

Abstract
Superhydrophobic monolithic porous polymer layers with a photopatterned hydrophilic channel have been prepared. These layers were used for two-dimensional thin layer chromatography of peptides. The 50 μm thin poly(butyl methacrylate-co-ethylene dimethacrylate) layers supported onto 4.0 × 3.3 cm glass plates were prepared using UV-initiated polymerization in a simple glass mold. Photografting of a mixture of 2-acrylamido-2-methyl-1-propanesulfonic acid and 2-hydroxyethyl methacrylate carried out through a mask afforded a 600 μm wide virtual channel along one side of the layer. This channel, which contains ionizable functionalities, enabled the first dimension separation in ion exchange mode. The aqueous mobile phase migrates only through the channel due to the large difference in surface tension at the interface of the hydrophilic channel and the superhydrophobic monolith. The unmodified part of the layer featuring hydrophobic chemistry was then used for the reversed phase separation in the orthogonal second dimension. Practical application of our technique was demonstrated with a rapid 2D separation of a mixture of model peptides differing in hydrophobicity and isoelectric point using a combination of ion-exchange and reversed phase modes. In the former mode, the peptides migrated 11 mm in less than 1 min. Detection of fluorescently labeled peptides was achieved through UV light visualization. Separation of the native peptides was monitored directly using a desorption electrospray ionization (DESI) source coupled to a mass spectrometer. Unidirectional surface scanning with the DESI source was found suitable to determine both the location of each separated peptide and its molecular mass.
Keywords: Polymer monolith, Superhydrophobic layer, Poly(butyl methacrylate-co-ethylene dimethacrylate), interfacial tension, Thin layer chromatography, Peptides
Following its emergence1 in the 1930’s thin layer chromatography (TLC) became widely accepted in the 1950’s based on the pioneering works of Kirchner and Stahl.2–4 Typically, a sample is spotted close to one edge of a TLC plate, which is then placed in either a vertical or horizontal development chamber where it contacts the mobile phase. The latter driven through the plate by capillary force moves the components of the sample, which are then separated according to differences in their partitioning between the mobile and stationary phases. Colored compounds can be seen directly, while other molecules must be visualized using for example fluorescence or UV shadowing of a fluorescent sorbent layer.
The major advantages of TLC reside in the simplicity and speed of the technique, which became especially popular in organic chemistry laboratories as it provides a simple and inexpensive means to both monitor the progress of a reaction and purity of the targeted product. Quantitative analysis is also possible using a scanning densitometer. Although the low cost TLC plates are most often disposed after each analysis, they can also be kept as a portable “storage” to archive the complete separation since they contain all separated components. Several samples can be separated in parallel thus increasing throughput.4
The most widely used TLC plates have consisted of a sorbent layer such as silica or alumina particles on a flat support such as glass or plastic. Alternatively, cellulose, bonded phase layers, and other specialty TLC plates are also commercially available.
Typical early TLC plate had an a sorbent layer with a thickness of 100–400 μm formed from irregular ca. 10 μm silica particles held together by a binder. High performing layers marketed since the mid 1970’s are fabricated from 5–7 μm porous silica beads that afford a much higher performance.5 The 10 μm ultrathin layer chromatographic plates introduced in 2001 based on a monolithic silica structure are the newest commercial contribution to TLC plate technology.6 Led by the rebirth of interest in TLC, several new developments in the layer technology have recently appeared. For example, ultra thin layers have been prepared by depositing a porous nanostructured film of silica via glancing-angle vapor deposition.7 Ultra thin layers have also been obtained by deposition of electrospinned polyacrylonitrile nanofibers on aluminum foil.8 We have previously reported porous polymer monolithic layers 100–200 μm in thickness supported by glass plates and demonstrated their use for the unidimensional separation of peptides and proteins.9
TLC is easily amenable to two-dimensional (2D) separations, which significantly increase the zone capacity. Clearly, the simplest 2D implementation includes spotting the sample near a corner of the layer, developing the layer in one direction using the first mobile phase, drying the layer, and developing it in the second mobile phase after rotating it 90°. Poole recently outlined several techniques to generate orthogonality in 2D TLC.10 The simplest approach uses two different eluents with complementary selectivity.11–29 However, finding a system of two truly orthogonal solvents may be a challenging task.30,31 The separation selectivity can also be changed by impregnating the layer with an immiscible solvent prior to the second development.15,18–20,25,26 Yet another option is to apply two different stationary phases in a dual-phase or a coupled layer format. The former uses a strip of a sorbent such as underivatized silica with an adjacent layer of C18 silica, which allows the first development to be performed with a non-aqueous mobile phase readily removed by evaporation.19,20,32–34 This approach does not appear to significantly modify the properties of the sorbents, as evidenced by the good agreement between computer simulated and experimental separations.35 The alternative format would require that the initial development be performed on the bonded phase layer with an aqueous mobile phase, which is strongly adsorbed on the normal phase silica used for the second dimension thus deactivating that layer.31,36
In contrast, two different plates are used in the coupled layer TLC.36–45 The first dimension separation is carried out on a plate later cut into strips that are then connected to the second dimension plate, the spots are transferred, and the plate developed. Since this approach requires manipulation skills, the hallmark of TLC – its simplicity - is lost. Clearly, new formats are desirable to fully exploit the 2D nature of TLC.46,47
Levi and Guiochon developed metrics that can be used to evaluate the quality of a 2D separation achieved by any of the above methods.48 Their approach was later expanded by Nurok and coworkers to evaluate a very large set of possible 2D separations using computer simulated chromatograms based on experimental 1-D separations49; their results showed good agreement in the pattern of separated spots obtained both via computer simulation and in experimental 2D separations.35
Detection to visualize the results of the separation in 2D TLC is also important.10 While the “classical” detection methods such as staining or labeling provide information related to the position of the spot or retardation factor (Rf value), mass spectrometry adds another dimension to the separation by identifying the molar mass of the separated compounds.50 For example, the similar shape of TLC plate and MALDI MS targets has facilitated the coupling of these two methods. However, several studies have revealed the poor sensitivity of MALDI detection since only compounds located at the top surface of the thin layer could be ionized.51–58 A significant improvement in sensitivity was achieved using ultrathin layer chromatographic plates59–60 and further improvements resulted from the introduction of the desorption electrospray ionization (DESI) source 61–63 which can be operated in scanning mode.64–72 DESI has also been used for the direct detection of compounds from commercial TLC plates.73–79
The objective of this work was to develop a new 2D TLC plate format with a virtual hydrophilic ionizable channel photopatterned on one side of a plate consisting of a superhydrophobic thin layer of monolithic porous polymer attached to a glass support. Separation in the first dimension would proceed via ion exchange within the hydrophilic channel while the remainder of the plate would be used for reversed phase separation in the second dimension.
