

Abstract
It is widely anticipated that the coming year will be marked by the complete characterization of DNA sequence of protein coding regions of thousands of human individuals. A number of existing computational methods use comparative protein sequence analysis and analysis of protein structure to predict the functional effect of coding human alleles. Functional and structural analysis of coding allelic variants can inform various aspects of research on human genetic variation. In population and evolutionary genetics it helps estimating the strength of purifying selection against deleterious missense mutations and study the imprint of demographic history on deleterious genetic variation. In medical genetics it may assist in the interpretation of uncharacterized mutations in genes involved in monogenic and oligogenic diseases. It has a potnetial to facilitate medical sequencing studies searching for genes underlying Mendelian diseases or harboring rare alleles involved in complex traits.
Introduction
The number of known individual human genomes is rapidly growing. Multiple ongoing projects aim to sequence thousands of new genomes or “exomes” (protein coding fractions of genomes) in the near future. These projects are enabled by rapidly evolving sequencing technologies and motivated by interest in the genetic background of human phenotypic variation, especially of medical relevance. Additionally, they offer the intriguing possibility to uncover the demographic history of the human population and advance our understanding of evolutionary mechanisms.
Although most of the work on interpreting human allelic variation is confined to the domain of statistical and population genetics, a growing body of literature is devoted to the analysis of coding genetic variation from the perspective of protein function, structure and evolution. This work is interesting in its own right because of its focus on characterizing the protein function repertoire of an individual human. At the same time it can greatly assist human medical and evolutionary genetics by contrasting functionally significant variation with likely neutral, non-functional variation. Alleles that influence phenotypic variation and alleles that are targets of natural selection are expected to be functionally important. Therefore, functional considerations can be highly informative for a variety of genetics studies interpreting incoming data on numerous individual human genomes.
We review current computational methods that use evolutionary or structural approaches to predict the functional effect of protein allelic variants, also known as non-synonymous single nucleotide polymorphisms (SNPs or nsSNPs). We discuss results of recent evolutionary and population genetics studies that relied on known or predicted functional effects of allelic variants. We also review applications of computational evolutionary and structural methods to medical genetics and genomics.
Predicting the functional effect of nonsynonymous SNPs
Arguably, the most useful application of protein sequence and structure analysis to genetics is in methods for predicting the functional effects of allelic variants. Current prediction methods are not yet considered highly accurate, but they are already at the point of being useful for some applications. Most methods either rely solely on phylogenetic information or combine phylogenetic information with structural analysis, annotation, and sequence properties. Below, we review phylogenetic and structural aspects of the methods separately.
Predicting the effect of nonsynonymous SNPs from phylogeny
Two major assumptions underlie the use of phylogenetic approaches for predicting the functional effect of amino acid replacements. First, it is assumed that variants that destroy a protein’s biochemical function or cause a medically detrimental phenotype also cause a loss of evolutionary fitness, making them deleterious in the evolutionary sense. Such variants are subject to purifying selection. Second, it is assumed that a deleterious allelic variant in the current population is also deleterious in homologous genes in other living and extinct species, or in other words that the fitness landscape is constant. Under these assumptions, the functional effect of an amino acid change can be predicted from the pattern of amino acids observed in the corresponding position of a multiple sequence alignment of related sequences. An allele that does not fit the pattern observed in the phylogeny is predicted to be damaging, especially if it is in a conserved position. Many methods incorporate prior information such as amino acid substitution matrices, to account for the fact that the multiple sequence alignments are often too shallow to produce a confident prediction unaided [1,2,3,4].
Phylogenetic methods typically have two steps in making their predictions. The first step is to choose appropriate homologous sequences and construct a multiple sequence alignment. The choice of sequences is critical because very shallow alignments are uninformative, while deep alignments may include very distant sequences that will mislead predictions. The most straightforward way of constructing an alignment would be to include only orthologs. Most existing methods, however, also include paralogs. This may be justified because most damaging mutations are known to affect the stability of the protein structure, which is expected to be highly similar among paralogs. Currently, limiting the analysis to orthologs would frequently result in shallow alignments. However, this may change as a result of many new sequencing projects, and new methods may choose to limit the analysis to orthologs in closely related species if sufficiently diverse informative alignments can be generated.
The second step is to evaluate how well an allelic variant fits the amino acid pattern observed in the phylogeny. Existing methods use positional conservation measures, probabilistic scoring functions, or both of these. MAPP [5] and Align-GVGD [6, 7] use a different approach based on conservation of amino acid physico-chemical properties. Phylogenetic relationships between sequences are taken into account by using sequence weights (SIFT, PMut, MAPP) [3,4,5,8], a pre-computed species tree (LRT) [9], or other heuristic algorithms such as PSIC (PolyPhen-2 and SNAP) [10,11].
