cells (Figure 4A). On day 2, when DPSCs reached confluence on coverslips, the sites of Cx43 fluorescence were randomly distributed in the cytoplasms of cells all over the monolayer (Figure 4B), however, with no particular pattern observed between the cells. Cx43 fluorescence progressively increased with time of culture, reaching a maximum on day 4 (Figure 4C). Similarly, on the cell surface, random sites of Cx43 expression were first observed on day 2 and progressively increased in number with time of culture, again with no intercellular pattern observed (Figure 4D). No nonspecific binding of Alexa Fluor - conjugated anti-rabbit IgG was observed in control samples, which demonstrated the specificity of fluorescent staining for Cx43.
Figure 4.
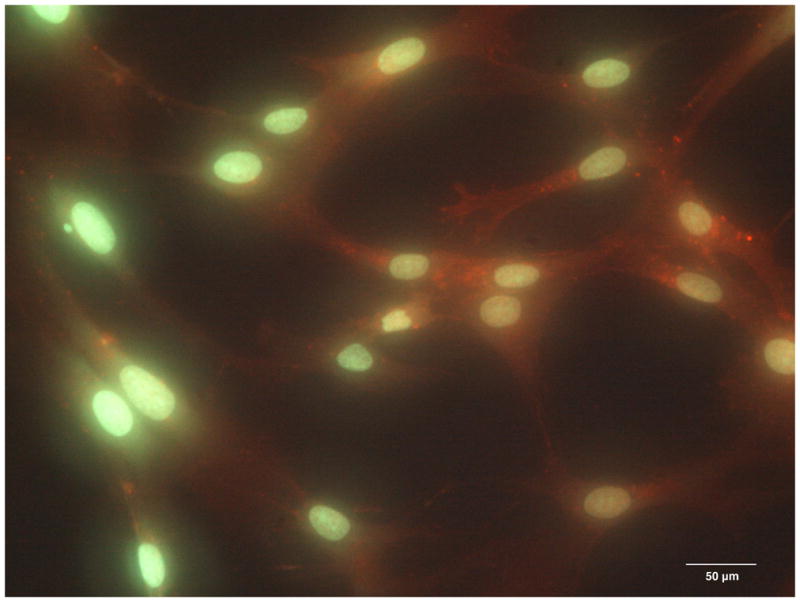
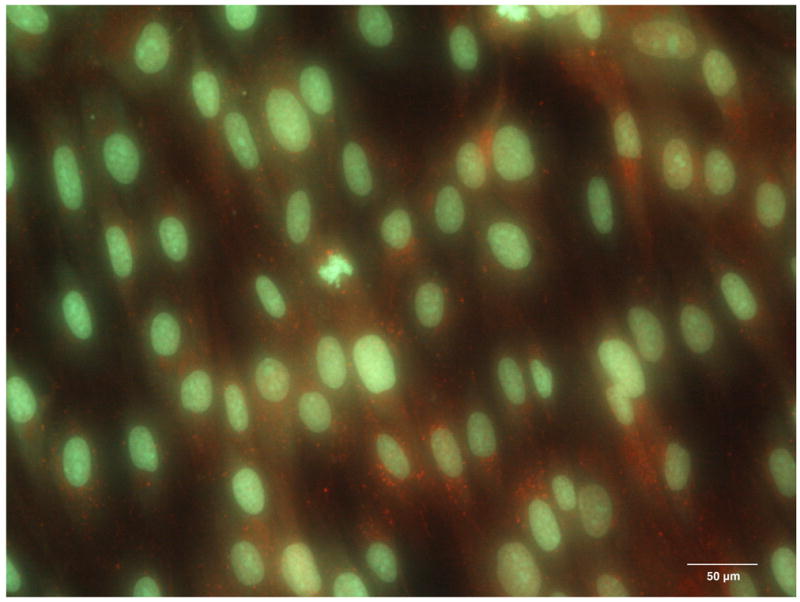
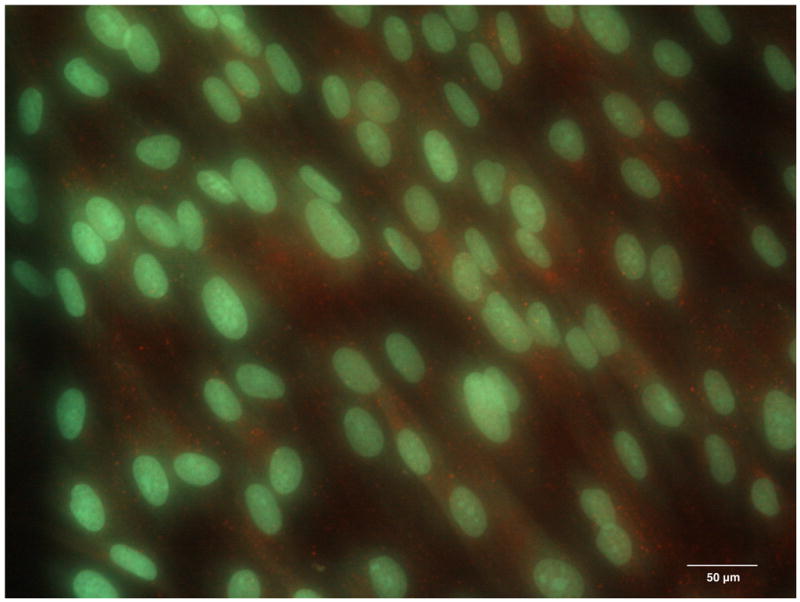
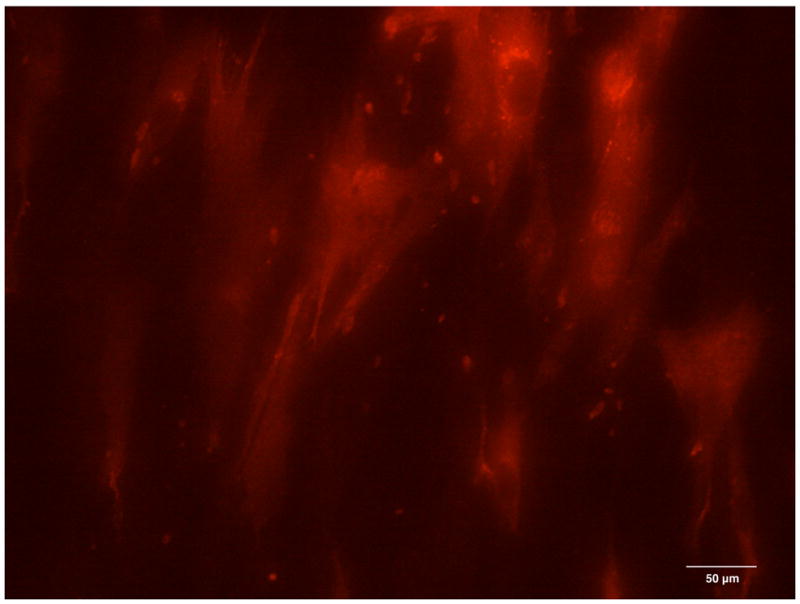