EXPERIMENTAL SECTION
Chemicals and Materials
2-Acrylamido-2-methyl-1-propanesulfonic acid (AMPS), butyl methacrylate (BMA), ethylene dimethacrylate (EDMA), 2-hydroxyethyl methacrylate (HEMA), benzophenone, 2,2Dimethoxy-2-phenylacetophenone, cyclohexanol, 1-decanol, , tert-butanol, 3-(trimethoxysilyl)propyl methacrylate, acetic acid, trifluoroacetic acid, ammonium acetate, acetone, acetonitrile, methanol, water, all of the highest available purity were purchased from Sigma-Aldrich (St. Louis, MO). The methacrylate monomers were purified by passing through a short column packed with basic alumina inhibitor remover (Prepacked Column for Removing Hydroquinone and Monomethyl Ether Hydroquinone, cat. # 306312-1EA, Sigma-Aldrich, St. Louis). Stock solutions of standard peptides (1 mg/mL) human angiotensin II, bradykinin acetate salt, leucine enkephalin acetate salt hydrate, and val-tyr-val (all from Sigma-Aldrich) were prepared in water. Fluorescamine (1 mg/mL) was dissolved in acetone and kept at 4°C. Peptides labeling was achieved by mixing their solution with an equal volume of fluorescamine solution. No decrease in fluorescence was observed even after several hours. For the TLC separation followed by UV detection, peptide mixture was prepared by mixing equal amounts of labeled peptides. For the TLC separation followed by DESI scanning, peptide mixture was prepared by mixing equal amounts of unlabeled peptides.
Borofloat glass plates 12 × 3.3 cm, 1.1 mm thick were purchased from S. I. Howard Glass Co. Inc. (Worcester, MA) and cut to three 4.0 × 3.3 cm pieces.
Surface Modification of Glass Plates
The glass plates were rinsed with water, activated with 1 mol/L sodium hydroxide for 30 min, washed with water, and 0.2 mol/L HCl for 30 min, rinsed with water again, and dried in stream of nitrogen. The glass surface was then functionalized using 20 vol% solution of 3-(trimethoxysilyl)propyl methacrylate in ethanol adjusted to pH 5 using acetic acid. This solution was placed for 30 min at the activated surface of one plate covered with another one, thus functionalizing two plates at the same time. This functionalization was repeated twice. The plates were then washed with acetone and dried with nitrogen.
Preparation of the Hydrophobic Monolithic Layer
The polymerization mixture comprising butyl methacrylate (24 wt.%), ethylene dimethacrylate (16 wt.%), 1-decanol + cyclohexanol (60 wt.%), and 2,2-dimethoxy-2-phenylacetophenone (1 wt% with respect to monomers) was de-aerated by purging with nitrogen for 10 min. This mixture was filled into an assembled mold consisting of two modified glass plates clamped face to face and separated with 50 μm thick FEP Teflon strips (American Durafilm Co.) placed between them along the longer sides of the glass plate to define the thickness of the polymer layer. The mold was filled with the polymerization mixture using capillary action and exposed to UV light for 15 min. Once the polymerization was completed, the mold was easily disassembled due to the difference in irradiation dose at the top and bottom of the mold (vide infra). The plate with attached polymer layer was immersed in methanol for 1 h to remove the porogens. Finally, the plates were dried in vacuum at 40°C for 30 min.
Photografting of Virtual Hydrophilic Monolithic Channel
The 50 μm thick hydrophobic layer was wetted with a solution containing 2 wt.% 2-acrylamido-2-methyl-1-propanesulfonic acid, 13 wt.% 2-hydroxyethyl methacrylate, and 0.22 wt.% benzophenone in 3:1 (v/v) tert-butanol-water mixture. The wetted layer was covered with a quartz plate and a mask with an opening defining the channel, and exposed to UV light for 7.5 min. The grafted plates were then washed with methanol and water to remove unreacted monomer, and dried in nitrogen.
Two-Dimensional TLC Separation of Peptides
An aqueous solution of a mixture of UV labeled peptides (50 nL) was spotted 4 mm from the beginning of the grafted hydrophilic channel. The separation in the first dimension was carried out using a mobile phase consisting of 30 vol.% acetonitrile in 0.2 mol/L aqueous ammonium acetate at pH 7. To avoid the mobile phase running over the plate instead of through the channel, a paper tissue wick saturated with the mobile phase was placed at one end of the virtual channel. The hydrophilic channel then pulled the aqueous mobile phase from the wick through the channel. A standard flat bottom development chamber containing 0.1 vol.% TFA in 40 vol.% aqueous acetonitrile, which was saturated with vapors before development, was used for the separation in the second dimension orthogonal to the development in the first dimension. After completion of the two-dimensional separation, the plate was air-dried and peptide spots were detected by illumination with UV light.
Instrumentation
An OAI Model 30 deep UV collimated light source (Optical Associates, Inc., San Jose, CA, USA) fitted with a 500-W HgXe lamp was used for UV exposures. The radiation power was adjusted to 12.0 mW/cm2, which affords 4.4 mW/cm2 after passing the cover glass plate. These values were measured using an OAI Model 306 UV power meter with a 260-nm probe head. Scanning electron micrographs were obtained using the Zeiss Gemini Ultra-55 analytical scanning electron microscope. The porous polymers were sputtered with gold using the BAL-TEC SCD 050 sputter coater. Static water contact angles were determined using an Easy Drop instrument (Krüss GmbH, Germany).