Two principal difficulties hamper development of phylogenetic methods for functional prediction of protein variants. The first is the existence of compensatory pathogenic deviations (CPDs). A highly damaging human allele can be benign in other species because of compensatory changes in other sites of the same protein or in its interaction partners. Such an allele may be observed in sequences included in the multiple sequence alignment, causing a phylogenetic method to erroneously predict it as benign. The existence of CPDs violates the assumption of a constant fitness landscape at a single amino acid site. A number of computational and experimental studies have demonstrated high prevalence of CPDs [12,13,14]. Some methods (e.g. PolyPhen-2 [10]) address this problem by considering conservation in close homologs less significant than conservation in remote homologs, since remote homologs are likely to have more CPDs. As compensatory mutation and co-evolution become better understood, it may be possible to to overcome this problem more directly by predicting compensatory mutations or co-evolved locations [15,16,17].
The second difficulty is in the inability of phylogenetic techniques to distinguish between strongly deleterious mutations and moderately deleterious mutations. This distinction is highly important in some applications, most notably in medical genetic diagnostics. The problem here is that a sequence variant associated with a fitness loss of as little as 0.1% or even 0.01% has almost no chance to become completely fixed in any population of realistic size, though it may segregate within the population at frequencies as high as 3–5% [18]. The corresponding amino acid position will be completely conserved in the phylogeny. Such a variant may be predicted as damaging by phylogenetic methods, despite having a very weak effect and most likely no medical significance.
Phylogeny is currently the most useful source of information for predicting the effect of nonsynonymous SNPs, and its value will increase with the number of known sequences and, possibly, new methods incorporating models of molecular evolution.
Predicting the effect of nonsynonymous SNPs from structural features
Another approach to predicting the effects of nonsynonymous SNPs is to use structural information. As mentioned above, structurally destabilizing mutations are extremely common among disease mutations [19], with one study identifying up to 74% of disease mutations as structurally destabilizing [20]. A more recent study suggests that destabilizing variants are common even among variants that are not recognized as disease-causing, and nearly half of variants present in the human population may be structurally destabilizing [21].
Predicting the change in stability caused by a mutation means estimating the change it will cause in folding free energy of the protein, or ΔΔG. The most theoretically straightforward way to estimate ΔΔG is to score the contribution of each residue to folding using a physics-based energy function, or even a full Molecular Dynamics simulation. In practice, however, this kind of scoring is normally very computationally intensive. This difficulty has led to the development a number of not purely physical energy functions that require less computation. These include purely statistical energy functions, which are constructed by using statistical methods to analyze structural features of known destabilizing mutations. They also include empirical or knowledge-based energy functions, which use physical modeling as a starting point but weight the parameters of these models using empirical data [22].
All three classes of energy functions are still advancing. A recently developed statistical method, PoPMuSiC-2.0, uses a neural network to combine an ensemble of 24 statistical potentials, each one optimized for a different level of solvent accessibility [23]. Another, AutoMute, converts a protein structure to a geometric network of neighboring amino acids, and uses this representation to train a machine learning algorithm on mutations with known effects on stability [24]. Empirical energy functions also continue to be developed. One recent example focuses on predicting the strength of pairwise atomic interactions [25]. Another recent study uses an empirical energy function to identify the folding cores of proteins [26]. The rapid growth of computing resources also makes purely physics-based methods more attractive than they once were. Recent attempts to predict folding stability of proteins have made use of optimized physical energy functions [27], theoretical advances in physical models [28], and even straightforward Molecular Dynamics simulations [29]. A variety of older energy functions also remain in use and in development, with two of the most prominent being FoldX [22] and CUPSAT [30].
It is also possible to abandon energy functions altogether and infer the protein’s structural properties from its sequence. Emidio Capriotti and colleagues successfully trained two different machine learning systems to predict ΔΔG from only sequence information [31,32]. The authors of these studies suggest that these sequence-based methods can be combined with structural methods for higher accuracy, and two available tools, I-Mutant 2.0 [33] and MUPro [34], use this strategy. More recently, sequence-based methods have been used to predict the effects of multiple mutations [35,36]. This may be an area where the sequence-based approach has an advantage over structural approaches. The combined structural effect from two distinct mutations may be extremely complicated and difficult to predict, while a doubly mutated sequence is not very much more difficult to analyze than a singly mutated sequence. One disadvantage of the sequence-based approach in either case is that the inner workings of the trained classifier tend to be opaque, so these systems provide little physical insight about the mechanism of stabilization or destabilization.
All of these methods report similar accuracy of prediction: their predicted ΔΔG is reported to correlate with empirically measured ΔΔG with a correlation coefficient of approximately 0.8. These values are likely to be inflated, though: when Potapov et al. tested several widely-used potentials, they found that, though all of the methods tested claim correlation coefficients close to 0.8, the actual values ranged from 0.26 to 0.59 [37]. Methods that attempt to predict mutations as destabilizing or non-destabilizing, or predict the sign of ΔΔG, claim accuracies in the range of 80-85%. Though Potapov et al did not address this question directly, their results suggest that these tools may be better at this task than at direct prediction of ΔΔG. It is frustrating that predictions of ΔΔG are so inaccurate, since, as mentioned above, an accurate prediction would theoretically be extremely effective at predicting the functional effects of SNPs. Currently, phylogenetic methods are much more accurate than structural predictions.
