

Abstract
During pregnancy, the energy requirements of the fetus impose changes in maternal metabolism. Increasing insulin resistance in the mother maintains nutrient flow to the growing fetus, while prolactin and placental lactogen counterbalance this resistance and prevent maternal hyperglycemia by driving expansion of the maternal population of insulin-producing β-cells1–3. However, the exact mechanisms by which the lactogenic hormones drive β-cell expansion remain uncertain. Here we show that serotonin acts downstream of lactogen signaling to drive β-cell proliferation. Serotonin synthetic enzyme Tph1 and serotonin production increased sharply in β-cells during pregnancy or after treatment with lactogens in vitro. Inhibition of serotonin synthesis by dietary tryptophan restriction or Tph inhibition blocked β-cell expansion and induced glucose intolerance in pregnant mice without affecting insulin sensitivity. Expression of the Gαq-linked serotonin receptor Htr2b in maternal islets increased during pregnancy and normalized just prior to parturition, while expression of the Gαi-linked receptor Htr1d increased at the end of pregnancy and postpartum. Blocking Htr2b signaling in pregnant mice also blocked β-cell expansion and caused glucose intolerance. These studies reveal an integrated signaling pathway linking β-cell mass to anticipated insulin need during pregnancy. Modulators of this pathway, including medications and diet, may affect the risk of gestational diabetes4.
Although lactogenic hormones regulate β-cell mass during pregnancy, they cannot alone explain all of the maternal changes in β-cell mass2,5–8. Despite persistent elevations of placental lactogen and prolactin through the end of pregnancy and the end of lactation, respectively, β-cell proliferation peaks at midgestation in rodents and drops to the normal non-pregnant rate or below by parturition, prompting rapid normalization of β-cell mass postpartum2,9,10 (Supplementary Fig. S1). Therefore, to identify other genes potentially involved in regulating maternal β-cell mass, we compared the global gene expression patterns in islets from non-pregnant and pregnant (G13 15) female mice by high-throughput sequencing of cDNA (Supplementary Table S1 and Supplementary Fig. S2.) and by hybridization to oligonucleotide microarrays (Supplementary Tables S2).
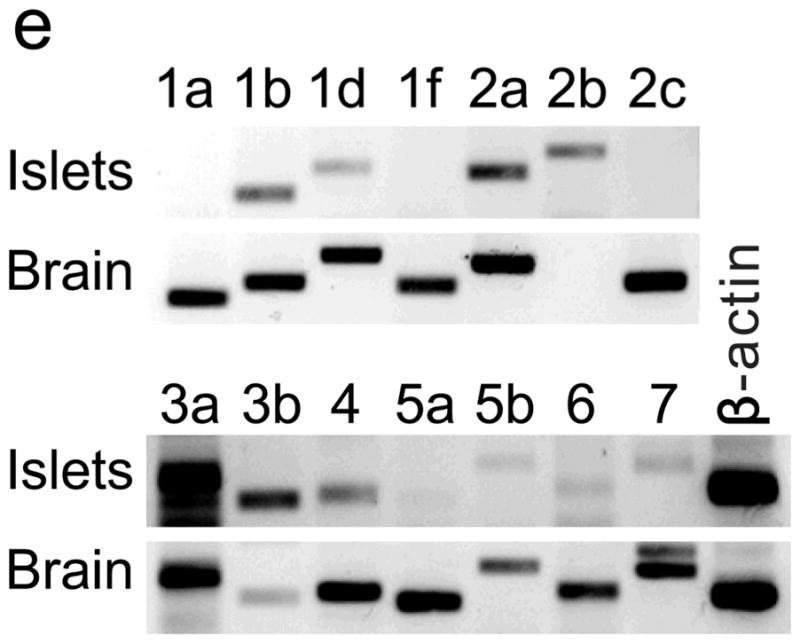
Among the genes most significantly induced during pregnancy were Tph1 and Tph2, which encode the two isoforms of tryptophan hydroxylase, the rate-limiting enzyme in the synthesis of serotonin (5-hydroxytryptamine, 5-HT). We have shown that β-cells share with serotonergic neurons a common gene expression program and the ability to synthesize, store and secrete serotonin (Y. Ohta and M. S. G., unpublished observations). Indeed, several other serotonergic transcripts were significantly increased in islets from pregnant mice, including those encoding aromatic L-amino acid decarboxylase, the enzyme that catalyzes the second and final step in serotonin synthesis, and vesicular monoamine transporter VMAT1 (Supplementary Table S1).
Real-time RT-PCR for Tph1 and Tph2 confirmed the genomic analyses (Fig. 1a). Expression of Tph1 and Tph2 increased from the non-pregnant baseline by gestational day 6 (G6) and peaked at G12 with 527- and 7-fold increases respectively, while levels in gut, heart and brain did not change (Supplementary Fig. S3). Tph1 expression remained high postpartum until the end of lactation, followed by a return to pre-pregnancy levels. Specific antiserum against Tph1 demonstrated the induction of Tph1 protein in pregnant mouse islets by western blot11 (Fig. 1b). High performance liquid chromatography (HPLC) detected low tissue levels of 5-HT in islets of non-pregnant mice, which increased 420-fold in islets from pregnant animals, but did not change in gut (Fig. 1c, d).