TLC/DESI-MS
The Omni Spray ion source (Prosolia, Indianapolis, IN) was interfaced with a quadrupole/TOF mass spectrometer (MicroTOFq, Bruker Daltonics, Billerica, MA). The sampling capillary was extended from the main body of the instrument by addition of a 28 mm long stainless steel tube (3 mm i.d., 3.4 mm o.d.). Mass spectra were acquired in the positive ion mode. The DESI emitter charged with a 2.5 kV spray voltage applied to the spray head was mounted 2 mm from the plate surface at an angle of 60° relative to the surface, and about 3 mm back from the area where the DESI plume jet impacted the surface. The sampling capillary was positioned just above the surface to be analyzed. The nebulizer gas (nitrogen) pressure was set at 0.69 MPa (100 psi). The electrospray solvent 0.1 vol.% acetic acid in 50/50 (v/v) methanol/water was delivered by a syringe pump at a flow rate of 5 μL/min.
Unidirectional scanning developed by Kertesz and Van Berkel70 was used for detection. Briefly, each selected lane on the surface of the plate was scanned in the same direction. The plate was placed on an insulated sample holder mounted on a manually controlled x/y/z stage. Fig. 1 shows that the first lane was scanned by moving the plate in a direction parallel to the x-axis, which represents the direction of the separation in the 1st dimension, i.e. in the grafted channel, from low to high Rf . At the end of the first lane, the surface was lowered by 2 mm and moved back to the beginning of the lane. Then the surface was moved parallel to the y-axis by 0.3 mm, raised by 2 mm and scanning of the second lane was carried out. The y-axis represents the direction of the separation in 2nd dimension. A CCD camera and a monitor were used to aid positioning of the DESI emitter and sampling capillary. Scanning was performed by moving the TLC plate at an average speed of about 0.2 mm/s with the stationary spray emitter and sampling capillary perpendicular to the scanning direction.
Figure 1.
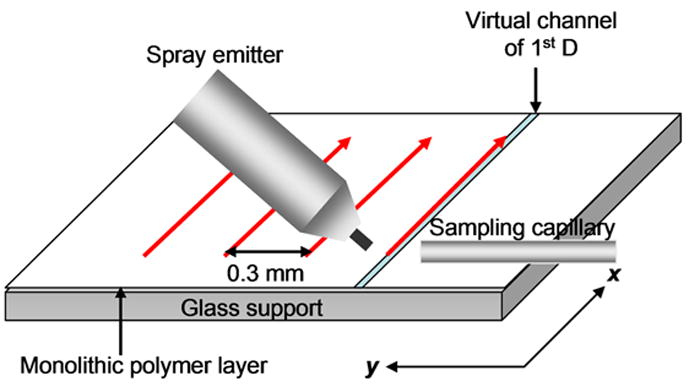
Schematic illustration of DESI scanning of surface of poly(butyl acrylate-co-ethylene dimethacrylate) monolithic layer.
RESULTS AND DISCUSSION
Superhydrophobic Porous Polymer Layer
We have recently demonstrated that poly(butyl methacrylate-co-ethylene dimethacrylate) monoliths80–84 can be readily prepared in thin layer format using photoinitiated polymerization. We found that some of these monolithic layers possessed superhydrophobic properties, i.e. they exhibited a contact angle for water in excess of 150°.85 Based on the theory of superhydrophobicity,86–87 we attributed this property to an interplay of both chemistry and morphology/topography of the layer.
As described in the experimental section, all polymerizations were carried out in a simple mold consisting of two glass plates separated by a Teflon film strip used to define the thickness of the monolithic layer. In initial experiments, the top glass plate, targeted for support of the thin layer, was functionalized with 3-(trimethoxysilyl)propyl methacrylate while the bottom plate was not. Thus, the monolithic layer formed in the mold became covalently attached to the surface of the top plate. We had speculated that the bottom glass plate should not be functionalized to avoid adhesion of the layer to that plate and facilitate disassembly of the mold. The polymer layer prepared under these conditions (Fig. 2) has a smooth surface formed from microglobules well assembled at the polymer-glass interface, but its water contact angle is only 77° as it lacks the topographical features of dual scale roughness required for superhydrophobicity. The situation changes when a silylated bottom plate is used. Because of the thickness of the layer, the intensity of the UV light first penetrating through the top plate and the polymerizing mixture is attenuated before it reaches the bottom plate. Therefore, the polymerization rate is slower in the vicinity of the bottom plate leading to less firm attachment to that plate. Upon disassembling the mold, most of the polymer adheres to the top plate while only a light coating of polymer remains attached to the bottom plate. As a result, the internal structure of the monolithic layer is revealed at the surface (Fig. 2), which then features the desired dual scale roughness and the water contact angle increases to a superhydrophobic value of 154°.
Figure 2.

SEM images of the 125μm thick porous poly(butyl acrylate-co-ethylene dimethacrylate) monolithic layers with surface exhibiting dual roughness (left panel) and with smooth surface (right panel) prepared in mold with surfaces of both top + bottom glass plate and the top glass plate only silanized, respectively. The layers were prepared from polymerization mixture #3 Photographs of water droplets on the surfaces are shown in inserts.
In previous work with superhydrophobic monoliths85, we had used polymerization mixtures containing 30% butyl methacrylate, 20% ethylene dimethacrylate, and 50% of porogenic solvents (cyclohexanol + decanol). As a result, the porosity of these layers was close to 50%. However, for chromatographic applications a higher porosity is desirable to accommodate a larger volume of the mobile phase. Therefore, we prepared a series of 50 μm thin layers from mixtures consisting of 24% butyl methacrylate, 16% ethylene dimethacrylate, with 60% cyclohexanol and decanol in varying proportions (Table 1). All these mixtures afforded polymers with porosity near 60%. The SEM micrographs shown in Fig. 3 reveal that an increase in the percentage of cyclohexanol in the polymerization mixture leads to significant changes in the morphology of the monolithic layer. The size of both microglobules and pores decreases as the percentage of cyclohexanol increases. Table 1 documents that these changes in morphology affect the microscale roughness and also lead to a decrease in the water contact angle from superhydrophobic (160°) to hydrophobic (135°).
Table 1.