One common way of dealing with this inaccuracy is to replace detailed predictions of protein stability with heuristic features that are considered correlated with stability. These features may focus on describing the mutation site, including its solvent accessibility, secondary structure, domain and functional annotations, and crystallographic B-factor; or they may focus on structural properties of the amino acids, such as its polarity, charge, and volume. Some methods, like SNPs3D [20] or TopoSNP [38], compute more detailed structural features, such as the formation of internal cavities or the loss of salt bridges. Many tools used for predicting the functional effects of SNPs use heuristics like these, often in combination with phylogenetic information.
Machine learning and classifiers
Many commonly used methods improve their performance by combining multiple different sources of data into a single classifier. This typically includes multiple different sources of both structural and phylogenetic information combined using machine learning techniques such as neural networks (PMut, SNAP) [8,11], support vector machines (SNPs3D, LS-SNP, PhD-SNP) [39,40,41], Naïve Bayes (PolyPhen-2) [10], or specifically designed custom algorithms (MSRV) [42]. These combined classifiers, while still not highly accurate, have moderately high success rates at predicting damaging variants. In general, highly confident predictions of these methods — those made with more inputs or more informative inputs — are highly accurate, while less confident predictions are not much better than random guesses. This gives these methods a large middle region of low-confidence predictions in between the confident neutral and deleterious predictions (Fig. 1). A list of these classifiers, along with some similar methods that do not use machine learning, can be found in Table 1.
Figure 1. Discrimination between frequent polymorphisms and disease mutations with PolyPhen-2 score.
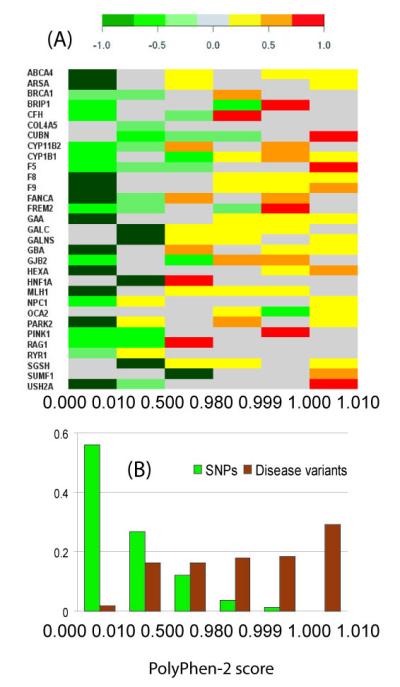
31 human genes were selected so that for each gene exists (i) at least one non-synonymous SNP listed in UniProt with minor allele frequency reaching at least 15% in one population; (ii) at least one disease-associated sequence variant annotated in the UniProt database; (iii) clinical genetic testing reviewed in NCBI GeneTests database (URL:http://www.ncbi.nlm.nih.gov/sites/GeneTests/review/) Hemoglobin beta was not included. The total set includes 499 disease variants and 82 SNPs (see Supplementary Table 1 for the detailed list). PolyPhen-2 score is based on the Naïve Bayes posterior probability with larger values reflecting the higher likelihood of a variant to be damaging [10]. The score value range was split into six bins including marginal values 0, 1 and four intermediate approximately equipopulated intervals. (A) The plot of the fraction of disease mutations minus the fraction of SNPs for each gene and score interval. As shown by the color code above, the green colors depict the prevalence of SNPs and the red colors the prevalence of disease mutations, respectively. (B) The histogram of SNPs and mutations populating each interval for all genes.
Table 1.
List of current computational methods for predicting the functional effects of non-synonymous alleles.