The expression of Connexin 43 in the cytoplasm of the DPSC cultures on day 1 (A), day 2 (B) and day 4 (C). The expression of Connexin 43 on the surface of DPSCs on day 2 of culture (D, red channel). Red fluorescence identifies Connexin 43; green fluorescence, DPSC nuclei and cytoplasms.
The incidence of IIF
On the cryostage, the incidence of IIF increased with decreasing extracellular ice nucleation temperature. In the absence of cryoprotectant, it reached 100% in DPSC suspensions upon nucleation at −12°C (Figure 5), and in DPSC monolayers upon nucleation at −6°C (Figure 6).
Figure 5.
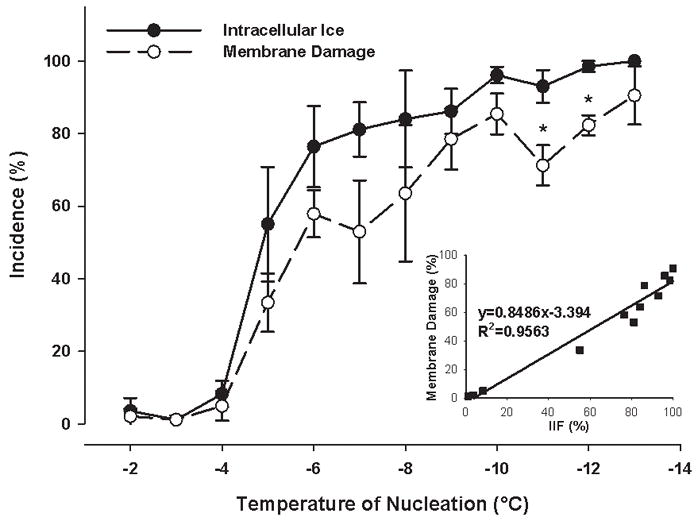
The incidence of both intracellular ice formation and membrane damage as a function of ice nucleation temperature and the correlation between intracellular ice formation and membrane damage in DPSC suspensions. No cryoprotective agent was present. Mean ± SEM (n = 3 – 7). * p < 0.05 vs. the incidence of intracellular ice formation at the given nucleation temperature (Mann-Whitney test).