Polymerization Mixtures Used for the Preparation of Hydrophobic Layers a)
| Mixture # | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| 1-decanol, wt. % | 60 | 50 | 40 | 30 | 20 | 10 | 0 |
| cyclohexanol, wt. % | 0 | 10 | 20 | 30 | 40 | 50 | 60 |
| contact angle, degrees | 156 | 152 | 154 | 152 | 151 | 139 | 136 |
Composition of complete polymerization mixture (in wt.%): butyl methacrylate 24, ethylene dimethacrylate 16, porogens (1-decanol + cyclohexanol) 60, 2,2-dimethoxy-2-phenylacetophenone 1 (with respect to monomers).
Figure 3.

SEM images of surfaces of the 50 μm thick hydrophobic poly(butyl acrylate-co-ethylene dimethacrylate) monolithic layers prepared using mixtures 1 to 7 and shape of water droplets on the corresponding surfaces. For compositions of the polymerization mixtures see Table 1.
Several factors such as hydrophobicity, porous properties, separation performance, and mechanical strength must be considered when selecting the layer with properties best matching the requirements of the intended chromatographic application. The hydrophobicity which is required for the separation of peptides in reversed phase mode using a mobile phase containing a high percentage of organic solvent is solely defined by the chemistry of the poly(butyl methacrylate-co-ethylene dimethacrylate) layer which remains constant for all plates prepared in this study. The monolithic polymer layers prepared from mixtures containing a higher percentage of cyclohexanol are more compact and less permeable, thus affecting the separation. For example, the time required for the separation of peptides using the layer prepared from mixture 5 was six times longer than that observed with the layer prepared from mixture 3. On the other hand, the mechanical strength of the layers weakens with an increase in the percentage of decanol. The layers prepared from mixtures 1 and 2 were too loose to keep the defined thickness and surface integrity during mold disassembly while layers based on mixtures 4 and 5 adhered more strongly to the bottom glass plate and the mold disassembly became progressively more difficult. Based on these observations, polymerization mixture 3 containing 40% dodecanol and 20% cyclohexanol was used for the preparation of the plates used in subsequent experiments.
Photografted Superhydrophilic Channel for Ion Exchange Chromatography
Photografting is a very powerful tool enabling the tailoring of the surface chemistry of porous polymer monoliths.82,88–89 Irradiation time at constant UV light power controls the extent of photografting ultimately affecting the surface water contact angle. Fig. 4 demonstrates that the water droplet contact angle after 7.5 min irradiation became close to 0°. This value means that the droplet wets the monolith instantaneously after placing it at the surface of the layer thus confirming its superhydrophilic character as defined in the literature.90
Figure 4.
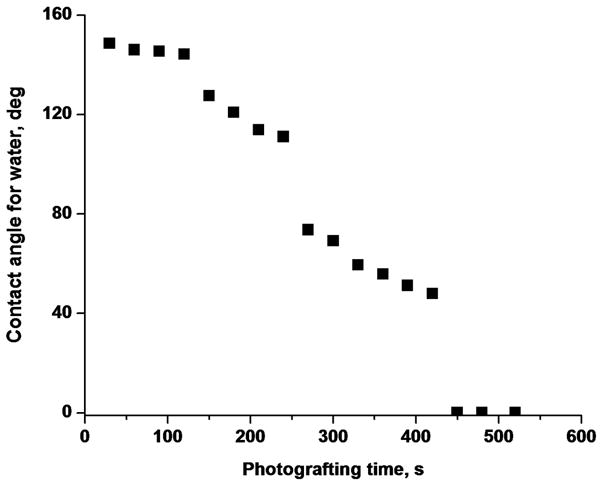
Effect of photografting time on water contact angle on the surface of poly(butyl acrylate-co-ethylene dimethacrylate) monolithic layer functionalized with the 2-acrylamido-2-methyl-1-propanesulfonic acid – 2-hydroxyethyl methacrylate mixture.
During the initial optimization experiments, we filled pores of the entire superhydrophobic layer with mixtures of 2-acrylamido-2-methyl-1-propanesulfonic acid, 2-hydroxyethylmethacrylate and benzophenone dissolved in tert-butanol-water. Photografting was then carried out by UV irradiation through a simple mask to afford a 0.6 mm wide channel across one side of the plate. A mixture of ionizable and neutral hydrophilic monomers was used since no migration of peptides was observed in a channel grafted with neat 2-acrylamido-2-methyl-1-propanesulfonic acid, most likely due to excessive coverage with sulfonic acid functionalities. We found that a solution containing of 2 wt.% 2-acrylamido- 2-methyl-1-propanesulfonic acid and 13 wt.% 2-hydroxyethyl methacrylate grafted for 7.5 min afforded channels with the best separation performance. Fig. 5 shows the optical microscopy image of the cross section of the patterned hydrophilic virtual channel filled with aqueous solution of red dye. The aqueous phase is retained within the three dimensional channel by surface tension at the interface, which prevents it from entering the adjacent superhydrophobic areas of the monolithic thin layer.
Figure 5.

Optical microscopic picture of cross section of the superhydrophilic channel filled with aqueous solution of red dye.
Two Dimensional Separation of Peptides
The superhydrophobic monolithic porous polymer layer with the photografted hydrophilic channel85 was used for the 2D separation of peptides. Migration in the first dimension was achieved using a mobile phase of 30 vol.% acetonitrile in aqueous 0.2 mol/L ammonium acetate at pH 7.0. Although migration of peptides could be achieved with the aqueous buffer alone, addition of the organic solvent accelerated the separation and provided for higher Rf values. A wick saturated with this mobile phase was placed on the top of the channel close to the location where the sample was applied. This approach was found more efficient than the typical approach to development in a chamber. The fluorescamine labeled peptides migrated in the channel over 16 mm in less than 1 minute. This separation yielded three distinct spots visualized using UV light. Separation in the second dimension was carried out with the dried plate turned by 90° in a chamber using 0.1 vol.% trifluoroacetic acid in 40 vol. % aqueous acetonitrile. A migration distance of 22 mm was achieved in less than 2 min. Fig. 6 shows the 2D separation of a mixture of fluorescently labeled peptides detected with UV light. Assignment of the spots results from Rf values calculated from 2D migrations of the individual peptides. It is worth noting that the val-tyr-val sample we used separated in two spots. However mass spectral analysis of the commercial tripeptide indicated that it contained molecules with the expected molecular mass of 380 and a dimer with a mass of 760, thus explaining the origin of the two spots.