| Method Name | Description | URL |
|---|---|---|
| Align-GVGD [6,7] |
Phylogenetic method using phyisco- chemical amino acid properties |
http://agvgd.iarc.fr/agvgd_input.php |
| LRT [9] | Phylogenetic method using estimated rate of evolution |
(no server) |
| LS-SNP [40] | Database of polymorphisms, classified by phylogenetic and structural features combined with machine learning |
http://modbase.compbio.ucsf.edu/LS-SNP/ |
| MAPP [5] | Phylogenetic method using patterns of physico-chemical properties of amino acid substitutions |
http://mendel.stanford.edu/sidowlab/downloads/MAPP/index.html |
| MSRV [42.73] | Various phylogenetic methods combined with a custom machine learning algorithm |
http://bioinfo.steadybj.com/msrv |
| nsSNPAnalyzer [74] |
Phylogenetic and structural features combined with machine learning |
http://snpanalyzer.utmem.edu/ |
| PANTHER [75,76] |
Database of polymorphisms, classified by a phylogenetic method using patterns of amino acid substitutions |
http://www.pantherdb.org/tools/csnpScoreForm.jsp |
| PhD-SNP [41] | Phylogenetic method using patterns of amino acid substitutions |
http://gpcr2.biocomp.unibo.it/cgi/predictors/PhD-SNP/PhDSNP.cgi |
| PMut [8] | Phylogenetic and structural features combined with machine learning |
http://mmb2.pcb.ub.es:8080/PMut/ |
| PolyPhen-2 [10] | Phylogenetic and structural features combined with machine learning |
http://genetics.bwh.harvard.edu/pph2 |
| SIFT [3,4,77] | Phylogenetic method using patterns of amino acid substitutions |
http://sift.jcvi.org/ |
| SNAP [11] | Phylogenetic and structural features combined with machine learning |
http://cubic.bioc.columbia.edu/services/SNAP/ |
| SNPs3D [20,39,78] |
Combination of 2 methods, one phylogenetic and one based on structural features |
http://www.snps3d.org/ |
| TopoSNP [38] | Database of polymorphisms, classified by structural features |
http://gila.bioengr.uic.edu/snp/toposnp/ |
Protein structure and function in evolutionary and population genetics
As mentioned above, it is believed that the majority of mutations with functional effect are also deleterious. Quantitative estimates of the proportion of new mutations that are deleterious and strength of selection against them are of great importance to a number of central problems of evolutionary genetics, and the prediction of the functional efffects of deleterious mutations is an important tool in making these estimates.
Functional and structural considerations helped to characterize natural selection against deleterious alleles. It has been shown that the fraction of nonsynonymous SNPs located in functionally and structurally important regions is higher than the corresponding fraction of nonsynonymous substitutions between species. This discrepancy indicates that purifying selection against deleterious alleles has occurred in our evolutionary history. Analysis of the allele frequency distribution of human SNPs also strongly suggests selection against strongly deleterious alleles in humans. Many studies have shown that SNPs predicted to be damaging have, on average, lower allele frequencies (Figure 2) [1,2,43,44,45,46,47].
Figure 2. Allele frequency distribution.

The figure shows the derived allele frequency distribution for the three groups of polymorphisms from dbSNP database: 6337 coding synonymous SNPs (light yellow), 4687 non-synonymous SNPs predicted as benign by PolyPhen-2 (green), and 1301 non-synonymous SNPs predicted damaging by PolyPhen-2 (orange). The frequencies were measured in HAPMAP-YRI population (120 individuals).
The analysis of allele frequency distribution of human SNPs has allowed estimation of the distribution of fitness effects of new amino acid mutations. Statistical approaches grounded in the diffusion models of population genetics were developed to disentangle the influence of demographic history and natural selection on the observed allele frequency distribution. These approaches assume that synonymous and non-coding variation are not subject to selective forces, and so the variation we observe in them is due to demographic history. All studies agree that the majority of new missense mutations are moderately deleterious, about a quarter of new missense mutations are effectively neutral, and the rest are strongly deleterious. [45,48,49,50,51,52]
Beyond evolutionary genetics, these results have implications for the genetics of human complex phenotypes. Based on the above estimates and theoretical considerations, it has been suggested that rare deleterious alleles may be important contributors to the genetics of common human phenotypes. [50,53,54] Ongoing and planned sequencing studies will test this model.
Functional considerations are also helpful in the analysis of demographic history and stratification of the human population. Lohmueller et al. analyzed the distribution of deleterious alleles in individuals of African and European descent and suggested that the observed differences at the population level reflect differences in demographic history, specifically the presence of a historic population bottleneck in Europeans [55]. Barreiro et al. showed that protein coding regions show less differentiation between human populations due to negative selection [56]. Both studies used computational predictions of the functional effect of nonsynonymous SNPs.
Medical applications
Functional prediction of variants has the potential to be extremely useful in medicine. Genetic tests are now performed for an increasing number of diseases, and decisions about treatment depend on whether or not a patient has a variant that is known to cause the disease in question. The classification of previously unknown variants revealed through these tests is becoming a large problem for physicians. The IARC Unclassified Genetic Variants Working Group discussed this problem in a special issue of Human Mutation [57]. Among other questions, this issue considers whether the computational methods discussed above can be useful in identifying disease mutations. The authors conclude that computational methods are not accurate enough to be relied on completely, but that they can be very useful in combination with other data [58]. They propose incorporating computational analyses, along with all other sources of data, into a unified prediction model that would assign each method a confidence according to its accuracy. This would allow medical professionals to use these methods while compensating for their low accuracy compared to traditional genetic methods [59]. These recommendations have not yet been implemented, and there is currently no framework in place for computational predictions to be used in medical applications.