Figure 1. 5-HT production in islets during pregnancy.

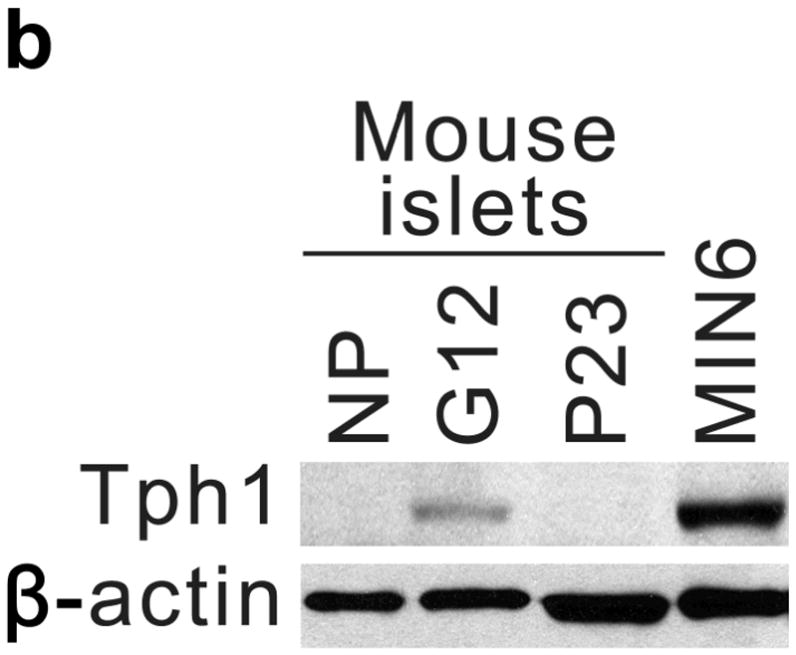
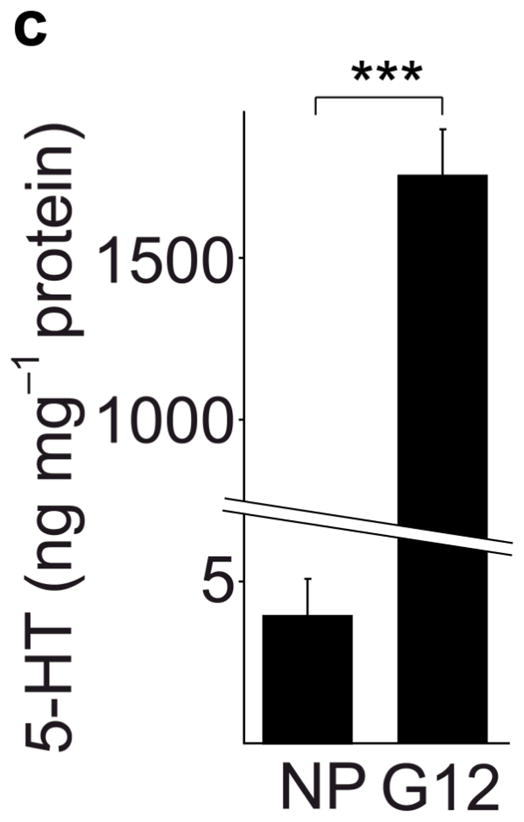
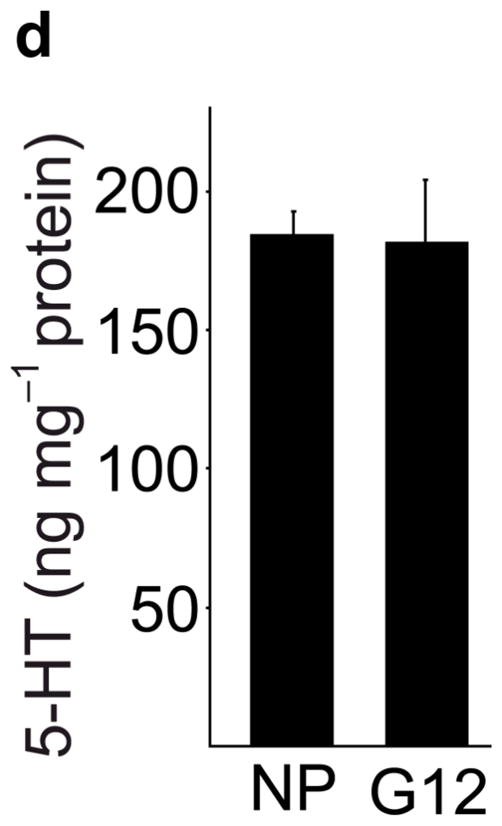
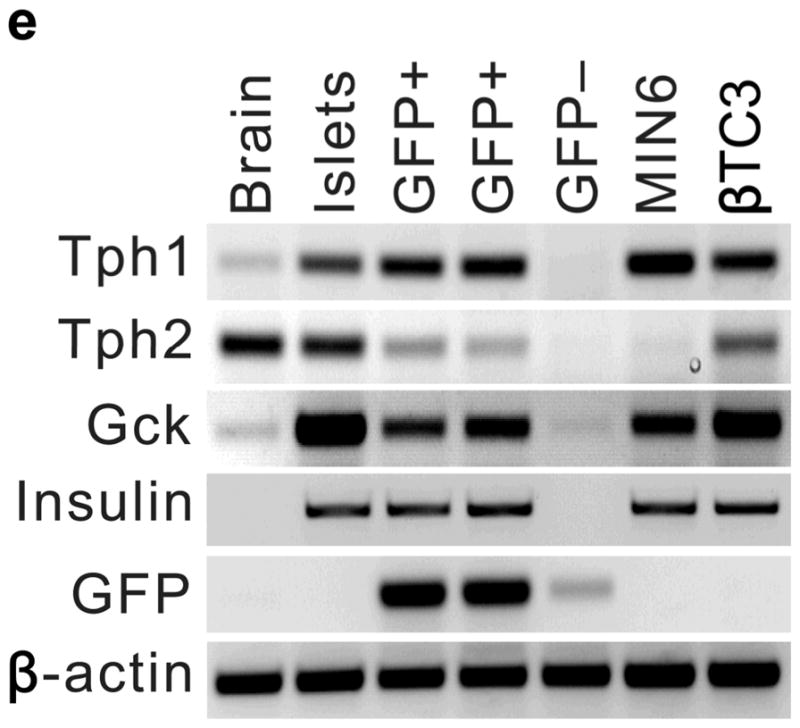



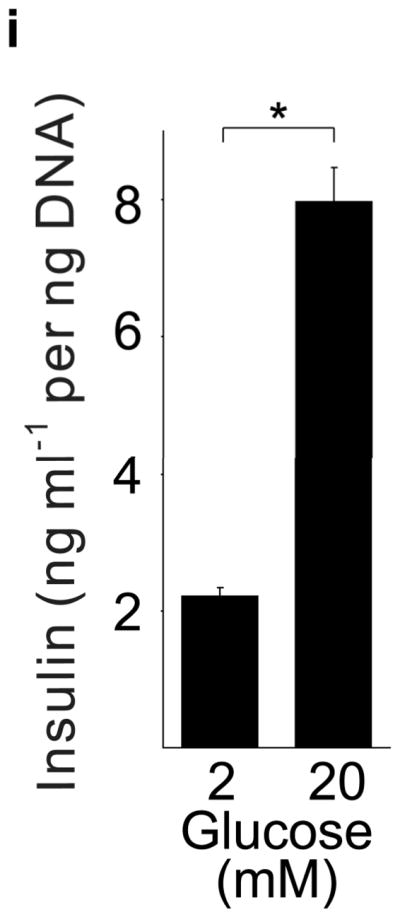
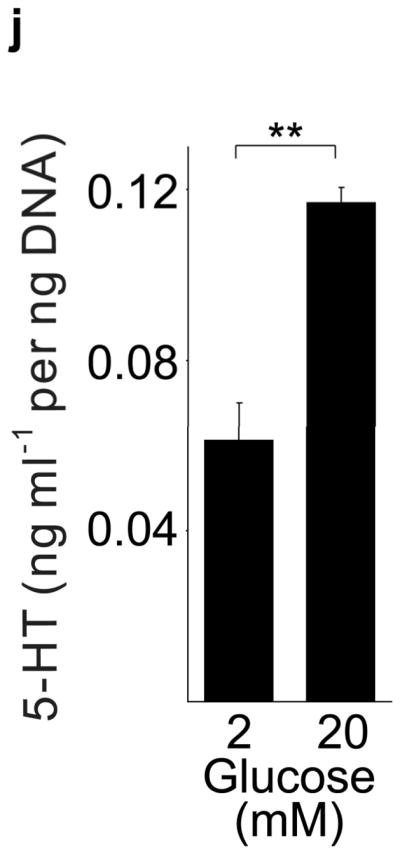
(a) mRNA levels for Tph1 and Tph2 measured by real time RT-PCR in islet RNA at the dates of gestation (G) or postpartum (P) indicated are shown relative to the levels in islets from non-pregnant female mice. n = 3 6 mice per data point. (b) Western blots were performed with Tph1 and β-actin antisera on protein extracted from islets of non-pregnant female (NP), pregnant (G12) and postpartum day 23 (P23) female mice and MIN6 insulinoma cells. (c, d) Tissue 5-HT concentrations were assayed by HPLC in islets (c) and duodenum (d) isolated from 3 non-pregnant (NP) and 3 pregnant (G12) female mice. (e) The mRNA shown were amplified by RT-PCR from mouse islets, β-cells (GFP+), non-β islet cells (GFP−), brain, and the β-cell lines MIN6 and βTC3. (f) Immunofluorescent staining labels pancreata from non-pregnant female (Non-preg) and G12 pregnant mice with insulin and glucagon in green and 5-HT in red. (g) Immunoperoxidase staining labels 5-HT and TPH1 in pancreata from non-pregnant (Non-preg), pregnant (37 weeks gestation) and postpartum day 2 human autopsies. (h) Immunohistochemical co-staining labels insulin (red, fluorescent) and TPH1 (black, peroxidase) in pregnant human autopsy pancreas. (i, j) Secreted insulin (i) and 5-HT (j) was assayed by ELISA (i) and HPLC (j), following a 1 hour incubation of islets isolated from pregnant (G13 G15) mice in 2 mM or 20 mM glucose. n = 5 groups of 40 islets. All data are presented as mean ± standard error. Statistical significance vs. islets cultured in 2mM glucose (i and j) or vs. non-pregnant control (a and c) was analyzed by Student’s t test: *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bar indicates 20 μm.