Figure 6.
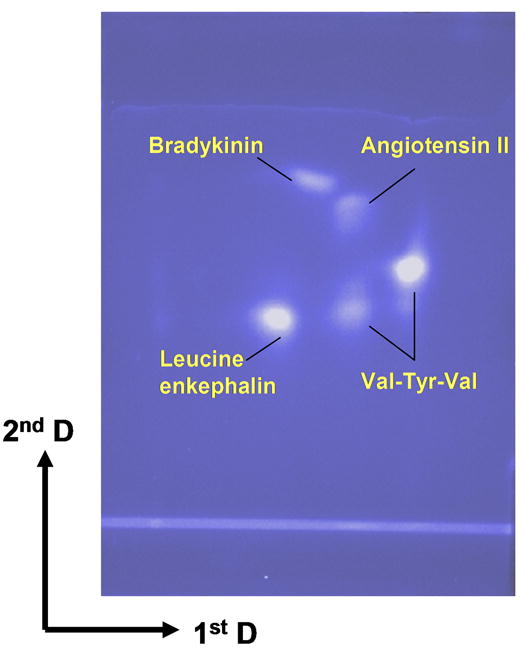
Two-dimensional TLC separation of a mixture of labeled peptides on 50 μm thick monolithic polymer layer with dual chemistry using UV detection.
Table 2 presents the properties of the peptides used in these experiments. They differ both in pI value and in hydrophobicity. The channel in our plate is designed to facilitate separation based on ion exchange interactions while the rest of the plate featuring hydrophobicity effects separation in the reverse phase mode. Indeed, the peptides first get separated in the channel according to their pI value while their hydrophobicity controls separation in the second dimension. Since acetonitrile accelerates the separation and therefore is part of mobile phases used in both dimensions, the separation modes may not be completely orthogonal. Yet, the spread of spots shown in Fig. 6 demonstrates that both separation mechanisms and selectivities are different.
Table 2.
Properties of Separated Peptides and their Retardation Factors Rf in the First and Second Dimension Separations
| peptide | mol. mass | pI a) | H.i.b) | Rf in 1st D | Rf in 2nd D |
|---|---|---|---|---|---|
| Val-Tyr-Val | 379.5 | 5.9 | 71.7 | 0.87 | 0.44 |
| leucine enkephalin | 555.6 | 5.9 | 52.0 | 0.41 | 0.34 |
| angiotensin II | 1046.2 | 6.8 | 28.5 | 0.66 | 0.56 |
| bradykinin | 1060.2 | 12.0 | 2.6 | 0.59 | 0.60 |
pI data were calculated using http://www.innovagen.se/custom-peptide-synthesis/peptide
Hydrophobicity index at pH 7.0 from http://www.sigma-genosys.com.
2D TLC/DESI MS of Peptides
Compounds separated in TLC are most often visualized via fluorescence. The more recent adoption of mass spectrometry in the arsenal of detection methods adds another dimension to the separation since it provides information about the molecular masses of the separated compounds. The simplest way to determine molecular mass of the labeled compounds would be to visualize them and then probe the spots only. However, imaging the entire surface of the plate via multiple, closely spaced line scanning is a more reliable way to detect all separated compounds whether labeled or not.75,78
Efficient ionization from plate to achieve good peptide signals in DESI mode requires the optimization of numerous variables including geometric parameters (incident angle, emitter-to-surface distance, sampling capillary-to-surface distance, emitter-to-sampling capillary distance), spray parameters (voltage, nebulizer gas rate, solvent flow rate, spray solvent composition) and mass spectrometer conditions. A solvent flow rate of 5μL/min and an emitter-to-sampling capillary distance of 3–4 mm were found suitable for efficient ionization and sampling from our monolithic porous polymer layers with rough surface. Other parameters were similar to those optimized elsewhere.62
Unidirectional scanning 70 was then used to detect the separated peptides. The lane scan along the virtual channel after the first dimension separation started from the spotting point and ended at the edge of the channel. The surface scan after 2D separation started 1 mm above the first dimensional channel and ended at the solvent front observed in the second dimension separation. Each lane was 16 mm long and 71 lanes were scanned in about 90 minutes. The 0.3 mm lane spacing used affords good resolution and avoids “oversampling”. The plate was lowered by 2 mm while changing the lane to minimize contamination with the sputtered material resulted from previous scans.
The peptide positions along the virtual channel constructed from extracted ion profiles using DESI are shown in center of Fig. 7 and represent the separation of peptides in the first dimension channel. The values at x-axis represent the length of the channel. The location of the peaks (order of separation) is equal to that observed for labeled peptides. The separation profile also indicates a certain overlap of spots of the separated peptides and the mass spectra of the individual spots are not very clean.
Figure 7.

DESI-MS scan of the first dimension separation achieved in the 30 mm long virtual channel grafted with the 2-acrylamido-2-methyl-1-propanesulfonic acid – 2-hydroxyethyl methacrylate mixture (central spectrum) and MS spectra of individual peptides: (1) leucine enkephalin , (2) bradykinin, (3) angiotensin II, (4) val-tyr-val.
Fig. 8 only shows mass spectra corresponding to individual peptides found during the scanning of the plate after 2D separation. The spectra are significantly cleaner thus demonstrating that the resolution improved after separation in the second dimension. Also, the order of the monitored peptides changed to leucine enkephalin, val-tyr-val, angiotensin II, and bradykinin and is different than that observed in the first dimension. Use of the MS signals found via the line by line scanning of the entire plate for plotting results of the separation in 2D space would be meaningless. Since we did not have available means to scan the plate using computer controlled x-y-z stage, creation of an accurate 2D map to visualize the location of spots is not possible. However, the occurrence of peptide ions once again matches the migration distance of these peptides during the development in the reverse phase mode monitored previously via fluorescence as presented in Fig. 6.