Another promising medical application uses functional prediction of variants to identify previously unknown genetic causes of Mendelian diseases. This method, developed by Sarah Ng and colleagues [44,60], begins by sequencing all protein-coding regions in several unrelated patients with the same disease. Assuming there is a single gene responsible for the disease in all patients, the sequencing should find a mutation in that gene in each patient. The list of genes that have mutations in all patients therefore becomes the list of candidate genes. The list can be further narrowed down by comparing against databases of neutral variation like dbSNP or HapMap. It can also be narrowed down using the computational prediction methods reviewed above, though this may require new statistical approaches to prevent causative genes from being overlooked due to a false negative in the prediction. Ng et al have used this method to identify causative genes for two diseases, one already known and one previously unknown [44,60].
With the production of several cancer genomes [61,62], cancer research seems also to be in a position to benefit from these methods. The study of SNPs in cancer is complicated by the presence of both “passenger” and “driver” mutations [63]. The typical cancer carries a large number of passenger mutations resulting from the highly mutagenic environment of cancer cells. These mutations may be loss of function and highly deleterious if appeared in germ line, but they are not the cause of the cancer. In contrast, the driver mutations, which actually caused the cancer, are not necessarily functionally impairing or structurally destabilizing: mutations in oncogenes (as opposed to tumor suppressors) are activating and highly advantageous to cell growth. These mutations therefore cannot be predicted using only the methods discussed above. The laboratory of Zemin Zhang has recently developed tools for predicting cancer driver mutations, built on top of general-purpose prediction programs like SIFT. These tools include CanPredict, which incorporates cancer-related scores into the SIFT algorithm [64], and B-SIFT, which modifies the SIFT algorithm to detect activating mutations in addition to damaging mutations [65].
One final area where these prediction methods may prove useful is in the analysis of rare alleles involved in human common diseases. Numerous candidate-gene-based, whole exome and whole genome sequencing studies aiming to identify the effect of rare nonsynonymous alleles on common phenotypes of complex inheritance are currently ongoing. The search for rare coding variants contributing to common diseases is motivated by theoretical arguments discussed above [50,53,54] and by highly successful candidate gene studies [66,67,68,69,70,71,72]. It is difficult to establish an association of a rare variant with a phenotype because statistical power is severely limited by low population frequency and also because the number of rare variants will require a very strict multiple test correction. A feasible way forward is to combine multiple rare variants observed in the same gene or pathway into a single statistical test. The association signal will be provided by functional variants in the gene or pathway, whereas neutral variants are a source of noise. Incorporating predictions or biochemical measurements of functional effect will potentially increase the power of these studies.
Conclusion
We are on the edge of knowing thousands and possibly millions of individual human genomes. Whether or not this massive amount of information will fulfill its promise to advance science and medicine depends on our ability to interpret sequencing data and identify the subset of functionally, phenotypically, and evolutionary relevant genetic variation. The perspective of protein function, structure, and evolution is of importance to all aspects of human genetic variation research, ranging from basic population and evolutionary genetics to genetics of complex traits, gene mapping, and clinical genetic diagnostics. We anticipate that this research area at the interface of molecular evolution, structural biology, and human genetics will be of growing importance in the next few years.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sunyaev S, Ramensky V, Koch I, Lathe W, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Human molecular genetics. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- 2.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic acids research. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome research. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic acids research. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stone EA, Sidow A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome research. 2005;15:978–986. doi: 10.1101/gr.3804205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, de Silva D, Zharkikh A, Thomas A. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. Journal of medical genetics. 2006;43:295–305. doi: 10.1136/jmg.2005.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic acids research. 2006;34:1317–1325. doi: 10.1093/nar/gkj518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrer-Costa C, Gelpí JL, Zamakola L, Parraga I, de la Cruz X, Orozco M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21:3176–3178. doi: 10.1093/bioinformatics/bti486. [DOI] [PubMed] [Google Scholar]
- “” 9.Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome research. 2009;19:1553–1561. doi: 10.1101/gr.092619.109. The authors develop a new phylogeny-based method for predicting the functional effects of non-synonymous alleles, and apply it to three individual human genomes. By comparing to PolyPhen and SIFT, they find that the overlap in prediction between different methods is small, and suggest using multiple methods to make more confident predictions.
- “ 10.Adzhubei I, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature Methods. 2010 doi: 10.1038/nmeth0410-248. (accepted) This paper introduces a new method PolyPhen-2 for functional prediction of SNPs. This method features a multiple sequence alignment pipeline, new predictive features, and a probabilistic classifier.