Figure 6.
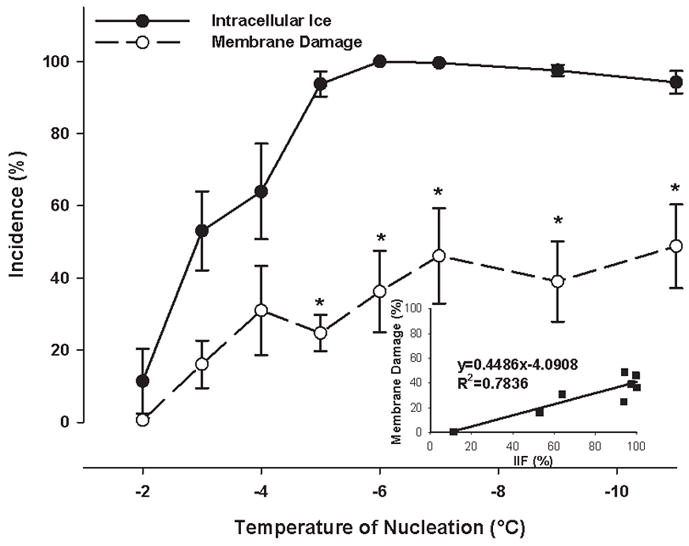
The incidence of both intracellular ice formation and membrane damage as a function of ice nucleation temperature and the correlation between intracellular ice formation and membrane damage in confluent DPSC monolayers. No cryoprotective agent was present. Mean ± SEM (n = 5 – 12). * p < 0.05 vs. the incidence of intracellular ice formation at the given nucleation temperature (Mann-Whitney test).
In the methanol bath, the incidence of IIF in monolayers increased with decreasing ice nucleation temperature in the same manner as on the cryostage. The incidence of IIF at most nucleation temperatures tested corresponded to that on the cryostage (Figure 7). A temperature of −7°C was chosen as the extracellular ice nucleation temperature to induce 100% IIF.
Figure 7.
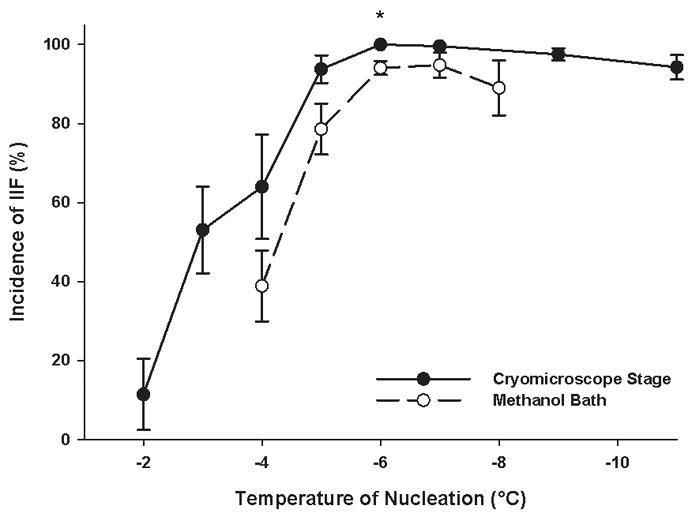
The incidence of intracellular ice formation in confluent DPSC monolayers as a function of ice nucleation temperature (both on the cryostage and in the methanol bath). Mean ± SEM (n = 7 – 18). * p < 0.05 vs. the incidence of intracellular ice formation in the methanol bath at the given nucleation temperature (Mann-Whitney test).
Viability of DPSCs after freezing
On the cryostage, the plotted slope of the incidences of membrane damage versus IIF was much smaller for monolayers (0.449, Figure 6) than for cell suspensions (0.849, Figure 5), meaning that similar incidences of IIF caused less membrane damage in monolayers than in cell suspensions. After 100% IIF, the incidence of membrane damage in DPSC suspensions was 85.9±1.7%, compared to 25.5±5.5% in DPSC monolayers (p<0.001, Figure 8).