Figure 8.

DESI-MS spectra of individual peptides observed during scan of the entire plate after two dimensional separation using monolithic polymer layer with dual chemistry. Peptides: (1) leucine enkephalin , (2) bradykinin, (3) angiotensin II, (4) val-tyr-val.
CONCLUSION
This study demonstrates the potential of superhydrophobic thin monolithic porous polymer layers in 2D chromatography. Preparation of the channel with properties orthogonal to the rest of the TLC plate is readily achieved via a photografting process that, with appropriate equipment, could readily be automated. The large difference in surface tension at the interface of the channel with the rest of the plate provides the means to retain the aqueous mobile phase within the channel. While we only have demonstrated here the 2D separation of a few peptides by combining ion exchange and reversed phase modes, our approach also enables combinations of other separation mechanisms for the separation of different families of compounds. Our current work is focused on the separation of more complex mixtures including intact proteins and their digests. The monolithic polymer plates have a resilient structure that is fully compatible with DESI MS analysis and future improvements in the area will include the use of a computer controlled x-y-z stage to scan the plate and create an accurate 2D map of the separated compounds.
Acknowledgments
This work was supported by a grant of the National Institute Institutes of Health (GM48364), which is gratefully acknowledged. F.S., I.A. and all analytical work performed at the Molecular Foundry, Lawrence Berkeley National Laboratory, were supported by the Office of Science, Office of Basic Energy Sciences, U.S. Department of Energy, under Contract No. DE-AC02-05CH11231. Financial support of Y.H. by China Scholarship Council affiliated with the Ministry of Education of China is also acknowledged.
References
- 1.Izmailov NA, Shraiber MS. Farmatsiya (Moscow) 1938:1–7. [Google Scholar]
- 2.Kirchner JG. Thin-Layer Chromatography. Wiley; New York: 1978. [Google Scholar]
- 3.Stahl E. Thin-Layer Chromatography. Springer; New York: 1969. [Google Scholar]
- 4.Poole CF. The Essence of Chromatography. Elsevier; Amsterdam: 2003. [Google Scholar]
- 5.Poole CF. J Chromatogr A. 1999;856:399–427. doi: 10.1016/s0021-9673(99)00430-6. [DOI] [PubMed] [Google Scholar]
- 6.Hauck HE, Schulz M. J Chromatogr Sci. 2002;40:550–552. doi: 10.1093/chromsci/40.10.550. [DOI] [PubMed] [Google Scholar]
- 7.Bezuidenhout LW, Brett MJ. J Chromatogr A. 2008;1183:179–185. doi: 10.1016/j.chroma.2007.12.089. [DOI] [PubMed] [Google Scholar]
- 8.Clark JE, Olesik SV. Anal Chem. 2009;81:4121–4129. doi: 10.1021/ac9004293. [DOI] [PubMed] [Google Scholar]
- 9.Bakry R, Bonn GK, Mair D, Svec F. Anal Chem. 2007;79:486–493. doi: 10.1021/ac061527i. [DOI] [PubMed] [Google Scholar]
- 10.Poole CF. J Chromatogr A. 2003;1000:963–984. doi: 10.1016/s0021-9673(03)00435-7. [DOI] [PubMed] [Google Scholar]
- 11.Wedge DE, Nagle DG. J NatProd. 2000;63:1050–1054. doi: 10.1021/np990628r. [DOI] [PubMed] [Google Scholar]
- 12.Holstege DM, Francis T, Puschner B, Booth MC, Galey FD. J Agric Food Chem. 2000;48:60–64. doi: 10.1021/jf990873g. [DOI] [PubMed] [Google Scholar]
- 13.Bathori M, Blunden G, Kalasz H. Chromatographia. 2000;52:815–817. [Google Scholar]
- 14.Aboul-Enein HY, Abu-Zaid S. Anal Let. 2001;34:2099–2110. [Google Scholar]
- 15.Soczewinski E, Hawryl MA, Hawryl A. Chromatographia. 2001;54:789–794. [Google Scholar]
- 16.Grishkovets VI. ChemNat Comp. 2001;37:57–60. [Google Scholar]
- 17.Hawryl MA, Soczewinski E. J Planar Chromatogr-Modern TLC. 2001;14:415–421. [Google Scholar]
- 18.Hawryl MA, Hawryl A, Soczewinski E. J Planar Chromatogr-Modern TLC. 2002;15:4–10. [Google Scholar]
- 19.Tuzimski T, Bartosiewicz A. Chromatographia. 2003;58:781–788. [Google Scholar]
- 20.Tuzimski T, Soczewinski E. J Planar Chromatogr-Modern TLC. 2003;16:263–267. [Google Scholar]
- 21.Bartnik M, Glowniak K, Dul R. J Planar Chromatogr-Modern TLC. 2003;16:206–210. [Google Scholar]
- 22.Medic-Saric M, Jasprica L, Mornar A, Smolcic-Bubalo A, Golja P. J Planar Chromatogr-Modern TLC. 2004;17:459–463. [Google Scholar]
- 23.Pyka A, Dolowy M. J Liquid Chromatogr. 2004;27:2031–2038. [Google Scholar]
- 24.Tuzimski T. J Planar Chromatogr-Modern TLC. 2004;17:46–50. [Google Scholar]