- 11.Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic acids research. 2007;35:3823–3835. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kondrashov AS, Sunyaev S, Kondrashov FA. Dobzhansky-Muller incompatibilities in protein evolution. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:14878–14883. doi: 10.1073/pnas.232565499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kulathinal RJ, Bettencourt BR, Hartl DL. Compensated deleterious mutations in insect genomes. Science (New York, NY) 2004;306:1553–1554. doi: 10.1126/science.1100522. [DOI] [PubMed] [Google Scholar]
- 14.Liao BY, Zhang J. Mouse duplicate genes are as essential as singletons. Trends in genetics : TIG. 2007;23:378–381. doi: 10.1016/j.tig.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 15.Pazos F, Valencia A. Protein co-evolution, co-adaptation and interactions. The EMBO Journal. 2008;27:2648–2655. doi: 10.1038/emboj.2008.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davis BH, Poon AF, Whitlock MC. Compensatory mutations are repeatable and clustered within proteins. Proceedings Biological sciences / The Royal Society. 2009;276:1823–1827. doi: 10.1098/rspb.2008.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chakrabarti S, Panchenko AR. Structural and functional roles of coevolved sites in proteins. PloS one. 5:e8591. doi: 10.1371/journal.pone.0008591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gillespie J. Population Genetics: A Concise Guide. The Johns Hopkins University Press; 2004. [Google Scholar]
- 19.Wang Z, Moult J. SNPs, protein structure, and disease. Human Mutation. 2001;17:263–270. doi: 10.1002/humu.22. [DOI] [PubMed] [Google Scholar]
- 20.Yue P, Li Z, Moult J. Loss of protein structure stability as a major causative factor in monogenic disease. Journal of Molecular Biology. 2005;353:459–473. doi: 10.1016/j.jmb.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 21.Allali-Hassani A, Wasney GA, Chau I, Hong BS, Senisterra G, Loppnau P, Shi Z, Moult J, Edwards AM, Arrowsmith CH, et al. A survey of proteins encoded by non-synonymous single nucleotide polymorphisms reveals a significant fraction with altered stability and activity. The Biochemical journal. 2009;424:15–26. doi: 10.1042/BJ20090723. [DOI] [PubMed] [Google Scholar]
- 22.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. Journal of Molecular Biology. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 23.Dehouck Y, Grosfils A, Folch B, Gilis D, Bogaerts P, Rooman M. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics. 2009;25:2537–2543. doi: 10.1093/bioinformatics/btp445. [DOI] [PubMed] [Google Scholar]
- 24.Masso M, Vaisman II. Accurate prediction of stability changes in protein mutants by combining machine learning with structure based computational mutagenesis. Bioinformatics. 2008;24:2002–2009. doi: 10.1093/bioinformatics/btn353. [DOI] [PubMed] [Google Scholar]
- 25.Ferrada E, Melo F. Effective knowledge-based potentials. Protein science : a publication of the Protein Society. 2009;18:1469–1485. doi: 10.1002/pro.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen M, Dousis AD, Wu Y, Wittung-Stafshede P, Ma J. Predicting protein folding cores by empirical potential functions. Archives of biochemistry and biophysics. 2009;483:16–22. doi: 10.1016/j.abb.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yin S, Ding F, Dokholyan NV. Eris: an automated estimator of protein stability. Nature Methods. 2007;4:466–467. doi: 10.1038/nmeth0607-466. [DOI] [PubMed] [Google Scholar]
- 28.Tan YH, Luo R. Protein stability prediction: a Poisson-Boltzmann approach. The journal of physical chemistry B. 2008;112:1875–1883. doi: 10.1021/jp709660v. [DOI] [PubMed] [Google Scholar]
- 29.Morra G, Colombo G. Relationship between energy distribution and fold stability: Insights from molecular dynamics simulations of native and mutant proteins. Proteins. 2008;72:660–672. doi: 10.1002/prot.21963. [DOI] [PubMed] [Google Scholar]
- 30.Parthiban V, Gromiha MM, Schomburg D. CUPSAT: prediction of protein stability upon point mutations. Nucleic acids research. 2006;34:W239–242. doi: 10.1093/nar/gkl190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Capriotti E, Fariselli P, Calabrese R, Casadio R. Predicting protein stability changes from sequences using support vector machines. Bioinformatics. 2005;21(Suppl 2):ii54–58. doi: 10.1093/bioinformatics/bti1109. [DOI] [PubMed] [Google Scholar]
- 32.Capriotti E, Fariselli P, Casadio R. A neural-network-based method for predicting protein stability changes upon single point mutations. Bioinformatics. 2004;20(Suppl 1):i63–68. doi: 10.1093/bioinformatics/bth928. [DOI] [PubMed] [Google Scholar]
- 33.Capriotti E, Fariselli P, Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic acids research. 2005;33:W306–310. doi: 10.1093/nar/gki375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng J, Randall A, Baldi P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins. 2006;62:1125–1132. doi: 10.1002/prot.20810. [DOI] [PubMed] [Google Scholar]
- 35.Montanucci L, Fariselli P, Martelli PL, Casadio R. Predicting protein thermostability changes from sequence upon multiple mutations. Bioinformatics. 2008;24:i190–195. doi: 10.1093/bioinformatics/btn166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang LT, Gromiha MM. Reliable prediction of protein thermostability change upon double mutation from amino acid sequence. Bioinformatics. 2009;25:2181–2187. doi: 10.1093/bioinformatics/btp370. [DOI] [PubMed] [Google Scholar]
- “ 37.Potapov V, Cohen M, Schreiber G. Assessing computational methods for predicting protein stability upon mutation: good on average but not in the details. Protein engineering, design & selection : PEDS. 2009;22:553–560. doi: 10.1093/protein/gzp030. The authors test several popular computational methods for predicting ΔΔG of protein mutations against experimental data. They find that these methods typically exhibit a trend in the right direction, but are not highly accurate at predicting ΔΔG for individual mutations.