RT-PCR amplified both Tph1 and Tph2 mRNA from β-cells purified from mice expressing eGFP in β-cells (MIP-GFP mice12) (Fig. 1e). Immunohistochemical staining detected 5-HT and TPH1 co-localized with insulin in islets from pregnant mice (Fig. 1f) and pregnant and post partum humans (Fig. 1g, h; Supplementary Fig. S3d). In a test of the ability of β-cells to secrete this stored 5-HT, glucose stimulation induced secretion of insulin and 5-HT from islets isolated from pregnant mice (Fig. 1i, j).
Taken together, these data demonstrate that β-cells dramatically increase synthesis, storage and secretion of 5-HT during pregnancy. This increase may modestly boost circulating 5HT levels (Supplementary Fig. S4), but likely increases local islet levels of 5-HT much more. Therefore, we tested whether 5-HT affects β-cell function and glucose metabolism during pregnancy. Because tryptophan is an essential amino acid and the Km of Tph for tryptophan is higher than tissue tryptophan concentrations, dietary restriction of tryptophan can sharply drop 5-HT levels13. We found that mice fed a tryptophan-free diet from G6 to G12 and tested at G13 (Fig. 2a) developed severe glucose intolerance compared to mice fed either a normal or histidine-free diet (Fig. 2b). Furthermore, a less restricted diet, with 20% of normal tryptophan content, or treatment with the Tph inhibitor 4-chloro-DL-phenylalanine methyl ester hydrochloride (PCPA) also induced glucose intolerance in pregnant mice (Fig. 2c), but not in non-pregnant female mice (Fig. 2d). These results support the conclusion that 5-HT production is necessary for the maintenance of glucose homeostasis during pregnancy.
Figure 2. 5-HT signaling and glucose metabolism in pregnancy.








(a) The treatment schedules for mouse experiments are shown. (b d) Blood glucose levels were measured after intraperotoneal injection of 2 g kg−1 glucose in pregnant mice (G13) fed a tryptophan-free (“Trp-free”, b), histidine-free (“His-free”, b) or low tryptophan (“Low Trp”, c) diet or treated with PCPA (300 mg kg−1 day−1)(c) or non-pregnant female mice (d) treated as shown. n = 5 18 mice per treatment group. (e) The Htr mRNAs shown were amplified by RT-PCR from mouse islet and brain RNA. (f) The mRNAs shown were amplified by RT-PCR from mouse islets, β-cells (GFP+), non-β islet cells (GFP−), brain, and β-cell lines MIN6 and βTC3. (g, h) mRNA levels for Htr2a and Htr2b (g) and Htr1b and Htr1d (h) measured by real time RT-PCR in islet RNA harvested at the gestational dates indicated are shown relative to the levels in islets from non-pregnant female mice. n = 3–6 mice per data point. (i) Blood glucose levels were measured after intraperitoneal injection of glucose (2 g kg−1)in pregnant mice treated with SB204741 (1 mg kg−1 day−1), Methysergide (3 mg kg−1 day−1) or Ketanserin (1 mg−1 kg−1 day−1). n = 5 18 mice per treatment group. (j) Pregnant mice were treated as in Fig. 2a with tryptophan-free diet, PCPA or SB204741 and fasted for 6 hours at G13 before measuring insulin in plasma collected before and 30 minutes after intraperitoneal injection of 2 g/kg glucose. n = 4 7 mice per group. All data are presented as mean ± standard error. Statistical significance vs. untreated control (b, c, i and j) or vs. non-pregnant control (g and h) was analyzed by Student’s t test: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