- 25.Hawryl MA, Waksmundzka-Hajnos M, Inglot T. J Liquid Chromatogr. 2005;28:2245–2259. [Google Scholar]
- 26.Waksmundzka-Hajnos M, Petruczynik A, Hajnos MT, Tuzimski T, Hawry A, Bogucka-Kocka A. J Chromatogr Sci. 2006;44:510–517. doi: 10.1093/chromsci/44.8.510. [DOI] [PubMed] [Google Scholar]
- 27.Salo PK, Vilmunen S, Salomies H, Ketola RA, Kostiainen R. Anal Chem. 2007;79:2101–2108. doi: 10.1021/ac0620359. [DOI] [PubMed] [Google Scholar]
- 28.Daszykowski M, Hawryl M, Waksmundzka-Hajnos M, Walczak B. Acta Chromatogr. 2008;20:283–307. [Google Scholar]
- 29.Pasilis SP, Kertesz V, Van Berkel GJ, Schulz M, Schorcht SJ. Mass Spectrom. 2008;43:1627–1635. doi: 10.1002/jms.1431. [DOI] [PubMed] [Google Scholar]
- 30.Poole CF, Poole SK. J Chromatogr A. 1995;703:573–612. [Google Scholar]
- 31.Ciesla L, Waksmundzka-Hajnos M. J Chromatogr A. 2009;1216:1035–1052. doi: 10.1016/j.chroma.2008.12.057. [DOI] [PubMed] [Google Scholar]
- 32.Tuzimski T, Soczewinski E. JChromatogr A. 2002;961:277–283. doi: 10.1016/s0021-9673(02)00693-3. [DOI] [PubMed] [Google Scholar]
- 33.Gadzikowska M, Petruczynik A, Waksmundzka-Hajnos M, Hawryl M, Jozwiak G. J Planar Chromatogr-Modern TLC. 2005;18:127–131. [Google Scholar]
- 34.Tuzimski T, Wojtowicz J. J Liquid Chromatogr. 2005;28:277–287. [Google Scholar]
- 35.Habibi-Goudarzi S, Ruterbories KJ, Nurok D. J Planar Chromatogr. 1988;1:161–68. [Google Scholar]
- 36.Glensk M, Bialy Z, Jurzysta M, Cisowski W. Chromatographia. 2001;54:669–672. [Google Scholar]
- 37.Ciesla L, Bogucka-Kocka A, Hajnos M, Petruczynik A, Waksmundzka-Hajnos M. J Chromatogr A. 2008;1207:160–168. doi: 10.1016/j.chroma.2008.08.065. [DOI] [PubMed] [Google Scholar]
- 38.Ciesla L, Petruczynik A, Hajnos M, Bogucka-Kocka A, Waksmundzka-Hajnos M. J Planar Chromatogr-Modern TLC. 2008;21:447–452. doi: 10.1016/j.chroma.2008.08.065. [DOI] [PubMed] [Google Scholar]
- 39.Ciesla L, Petruczynik A, Hajnos M, Bogucka-Kocka A, Waksmundzka-Hajnos M. J Planar Chromatogr-Modern TLC. 2008;21:237–241. doi: 10.1016/j.chroma.2008.08.065. [DOI] [PubMed] [Google Scholar]
- 40.Hawryl MA, Waksmundzka-Hajnos M. J Planar Chromatogr-Modern TLC. 2006;19:92–97. [Google Scholar]
- 41.Tuzimski T. J Planar Chromatogr-Modern TLC. 2005;18:349–357. [Google Scholar]
- 42.Luczkiewicz M, Migas P, Kokotkiewicz A, Walijewska M, Cisowski W. J Planar Chromatogr-Modern TLC. 2004;17:89–94. [Google Scholar]
- 43.Glensk M, Sawicka U, Mazol I, Cisowski W. J Planar Chromatogr-Modern TLC. 2002;15:463–465. [Google Scholar]
- 44.Glensk M, Cisowski W. J Planar Chromatogr-Modern TLC. 2000;13:9–11. [Google Scholar]
- 45.Glensk M, Czekalska M, Cisowski W. J Planar Chromatogr-Modern TLC. 2001;14:454–456. [Google Scholar]
- 46.Sherma J. Anal Chem. 2008;80:4253–4267. doi: 10.1021/ac7023415. [DOI] [PubMed] [Google Scholar]
- 47.Sherma J. Anal Chem. 2006;78:3841–3852. doi: 10.1021/ac060098l. [DOI] [PubMed] [Google Scholar]
- 48.Busch KL. J Chromatogr A. 1995;692:275–290. doi: 10.1016/0021-9673(94)00769-6. [DOI] [PubMed] [Google Scholar]
- 49.Gonnord MF, Levi F, Guiochon G. J Chromatogr. 1983;271:1–6. [Google Scholar]
- 50.Nurok D, Habibi-Goudarzi S, Kleyle R. AnalChem. 1987;59:2424–28. [Google Scholar]
- 51.Gusev AI, Vasseur OJ, Proctor A, Sharkey AG, Hercules DM. Anal Chem. 1995;67:4565–4570. [Google Scholar]
- 52.Gusev AI, Proctor A, Rabinovich YI, Hercules DM. Anal Chem. 1995;67:1805–1814. [Google Scholar]
- 53.Nicola AJ, Gusev AI, Hercules DM. Appli Spectrosc. 1996;50:1479–1482. [Google Scholar]
- 54.Mehl JT, Gusev AI, Hercules DM. Chromatographia. 1997;46:358–364. [Google Scholar]
- 55.Isbell DT, Gusev AI, Taranenko NI, Chen CH, Hercules DM. Fresenius' J AnalChem. 1999;365:625–630. [Google Scholar]
- 56.Vermillion-Salsbury RL, Hoops AA, Gusev AI, Hercules DM. Intern J Environm Anal Chem. 1999;73:179–190. [Google Scholar]
- 57.Gusev AI. Fresenius' J Anal Chem. 2000;366:691–700. doi: 10.1007/s002160051563. [DOI] [PubMed] [Google Scholar]
- 58.Panchagnula V, Mikulskis A, Song L, Wang Y, Wang M, Knubovets T, Scrivener E, Golenko E, Krull IS, Schulz M, Heinz EH, Patton WF. J Chromatogr A. 2007;1155:112–123. doi: 10.1016/j.chroma.2007.04.029. [DOI] [PubMed] [Google Scholar]