Figure 8.
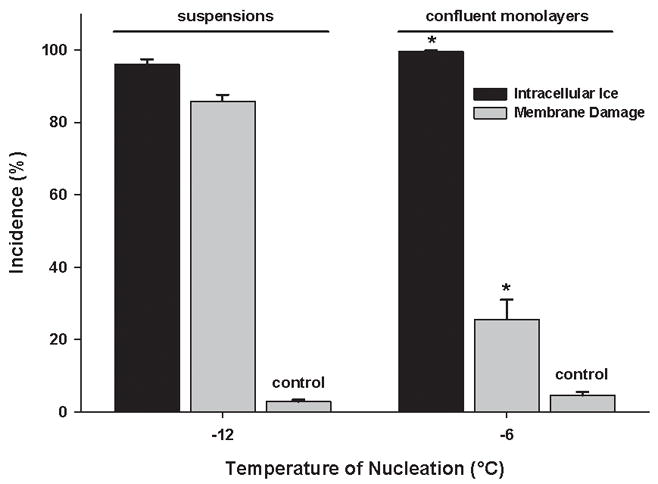
The incidence of membrane damage in both suspensions and confluent monolayers of DPSC after complete intracellular freezing. Unfrozen samples acted as controls. Mean ± SEM (n = 16 – 17). * p < 0.05 vs. suspensions (Mann-Whitney test).
In the methanol bath, only 14.8±3.3 % of DPSCs in monolayers were damaged after nucleation at −7°C and immediate thawing (Group 1, p<0.001 vs. unfrozen control). However, when monolayers were cooled to −80°C, either continuously (Group 4) or after isothermal nucleation (Groups 2 and 3), the incidence of membrane damage rose to 59.9±12.0% in Group 2 (p=0.024 vs. Group 1), 70.9±5.2% in Group 3 (p<0.001 vs. Group 1) and 46.8±10.3% in Group 4 (p=0.012 vs. Group 1, Figure 9). We also performed a colony forming assay for confluent monolayers after nucleation at −7°C and immediate thawing (Group 1) and, although the colony growth was observed in control samples, experimental samples showed no colony forming ability after 100% IIF (Table 1).
Figure 9.

The incidence of membrane damage in confluent DPSC monolayers resulting from different freezing profiles performed in the methanol bath. Group 1 – nucleation at −7°C, immediate thawing; Group 2 – nucleation at −7°C, fast two-step cooling; Group 3 – nucleation at −7°C, slow two-step cooling; Group 4 – no nucleation, slow two-step cooling. Unfrozen monolayers acted as the experimental control. Mean ± SEM (n = 8–9). * p < 0.05 vs. untreated control, ** p < 0.05 vs. Group 1 (Mann-Whitney test).
Table 1.
The yield of DPSC from confluent monolayers at different stages of cryopreservation.
| Experimental condition | DPSC yield | |
|---|---|---|
| Control | Experiment | |
| Untreated | 100.0 ± 0.0 % | N/A |
| Predigestiona | 119.6 ± 5.4 % * | 105. 8 ± 7.9 % |
| Post-digestionb | 2.8 ± 1.0 % * | 10.0 ± 1.7 % * |
| Five days in culturec | 0.8 ± 0.2 % * | 0.0 ± 0.0 % * |
Controls were incubated at room temperature during freeze-thawing of experimental samples. Experiment samples were nucleated at −7°C and then thawed.
Samples were treated with a cell dissociation buffer solution and the resulting harvested DPSCs with intact membranes were quantified.
The number of intact DPSCs, obtained after digestion, that produced colonies. Values are presented as the percentage of untreated controls, mean ± SEM (n = 3–24).
p < 0.05 vs. the untreated control (Mann-Whitney test).
Cell loss during cryopreservation processing
No cells were detached from coverslips after simply exposure to room temperature or after ice nucleation at −7°C and thawing. However, during harvesting of DPSCs from coverslips we were able to collect only 2.8±1.0% of cells in control group (exposure to room temperature) and 10.0±1.7% of cells in experimental group (ice nucleation at −7°C and thawing) (Table 1). In the control group, 30.7% of obtained DPSCs were able to proliferate and form colonies; however, no colony growth was observed in the experimental group (Table 1).