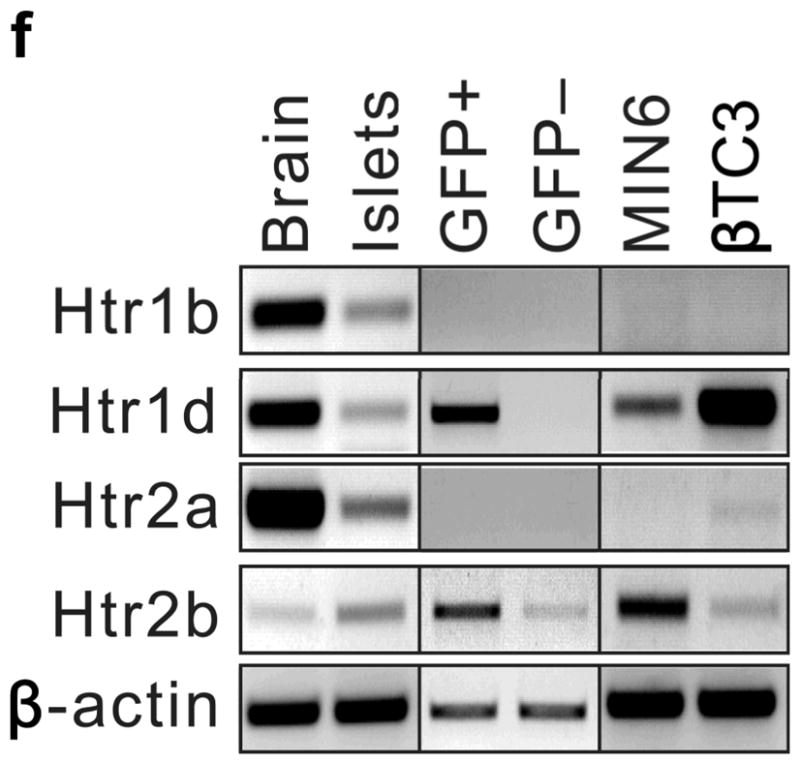
To determine how 5-HT might regulate islet function, we assayed the expression in islets of the 14 murine 5-HT receptors (Htr) by RT-PCR14. Several were expressed at relatively high levels in mouse islets (Fig. 2e), and transcripts encoding the Gαi-linked receptor Htr1d and the Gαq-linked receptor Htr2b were detected in purified β-cells (Fig. 2f). In pregnant mice, Htr2b expression increased significantly from G6 through G15 (Fig. 2g) and normalized at the end of gestation, while Htr1d expression increased at the end of gestation (G17) and postpartum (Fig. 2h). Interestingly, increased Htr2b expression closely correlated with the period of increased β-cell proliferation, and increased Htr1d expression correlated with the cessation of β-cell proliferation and regression of β-cell mass2,9.
To assess the contribution of 5-HT signaling, mice were treated with Htr antagonists (Fig. 2a). Nonspecific Htr antagonist Methysergide and selective Htr2b antagonist SB204741 induced glucose intolerance in pregnant mice (Fig. 2i), but not in non-pregnant female mice (SB204741, Fig. 2d) or when given acutely (Supplementary Fig. S5a), while selective Htr2a antagonist Ketanserin had no effect on glucose tolerance (Fig. 2i). Treatment with PCPA, SB204741, or a tryptophan-free diet all reduced insulin levels in pregnant mice (Fig. 2j), but insulin sensitivity was not reduced with PCPA or SB204741 (Supplementary Fig. S5b). To test genetically the requirement for Htr2b signaling during pregnancy, we assessed Htr2b null mice and observed glucose intolerance during pregnancy (Fig. 3a, b).
Figure 3. Htr2b signaling and β-cell proliferation in pregnancy.








(a, b) Blood glucose levels were measured after intraperitoneal injection of glucose (2 g kg−1) in pregnant (a) and non-pregnant (b) mice with the genotypes shown. n = 4 7 mice per group. (c, d) In non-pregnant and pregnant control mice, pregnant mice treated with SB204741 or PCPA, and pregnant Htr2b−/− mice, β-cell proliferation (c) was quantified by counting the percent of insulin + cells labeled with BrdU, and relative β-cell mass (d) was calculated as the area of insulin + cells / total pancreatic area. n = 4 6 mice per group. (e) Islets isolated from Htr2b+/+ (black) or Htr2b−/− (red) mice were transplanted into the kidney capsule of Htr2b+/+ mice. (f) Immunofluorescent staining for 5-HT (red) and insulin (green) was performed on donor islets with the genotypes shown transplanted under the kidney capsule in non-pregnant or G13 pregnant hosts. (g) Immunofluorescent staining for insulin (red) and BrdU (green) together with DNA staining with DAPI (blue) was performed on host pancreatic islets and on donor islets with the genotypes shown transplanted under the kidney capsule in non-pregnant and G13 pregnant mice. (h) β-Cell proliferation in transplanted islets with the genotypes shown was quantified by counting the percent of insulin + cells labeled with BrdU in non-pregnant (NP) and pregnant mice. n = 3 4 per group. All data are presented as mean ± standard error. Statistical significance vs. Htr2b+/+ control (a), vs. pregnant untreated Htr2b+/+ control (c and d) or vs. pregnant Htr2b+/+ control (h) was analyzed by Student’s t test: *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bar indicates 20 μm.