- 38.Stitziel NO, Binkowski TA, Tseng YY, Kasif S, Liang J. topoSNP: a topographic database of non-synonymous single nucleotide polymorphisms with and without known disease association. Nucleic acids research. 2004;32:D520–522. doi: 10.1093/nar/gkh104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yue P, Melamud E, Moult J. SNPs3D: candidate gene and SNP selection for association studies. BMC bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karchin R, Diekhans M, Kelly L, Thomas D, Pieper U, Eswar N, Haussler D, Sali A. LS-SNP: large-scale annotation of coding non-synonymous SNPs based on multiple information sources. Bioinformatics. 2005;21:2814–2820. doi: 10.1093/bioinformatics/bti442. [DOI] [PubMed] [Google Scholar]
- 41.Capriotti E, Calabrese R, Casadio R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics. 2006;22:2729–2734. doi: 10.1093/bioinformatics/btl423. [DOI] [PubMed] [Google Scholar]
- 42.Jiang R, Yang H, Zhou L, Kuo CC, Sun F, Chen T. Sequence-based prioritization of nonsynonymous single-nucleotide polymorphisms for the study of disease mutations. American journal of human genetics. 2007;81:346–360. doi: 10.1086/519747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ng PC, Levy S, Huang J, Stockwell TB, Walenz BP, Li K, Axelrod N, Busam DA, Strausberg RL, Venter JC. Genetic variation in an individual human exome. PLoS genetics. 2008;4:e1000160. doi: 10.1371/journal.pgen.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler EE, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boyko AR, Williamson SH, Indap AR, Degenhardt JD, Hernandez RD, Lohmueller KE, Adams MD, Schmidt S, Sninsky JJ, Sunyaev SR, et al. Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS genetics. 2008;4:e1000083. doi: 10.1371/journal.pgen.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gorlov IP, Gorlova OY, Sunyaev SR, Spitz MR, Amos CI. Shifting paradigm of association studies: value of rare single-nucleotide polymorphisms. American journal of human genetics. 2008;82:100–112. doi: 10.1016/j.ajhg.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williamson SH, Hernandez R, Fledel-Alon A, Zhu L, Nielsen R, Bustamante CD. Simultaneous inference of selection and population growth from patterns of variation in the human genome. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:7882–7887. doi: 10.1073/pnas.0502300102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eyre-Walker A, Woolfit M, Phelps T. The distribution of fitness effects of new deleterious amino acid mutations in humans. Genetics. 2006;173:891–900. doi: 10.1534/genetics.106.057570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yampolsky LY, Kondrashov FA, Kondrashov AS. Distribution of the strength of selection against amino acid replacements in human proteins. Human molecular genetics. 2005;14:3191–3201. doi: 10.1093/hmg/ddi350. [DOI] [PubMed] [Google Scholar]
- 50.Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. American journal of human genetics. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- “ 51.Kryukov GV, Shpunt A, Stamatoyannopoulos JA, Sunyaev SR. Power of deep, all-exon resequencing for discovery of human trait genes. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3871–3876. doi: 10.1073/pnas.0812824106. The authors use simulation to investigate the feasibility of identifying rare variants associated with human traits by exome resequencing studies. They conclude that that genes meaningfully affecting a human trait can be identified using the combined analysis of rare non-synonymous alleles. However, large sample sizes would be required to achieve substantial power.
- 52.Keightley P, Eyre-Walker A. Joint inference of the distribution of fitness effects of deleterious mutations and population demography based on nucleotide polymorphism frequencies. Genetics. 2007;177:2251–2261. doi: 10.1534/genetics.107.080663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? American journal of human genetics. 2001;69:124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nature Genetics. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lohmueller KE, Indap AR, Schmidt S, Boyko AR, Hernandez RD, Hubisz MJ, Sninsky JJ, White TJ, Sunyaev SR, Nielsen R, et al. Proportionally more deleterious genetic variation in European than in African populations. Nature. 2008;451:994–997. doi: 10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- “” 56.Barreiro LB, Laval G, Quach H, Patin E, Quintana-Murci L. Natural selection has driven population differentiation in modern humans. Nature genetics. 2008;40:340–345. doi: 10.1038/ng.78. The authors compute FST, a measure of population differentiation, using several different classes of variants from HapMap. They find that coding variants and variants predicted to be functional are more homogeneous between populations, while variants that have undergone positive selection differentiate between populations more clearly.