Discussion
Observations from DPSCs cryopreserved with 10% Me2SO, according to the standard method, were consistent with previous results [25], showing good post-thaw survival. This suggests that for cells in suspension the standard method is an effective cryopreservation procedure.
The expression of gap junction-forming protein Cx43 was observed in the cytoplasm of DPSCs beginning on the first day in culture, even before cells reached confluence. However, on the cell surface, random sites of Cx43 expression appeared only on day 2, once monolayers had reached confluence. Both intracellular and surface expression of Cx43 was not limited to the periphery of cells, but was evenly distributed throughout the cell monolayer. The absence of a defined intercellular pattern of Cx43 expression can be explained by the fact that Cx43 protein is synthesized in the granular endoplasmic reticulum on the surface of the cell nucleus, and then is transported to the periphery of the cell, eventually reaching the cell surface. Although the presence of gap junctions between DPSCs was not demonstrated directly, we thus showed that DPSCs in a confluent monolayer express gap junction-forming proteins on the cell surface.
The temperature at which 100% IIF occurred was higher in DPSC monolayers (−6°C) compared to DPSC suspensions (−12°C), which was consistent with previous observations that cell-to-cell contact increases the incidence of IIF [4–7,9,11,12]. Once IIF was achieved, cell preparation (suspension or monolayer) and final subzero temperature, to which cells were cooled, were both found to impact the incidence of membrane damage. Following 100% IIF on the cryostage, significantly more DPSCs had damaged membranes when in suspension (p<0.001) versus when arranged in monolayers. Specifically, of DPSC monolayers that were thawed immediately after 100% IIF, both on the cryostage versus in a methanol bath, approximately 74% and 85% of the DPSCs were undamaged respectively. However, when methanol bath-treated monolayers were cooled below the extracellular ice nucleation temperature that induced 100% IIF, then stored at −80 °C for 48 hours, the incidence of membrane damage increased significantly (p<0.05). The reason for this observed increase is not well understood. In the case when extracellular ice nucleation was induced directly using different freezing profiles, increased incidence of membrane damage during cooling below ice nucleation temperature may be due to altered kinetics of IIF, the different mechanical properties of cell culture substrate at lower temperatures or changes in the size of ice crystals as a function of cooling rate. It is worth mentioning however, that even at the highest incidence of membrane damage, about 30% of DPSCs in the monolayer remained intact, which is quite significant considering the absence of any conventional cryoprotectant during the freezing process.
Since confluent monolayers in Group 1 (nucleated at −7°C and immediately thawed) showed the lowest incidence of membrane damage, we chose this group to test monolayer colony forming ability following 100% IIF. Despite a low incidence of membrane damage, DPSCs showed no colony forming ability after 100% IIF. The observed discrepancy between membrane integrity and colony forming ability may result from IIF-induced damage to those inner structures of the cell responsible for its proliferation and normal function, at the same time leaving the membrane undamaged. Also, given that the control group showed that less than one third of intact cells were able to proliferate, a higher seeding density may be required for cells exposed to 100 % IIF, in order to observe any colony growth. It should further be noted that this discrepancy in cell viability as measured by membrane integrity and colony forming assay is a common observation. For example, it was shown that only one in eight intact CD34+ cells in stem cell transplants is capable of developing into a granulocyte-macrophage colony-forming unit after cryopreservation [26].
The experimental results also showed evidence of a decrease in the number of cells in DPSC monolayers during the different stages of the cryopreservation process. Although the data showed an absence of cell detachment in DPSC monolayers following extracellular ice nucleation at −7°C (100% IIF) and subsequent thawing, visual assessment of monolayers that were subsequently cooled to −80°C revealed significant cell detachment. This may reflect the negative impact of lower temperatures on the binding between monolayer and glass substrate, or on cell-cell or cell-substrate adhesion. Significantly fewer cells with intact membrane were collected from coverslips post-digestion than were initially present on them. In control monolayers incubated at room temperature, large regions of undetached cells remained on the coverslip after application of the cell dissociation buffer, likely due to the strong attachment of cells to substrate in the unfrozen monolayer. In monolayers that underwent 100% IIF, cell-substrate attachment was not as strong, and almost all of the cells were easily dislodged from the coverslip. Together with the fact that only about 30% of control cells were able to proliferate and form colonies, this should be taken into account for accurate calculation of cell dose if monolayers are cryopreserved for clinical use as in transplants.