Given the known role of Htr2b signaling in driving cell proliferation in other tissues15–21, we tested whether 5-HT signaling contributes to increased β-cell proliferation during pregnancy. Treatment with PCPA or SB204741 as well as Htr2b loss reduced the normal increase in β-cell proliferation in pregnant mice at G13 as assayed by 5-bromo-2-deoxyuridine (BrdU) incorporation (Fig. 2a, 3c), and prevented the pregnancy-induced expansion of β-cell mass (Fig 3d), but did not reduce insulin secretion acutely from islets in vitro (Supplementary Fig. S5c). In addition, both PCPA and SB204741 attenuated the growth of the MIN6 β-cell tumor line but not of NIH-3T3 fibroblasts (Supplementary Fig. S6a). Therefore, expansion of β-cell mass during pregnancy requires both Tph1-driven 5-HT production and signaling through Htr2b.
To determine whether the requirement for Htr2b signaling resides specifically in the islets, we transplanted islets isolated from Htr2b+/+ or Htr2b−/− mice under the kidney capsule of Htr2b+/+ mice (Fig. 3e) and assessed β-cell proliferation in the transplanted islets during pregnancy. Although this approach removes the islets from their normal anatomic context, it avoids the use of cre-recombinase, which when expressed under the control of pancreatic promoters often recombines targeted genes in other tissues as well22. In transplanted β-cells, pregnancy induced 5-HT production regardless of the presence of Htr2b (Fig. 3f). However, while both the host and transplanted Htr2b+/+ β-cells similarly increased BrdU incorporation during pregnancy, fewer transplanted Htr2b−/− β-cells incorporated BrdU, demonstrating a local requirement for Htr2b signaling in islets during pregnancy (Fig. 3g, h).
Because pregnancy hormones prolactin and placental lactogen stimulate β-cell proliferation2,3,5,6, and prolactin induces Tph1 expression in the mammary gland23, we tested the ability of lactogenic hormones to induce Tph1 expression in islets isolated from pregnant mice at G3. Moderate concentrations of prolactin (200 ng ml−1) increased Tph1 expression 28-fold, while high concentrations of prolactin (1,000 ng ml−1) corresponding to midgestation lactogen levels increased Tph1 expression by 147-fold (Fig. 4a) without affecting expression of Tph2, Htr1d or Htr2b (Fig. 4b–d). Human placental lactogen induced Tph1 expression to similar levels in G3 mouse islets, but non-pregnant islets responded less robustly (Supplementary Fig. S6d), consistent with evidence that prolactin receptor expression and signaling increase in islets early during pregnancy24 (Supplementary Table S1).
Figure 4. 5-HT and lactogen induced β-cell proliferation.








(a d) mRNA levels for Tph1 (a), Tph2 (b), Htr1d (c) and Htr2b (d) were measured by real-time RT-PCR in RNA from islets isolated from G3 pregnant mice and cultured with the concentration of mouse prolactin (PRL) indicated for 72 hours. n = 3 6 per group. (e) β-Cell proliferation was quantified by counting the percent of insulin + cells labeled with BrdU in mouse islets cultured with prolactin (1,000 ng ml−1, PRL), 5-HT (10 μM) or no added hormone (control) for 4 days. n = 5 10 groups of islets per data point. (f, g) Levels of cyclin mRNAs were measured by real-time RT-PCR in RNA (f) from islets harvested at the dates indicated and shown relative to the levels in control islets from non-pregnant female mice or (g) from islets cultured with 5-HT (10 μM) for the time indicated and shown relative to the levels in control islets cultured without 5-HT for 24 hours. n = 4 10 groups of islets per data point. (h) A proposed model is shown for 5-HT regulation of β-cell proliferation during pregnancy. All data are presented as mean ± standard error. Statistical significance vs. untreated control (a, e and g) or vs. non-pregnant control (f) was analyzed by Student’s t test: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Next we tested whether 5-HT can independently induce islet cells to proliferate. Treatment of mouse islets with prolactin (1,000 ng ml−1) or 5-HT (10 μM) induced similar increases in proliferation as measured by BrdU incorporation (Fig. 4e) and Ki67 staining (Supplementary Fig. S6e). Finally, treatment of islets with 5-HT (10 μM) in vitro altered cyclin gene expression in a pattern similar to that seen in islets from pregnant mice (Fig. 4f, g).