- 59.Salo PK, Salomies H, Harju K, Ketola RA, Kotiaho T, Yli-Kauhaluoma J, Kostiainen R. J Am Soc Mass Spectrom. 2005;16:906–915. doi: 10.1016/j.jasms.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 60.Salo PK, Vilmunen S, Salomies H, Ketola RA, Kostiainen R. AnalChem. 2007;79:2101–2108. doi: 10.1021/ac0620359. [DOI] [PubMed] [Google Scholar]
- 61.Takats Z, Wiseman JM, Gologan B, Cooks RG. Science. 2004;306:471–473. doi: 10.1126/science.1104404. [DOI] [PubMed] [Google Scholar]
- 62.Takats Z, Wiseman JM, Cooks RG. J Mass Spectrom. 2005;40:1261–1275. doi: 10.1002/jms.922. [DOI] [PubMed] [Google Scholar]
- 63.Cooks RG, Ouyang Z, Takats Z, Wiseman JM. Science. 2006;311:1566–1570. doi: 10.1126/science.1119426. [DOI] [PubMed] [Google Scholar]
- 64.Wiseman JM, Ifa DR, Song QY, Cooks RG. Angew Chem Intern Ed. 2006;45:7188–7192. doi: 10.1002/anie.200602449. [DOI] [PubMed] [Google Scholar]
- 65.Pasilis SP, Kertesz V, Van Berkel GJ. Anal Chem. 2007;79:5956–5962. doi: 10.1021/ac070527v. [DOI] [PubMed] [Google Scholar]
- 66.Ifa DR, Wiseman JM, Song QY, Cooks RG. Intern J Mass Spectrom. 2007;259:8–15. [Google Scholar]
- 67.Ifa DR, Gumaelius LM, Eberlin LS, Manicke NE, Cooks RG. Analyst. 2007;132:461–467. doi: 10.1039/b700236j. [DOI] [PubMed] [Google Scholar]
- 68.Wiseman JM, Ifa DR, Zhu YX, Kissinger CB, Manicke NE, Kissinger PT, Cooks RG. Proceed Nat Acad Sci USA. 2008;105:18120–18125. doi: 10.1073/pnas.0801066105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wiseman JM, Ifa DR, Venter A, Cooks RG. Nature Protocols. 2008;3:517–524. doi: 10.1038/nprot.2008.11. [DOI] [PubMed] [Google Scholar]
- 70.Kertesz V, Van Berkel GJ. Anal Chem. 2008;80:1027–1032. doi: 10.1021/ac701947d. [DOI] [PubMed] [Google Scholar]
- 71.Kertesz V, Van Berkel GJ. Rapid Commun Mass Spectrom. 2008;22:2639–2644. doi: 10.1002/rcm.3662. [DOI] [PubMed] [Google Scholar]
- 72.Green FM, Stokes P, Hopley C, Seah MP, Gilmore IS, O'Connor G. Anal Chem. 2009;81:228. doi: 10.1021/ac802440w. [DOI] [PubMed] [Google Scholar]
- 73.Van Berkel GJ, Ford MJ, Deibel MA. Anal Chem. 2005;77:1207–1215. doi: 10.1021/ac048217p. [DOI] [PubMed] [Google Scholar]
- 74.Kauppila TJ, Talaty N, Salo PK, Kotiah T, Kostiainen R, Cooks RG. Rapid Commun Mass Spectrom. 2006;20:2143–2150. doi: 10.1002/rcm.2566. [DOI] [PubMed] [Google Scholar]
- 75.Van Berkel GJ, Kertesz V. Anal Chem. 2006;78:4938–4944. doi: 10.1021/ac060690a. [DOI] [PubMed] [Google Scholar]
- 76.Van Berkel GJ, Tomkins BA, Kertesz V. Anal Chem. 2007;79:2778–2789. doi: 10.1021/ac0622330. [DOI] [PubMed] [Google Scholar]
- 77.Janecki DJ, Novotny AL, Woodward SD, Wiseman JM, Nurok D. J Planar Chromatogr-Modern TLC. 2008;21:11–14. [Google Scholar]
- 78.Pasilis SP, Kertesz V, Van Berkel GJ, Schulz M, Schorcht S. J Mass Spectrom. 2008;43:1627–1635. doi: 10.1002/jms.1431. [DOI] [PubMed] [Google Scholar]
- 79.Pasilis SP, Kertesz V, Van Berkel GJ, Schulz M, Schorcht S. Anal Bioanal Chem. 2008;391:317–324. doi: 10.1007/s00216-008-1874-6. [DOI] [PubMed] [Google Scholar]
- 80.Petro M, Svec F, Fréchet JMJ. Biotech Bioeng. 1996;49:355–363. doi: 10.1002/(SICI)1097-0290(19960220)49:4<355::AID-BIT1>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 81.Hilder EF, Svec F, Fréchet JMJ. J Chromatogr A. 2004;1044:3–22. doi: 10.1016/j.chroma.2004.04.057. [DOI] [PubMed] [Google Scholar]
- 82.Eeltink S, Hilder EF, Geiser L, Svec F, Fréchet JMJ, Rozing GP, Schoenmakers PJ, Kok WT. J Sep Sci. 2007;30:407–13. doi: 10.1002/jssc.200600316. [DOI] [PubMed] [Google Scholar]
- 83.Irgum K, Viklund C, Svec F, Fréchet JMJ. Biotech Progr. 1997;13:597–600. doi: 10.1021/bp9700667. [DOI] [PubMed] [Google Scholar]
- 84.Pucci V, Raggi M-A, Svec F, Fréchet JMJ. J Sep Sci. 2004;27:779–788. doi: 10.1002/jssc.200401828. [DOI] [PubMed] [Google Scholar]
- 85.Levkin PA, Svec F, Fréchet JMJ. Adv Funct Mater. 2009;19:1993–1998. doi: 10.1002/adfm.200801916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li XM, Reinhoudt D, Crego-Calama M. Chem Soc Rev. 2007;36:1350–1368. doi: 10.1039/b602486f. [DOI] [PubMed] [Google Scholar]
- 87.Zhang X, Shi F, Niu J, Jiang YG, Wang ZQ. J Mater Chem. 2008;18:621–633. [Google Scholar]
- 88.Rohr T, Hilder EF, Donovan JJ, Svec F, Fréchet JMJ. Macromolecules. 2003;36:1677–1684. [Google Scholar]
- 89.Stachowiak TB, Svec F, Fréchet JMJ. Chem Mater. 2006;18:5950–5957. [Google Scholar]
- 90.Cebeci FC, Wu ZZ, Zhai L, Cohen RE, Rubner MF. Langmuir. 2006;22:2856–2862. doi: 10.1021/la053182p. [DOI] [PubMed] [Google Scholar]