There were some limitations of this initial study worth noting. First, due to the intrinsic differences in freezing response between freshly isolated and cultured cells [14], the response to IIF of DPSCs which were cultured for several passages may not accurately reflect that of DPSCs which have been freshly isolated from dental pulp. Freshly isolated DPSCs will be a better experimental model and will eliminate the possible effect of culture conditions on cell properties. Second, there may be donor specific differences in survivability of DPSCs isolated from teeth, which would be overcome by including DPSCs from different sources during experimentation. Third, the volume of samples used for testing was very small and is not representative of that required for clinical applications. Since the freezing profile of the sample differs according to its volume, further scale-up work is required before clinical implementation.
The effects of IIF on cell viability need to be researched further. In particular, we would like to investigate the detrimental effect of IIF on cell ability to proliferate and to better understand the nature of damage to DPSC monolayers that takes place during cooling below extra- and intracellular ice nucleation temperatures. The hypothesized involvement of gap junctions in the propagation of intracellular ice between adjacent cells in a confluent monolayer also needs to be tested directly. In addition, improving cell attachment to the substrate will reduce cell loss during cryopreservation and therefore allow a higher number of DPSCs to be recovered and available for transplantation.
We conclude that confluent monolayers of DPSCs express the gap junction-forming protein Cx43, and upon IIF retain membrane integrity, however lose the ability to proliferate. The present findings offer improved conceptual/theoretical understanding by adding further evidence against the traditional concept of cell membrane injury due to IIF. They also have important practical implications for transplantation – introducing the possibility of DPSC cryopreservation without the use of cryoprotectants.
Acknowledgments
The authors would like to acknowledge the technical support of Dr. Locksley McGann and his research group, and the insights offered by Dr. Peter Mazur in the preparation of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Acker JP. Innocuous Intracellular ice Formation: Mechanisms and Implications. 2000:1–196. [Google Scholar]
- 2.Acker JP, Croteau IM. Pre- and post-thaw assessment of intracellular ice formation. J Microsc. 2004;215:131–138. doi: 10.1111/j.0022-2720.2004.01375.x. [DOI] [PubMed] [Google Scholar]
- 3.Acker JP, Elliott JA, McGann LE. Intercellular ice propagation: experimental evidence for ice growth through membrane pores. Biophys J. 2001;81:1389–1397. doi: 10.1016/S0006-3495(01)75794-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Acker JP, Larese A, Yang H, Petrenko A, McGann LE. Intracellular ice formation is affected by cell interactions. Cryobiology. 1999;38:363–371. doi: 10.1006/cryo.1999.2179. [DOI] [PubMed] [Google Scholar]
- 5.Acker JP, McGann LE. Protective effect of intracellular ice during freezing? Cryobiology. 2003;46:197–202. doi: 10.1016/s0011-2240(03)00025-7. [DOI] [PubMed] [Google Scholar]
- 6.Acker JP, McGann LE. Innocuous intracellular ice improves survival of frozen cells. Cell Transplant. 2002;11:563–571. [PubMed] [Google Scholar]
- 7.Acker JP, McGann LE. Cell-cell contact affects membrane integrity after intracellular freezing. Cryobiology. 2000;40:54–63. doi: 10.1006/cryo.1999.2221. [DOI] [PubMed] [Google Scholar]
- 8.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Cell junctions, cell adhesion, and the extracellular matrix. In: Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P, editors. Molecular Biology of the Cell, Garland Science. Taylor & Francis Group, LLC; New York, NY: 2008. pp. 1131–1204. [Google Scholar]
- 9.Armitage WJ, Juss BK. Freezing monolayers of cells without gap junctions. Cryobiology. 2003;46:194–196. doi: 10.1016/s0011-2240(03)00017-8. [DOI] [PubMed] [Google Scholar]