Taken together, these studies suggest a simple model for the regulation of β-cell mass during pregnancy (Fig. 4h). During pregnancy, lactogenic signaling induces Tph1 expression and 5-HT synthesis in islets. 5-HT in turn functions in a paracrine/autocrine fashion through Gαq-coupled Htr2b to stimulate β-cell proliferation. Shortly before parturition, expression of Htr2b decreases and expression of Gαi-coupled Htr1d increases, generating an inhibitory signal capable of reducing β-cell proliferation and β-cell mass (M. Berger and M. S. G., unpublished data), so that β-cell mass returns rapidly to pre-pregnancy levels. While this model focuses on the regulation of β-cell mass, increasing evidence suggests that β-cell mass influences insulin production capacity and thus the risk of diabetes25–27. In addition, serotonin signaling may also directly impact insulin secretion independent of its effect on β-cell mass. Finally, similar shifts in receptor expression could possibly contribute to other peripartum changes, such as the mobilization of calcium from bone18 that occurs with lactation.
Modulators of this pathway, including drugs, diet and genetic inheritance, could impact the risk of gestational diabetes, and possibly the long term risk of developing type 2 diabetes. The dual roles of serotonin in regulating mood and β-cell mass provide a possible link that could explain the association of depression with both type 2 diabetes28 and gestational diabetes29, as well as the diabetogenic effects of some classes of psychiatric medications30. A more complete understanding of the function of this pathway may suggest improved methods for both preventing and treating diabetes.
Methods
C57BL/6J mice were housed on a 12-h light/dark cycle in climate-controlled, pathogen-free barrier facilities. The Institutional Animal Care and Use Committees at UCSF or Juntendo University approved all studies involving mice. Mating was confirmed by the presence of a vaginal plug the next morning, designated day 0 of gestation (G0). The control amino acid diet contained 0.2% tryptophan and 0.4% histidine, and the low tryptophan diet contained 0.04% tryptophan. All drugs were administered by daily intraperitoneal injection except for BrdU, which was supplied in the drinking water at 1 mg ml−1.
Pancreatic islets were isolated from female mice by collagenase digestion, hand-picked and pooled. Glucose and insulin tolerance and insulin secretion tests were performed on fasting non-pregnant female or pregnant (G13) mice. Glucose and 5-HT levels were measured on whole blood, and insulin on plasma. Counting of BrdU positive β-cells and morphometric analyses were performed by serially sectioning the entire pancreas followed by staining and counting every 20th section. A minimum of 3,000 insulin positive cells were counted per pancreas. Human pancreatic tissue was obtained at autopsy at the UCSF Medical Center and Hirosaki University affiliated teaching hospitals. Details of tissue processing and staining, and RT-PCR assays are provided in Supplementary Materials.
All high-throughput sequencing data in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE21860 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE21860).
Supplementary Material
Acknowledgments
We thank G. Grodsky, W. Rutter, and members of the German laboratory for helpful discussions; F. Schaufle and the UCSF DERC Microscopy Core Laboratory; G. Szot and the UCSF DERC Islet Core; Y. Zhang, Z. Li and S. Zhao for technical assistance with mouse husbandry and genotyping; N. Daimaru, E. Magoshi, and K. Nakamura in Juntendo University for technical assistance; K. Takahashi, W. Inaba, M. Tsujii, and S. Nakayama in Hirosaki University for immunohistochemical studies; Y. Katayama (HU) and P. Ursell, (UCSF) for providing human pancreatic samples; and D. Kuhn, L. Maroteaux and M. Hara for generously providing the anti-Tph1 antisera, Htr2b-targeted mice and MIP-GFP mice, respectively. This work was supported by grants from the Larry L. Hillblom Foundation, the Juvenile Diabetes Research Foundation, the American Diabetes Association, US National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases, and the Ministry of Education, Sports and Culture of Japan.
Footnotes
Author Contributions
H.K., H.W. and M.S.G. designed research; H.K., Y.T, F.C.L., E.C., T.U., H.M., Y.F., T.M., Y.K., G.H., M.H., K.Y., N.K., and J.W. performed research; H.K., F.C.L., R.K., S.Y., H.W., L.H.T and M.S.G. analyzed data; and H.K., F.C.L., H.W. and M.S.G. wrote the paper.