- “ 57.Tavtigian SV, Greenblatt MS, Goldgar DE, Boffetta P, Group IUGVW Assessing pathogenicity: overview of results from the IARC Unclassified Genetic Variants Working Group. Human mutation. 2008;29:1261–1264. doi: 10.1002/humu.20903. This paper introduces the unclassified genetic variants special issue of Human Mutation, which discusses in detail the problem of unclassified variants and proposes procedures for using functional prediction of variants in medical applications.
- 58.Tavtigian SV, Greenblatt MS, Lesueur F, Byrnes GB, Group IUGVW In silico analysis of missense substitutions using sequence-alignment based methods. Human mutation. 2008;29:1327–1336. doi: 10.1002/humu.20892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goldgar DE, Easton DF, Byrnes GB, Spurdle AB, Iversen ES, Greenblatt MS, Group IUGVW Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Human mutation. 2008;29:1265–1272. doi: 10.1002/humu.20897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- “” 60.Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et al. Exome sequencing identifies the cause of a mendelian disorder. Nature genetics. 2009 doi: 10.1038/ng.499. The authors use the exome sequencing technique introduced in their earlier paper [44] and described in this review to identify the cause of a Mendelian disorder. This is the first successful use of exome sequencing to identify a previously unknown disease gene using rare mutations.
- 61.McLendon R, Friedman A, Bigner D, Van Meir E, Brat D, Mastrogianakis GM, Olson J, Mikkelsen T, Lehman N, Aldape K, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haber DA, Settleman J. Cancer: drivers and passengers. Nature. 2007;446:145–146. doi: 10.1038/446145a. [DOI] [PubMed] [Google Scholar]
- 64.Kaminker JS, Zhang Y, Watanabe C, Zhang Z. CanPredict: a computational tool for predicting cancer-associated missense mutations. Nucleic acids research. 2007;35:W595–598. doi: 10.1093/nar/gkm405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- “ 65.Lee W, Zhang Y, Mukhyala K, Lazarus RA, Zhang Z. Bi-directional SIFT predicts a subset of activating mutations. PloS one. 2009;4:e8311. doi: 10.1371/journal.pone.0008311. The authors modify the SIFT algorithm to report not only mutations with poor conservation scores, which are presumed to be damaging, but also those with higher conservation scores than the wild-type sequence, which are potentially activating mutations. They show that this approach does correctly predict a subset of gain-of-function mutations.
- 66.Cohen JC, Kiss RS, Pertsemlidis A, Marcel YL, McPherson R, Hobbs HH. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–72. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- 67.Cohen JC, Boerwinkle E, Mosley TH, Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. New Engl J Med. 2006;354:1264–1272. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 68.Romeo S, Pennacchio LA, Fu Y, Boerwinkle E, Tybjaerg-Hansen A, Hobbs HH, Cohen JC. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat Genet. 2007;39:513–516. doi: 10.1038/ng1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ahituv N, Kavaslar N, Schackwitz W, Ustaszewska A, Martin J, Hebert S, Doelle H, Ersoy B, Kryukov G, Schmidt S, et al. Medical sequencing at the extremes of human body mass. Am J Hum Genet. 2007;80:779–91. doi: 10.1086/513471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ji W, Foo JN, O’Roak BJ, Zhao H, Larson MG, Simon DB, Newton-Cheh C, State MW, Levy D, Lifton RP. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet. 2008;40:592–9. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–9. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Romeo S, Yin W, Kozlitina J, Pennacchio LA, Boerwinkle E, Hobbs HH, Cohen JC. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J Clin Invest. 2009;119:70–79. doi: 10.1172/JCI37118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jiang R, Yang H, Sun F, Chen T. Searching for interpretable rules for disease mutations: a simulated annealing bump hunting strategy. BMC bioinformatics. 2006;7:417. doi: 10.1186/1471-2105-7-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bao L, Zhou M, Cui Y. nsSNPAnalyzer: identifying disease-associated nonsynonymous single nucleotide polymorphisms. Nucleic acids research. 2005;33:W480–482. doi: 10.1093/nar/gki372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome research. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thomas PD, Kejariwal A. Coding single-nucleotide polymorphisms associated with complex vs. Mendelian disease: evolutionary evidence for differences in molecular effects. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:15398–15403. doi: 10.1073/pnas.0404380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- “ 77.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. This paper briefly describes the algorithm used by one of the most popular methods for functional prediction of SNPs, SIFT, and provides detailed instructions for using the SIFT server.
- 78.Yue P, Moult J. Identification and analysis of deleterious human SNPs. Journal of Molecular Biology. 2006;356:1263–1274. doi: 10.1016/j.jmb.2005.12.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.