- 10.Ballini A, De Frenza G, Cantore S, Papa F, Grano M, Mastrangelo F, Tete S, Grassi FR. In vitro stem cell cultures from human dental pulp and periodontal ligament: new prospects in dentistry. Int J Immunopathol Pharmacol. 2007;20:9–16. doi: 10.1177/039463200702000102. [DOI] [PubMed] [Google Scholar]
- 11.Berger WK, Uhrík B. Freeze-induced shrinkage of individual cells and cell-to-cell propagation of intracellular ice in cell chains from salivary glands. Experientia. 1996;52:843–850. doi: 10.1007/BF01938868. [DOI] [PubMed] [Google Scholar]
- 12.Irimia D, Karlsson JO. Kinetics and mechanism of intercellular ice propagation in a micropatterned tissue construct. Biophys J. 2002;82:1858–1868. doi: 10.1016/S0006-3495(02)75536-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jo YY, Lee HJ, Kook SY, Choung HW, Park JY, Chung JH, Choung YH, Kim ES, Yang HC, Choung PH. Isolation and characterization of postnatal stem cells from human dental tissues. Tissue Eng. 2007;13:767–773. doi: 10.1089/ten.2006.0192. [DOI] [PubMed] [Google Scholar]
- 14.Karlsson JO, Toner M. Long-term storage of tissues by cryopreservation: critical issues. Biomaterials. 1996;17:243–256. doi: 10.1016/0142-9612(96)85562-1. [DOI] [PubMed] [Google Scholar]
- 15.Kerkis I, Kerkis A, Dozortsev D, Stukart-Parsons GC, Gomes Massironi SM, Pereira LV, Caplan AI, Cerruti HF. Isolation and characterization of a population of immature dental pulp stem cells expressing OCT-4 and other embryonic stem cell markers. Cells Tissues Organs. 2006;184:105–116. doi: 10.1159/000099617. [DOI] [PubMed] [Google Scholar]
- 16.Lelkens CCM, Koning JG, de Kort B, Floot IBG, Noorman F. Experiences with frozen blood products in the Netherlands military. Transfus Apheresis Sci. 2006;34:289–298. doi: 10.1016/j.transci.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 17.Mauth C, Huwig A, Graf-Hausner U, Roulet J-F. Restorative applications for dental pulp therapy. In: Ashammakhi N, Reis RL, Chiellini E, editors. Topics in Tissue Engineering. 2007. pp. 1–32. [Google Scholar]
- 18.Mazur P, Leibo SP, Chu EH. A two-factor hypothesis of freezing injury. Evidence from Chinese hamster tissue-culture cells. Exp Cell Res. 1972;71:345–355. doi: 10.1016/0014-4827(72)90303-5. [DOI] [PubMed] [Google Scholar]
- 19.McGrath JJ, Cravalho EG, Huggins CE. An experimental comparison of intracellular ice formation and freeze-thaw survival of HeLa S-3 cells. Cryobiology. 1975;12:540–550. doi: 10.1016/0011-2240(75)90048-6. [DOI] [PubMed] [Google Scholar]
- 20.Mullen SF, Critser JK. The science of cryobiology. Cancer Treat Res. 2007;138:83–109. doi: 10.1007/978-0-387-72293-1_7. [DOI] [PubMed] [Google Scholar]
- 21.Perry BC, Zhou D, Wu X, Yang F, Byers MA, Chu T-G, Hockema JJ, Woods EJ, Goebel WS. Collection, cryopreservation, and characterization of human dental pulp-derived mesenchymal stem cells for banking and clinical use. Tissue Eng Part C Methods. 2008;14:149–156. doi: 10.1089/ten.tec.2008.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pijnappels DA, Schalij MJ, van Tuyn J, Ypey DL, de Vries AAF, van der Wall EE, van der Laarse A, Atsma DE. Progressive increase in conduction velocity across human mesenchymal stem cells is mediated by enhanced electrical coupling. Cardiovasc Res. 2006;72:282–291. doi: 10.1016/j.cardiores.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 23.Prickett RC, Elliott JAW, McGann LE. Application of the osmotic virial equation in cryobiology. Cryobiology. 2009 doi: 10.1016/j.cryobiol.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 24.Suguro AMH. Characterization of human dental pulp-derived cell lines. Int Endod J. 2008;41:609–616. doi: 10.1111/j.1365-2591.2008.01409.x. [DOI] [PubMed] [Google Scholar]
- 25.Woods EJ, Perry BC, Hockema JJ, Larson L, Zhou D, Goebel WS. Optimized cryopreservation method for human dental pulp-derived stem cells and their tissues of origin for banking and clinical use. Cryobiology. 2009;59:150–157. doi: 10.1016/j.cryobiol.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang H, Acker JP, Cabuhat M, Letcher B, Larratt L, McGann LE. Association of post-thaw viable CD34+ cells and CFU-GM with time to hematopoietic engraftment. Bone Marrow Transplant. 2005;35:881–887. doi: 10.1038/sj.bmt.1704926. [DOI] [PubMed] [Google Scholar]