References
- 1.Van Assche FA, Aerts L, De Prins F. A morphological study of the endocrine pancreas in human pregnancy. Br J Obstet Gynaecol. 1978;85:818–820. doi: 10.1111/j.1471-0528.1978.tb15835.x. [DOI] [PubMed] [Google Scholar]
- 2.Parsons JA, Brelje TC, Sorenson RL. Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology. 1992;130:1459–1466. doi: 10.1210/endo.130.3.1537300. [DOI] [PubMed] [Google Scholar]
- 3.Huang C, Snider F, Cross JC. Prolactin receptor is required for normal glucose homeostasis and modulation of beta-cell mass during pregnancy. Endocrinology. 2009;150:1618–1626. doi: 10.1210/en.2008-1003. [DOI] [PubMed] [Google Scholar]
- 4.Buchanan TA, Xiang AH. Gestational diabetes mellitus. J Clin Invest. 2005;115:485–491. doi: 10.1172/JCI24531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freemark M, et al. Targeted deletion of the PRL receptor: effects on islet development, insulin production, and glucose tolerance. Endocrinology. 2002;143:1378–1385. doi: 10.1210/endo.143.4.8722. [DOI] [PubMed] [Google Scholar]
- 6.Vasavada RC, et al. Targeted expression of placental lactogen in the beta cells of transgenic mice results in beta cell proliferation, islet mass augmentation, and hypoglycemia. The Journal of biological chemistry. 2000;275:15399–15406. doi: 10.1074/jbc.275.20.15399. [DOI] [PubMed] [Google Scholar]
- 7.Sorenson RL, Brelje TC. Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Hormone and metabolic research. 1997;29:301–307. doi: 10.1055/s-2007-979040. [DOI] [PubMed] [Google Scholar]
- 8.Brelje TC, et al. Effect of homologous placental lactogens, prolactins, and growth hormones on islet B-cell division and insulin secretion in rat, mouse, and human islets: implication for placental lactogen regulation of islet function during pregnancy. Endocrinology. 1993;132:879–887. doi: 10.1210/endo.132.2.8425500. [DOI] [PubMed] [Google Scholar]
- 9.Karnik SK, et al. Menin Controls Growth of Pancreatic β-Cells in Pregnant Mice and Promotes Gestational Diabetes Mellitus. Science. 2007;318:806–809. doi: 10.1126/science.1146812. [DOI] [PubMed] [Google Scholar]
- 10.Ben-Jonathan N, LaPensee CR, LaPensee EW. What can we learn from rodents about prolactin in humans? Endocrine reviews. 2008;29:1–41. doi: 10.1210/er.2007-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakowski SA, et al. Differential tissue distribution of tryptophan hydroxylase isoforms 1 and 2 as revealed with monospecific antibodies. Brain Res. 2006;1085:11–18. doi: 10.1016/j.brainres.2006.02.047. [DOI] [PubMed] [Google Scholar]
- 12.Hara M, et al. Transgenic mice with green fluorescent protein-labeled pancreatic beta -cells. Am J Physiol Endocrinol Metab. 2003;284:E177–183. doi: 10.1152/ajpendo.00321.2002. [DOI] [PubMed] [Google Scholar]
- 13.Fadda F. Tryptophan-free diets: a physiological tool to study brain serotonin function. News Physiol Sci. 2000;15:260–164. doi: 10.1152/physiologyonline.2000.15.5.260. [DOI] [PubMed] [Google Scholar]
- 14.Hoyer D, et al. International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (Serotonin) Pharmacol Rev. 1994;46:157–203. [PubMed] [Google Scholar]
- 15.Lesurtel M, et al. Platelet-derived serotonin mediates liver regeneration. Science. 2006;312:104–107. doi: 10.1126/science.1123842. [DOI] [PubMed] [Google Scholar]
- 16.Nebigil CG, et al. Serotonin 2B receptor is required for heart development. Proc Natl Acad Sci U S A. 2000;97:9508–9513. doi: 10.1073/pnas.97.17.9508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wouters MM, et al. Exogenous serotonin regulates proliferation of interstitial cells of Cajal in mouse jejunum through 5-HT2B receptors. Gastroenterology. 2007;133:897–906. doi: 10.1053/j.gastro.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 18.Rothman RB, et al. Evidence for possible involvement of 5-HT(2B) receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation. 2000;102:2836–2841. doi: 10.1161/01.cir.102.23.2836. [DOI] [PubMed] [Google Scholar]
- 19.Fitzgerald LW, et al. Possible role of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with fenfluramine. Mol Pharmacol. 2000;57:75–81. [PubMed] [Google Scholar]
- 20.Collet C, et al. The serotonin 5-HT2B receptor controls bone mass via osteoblast recruitment and proliferation. FASEB J. 2008;22:418–427. doi: 10.1096/fj.07-9209com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Lucchini S, Ori M, Cremisi F, Nardini M, Nardi I. 5-HT2B-mediated serotonin signaling is required for eye morphogenesis in Xenopus. Mol Cell Neurosci. 2005;29:299–312. doi: 10.1016/j.mcn.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 22.Gannon M, Shiota C, Postic C, Wright CV, Magnuson M. Analysis of the Cre-mediated recombination driven by rat insulin promoter in embryonic and adult mouse pancreas. Genesis. 2000;26:139–142. doi: 10.1002/(sici)1526-968x(200002)26:2<139::aid-gene12>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 23.Matsuda M, et al. Serotonin regulates mammary gland development via an autocrine-paracrine loop. Dev Cell. 2004;6:193–203. doi: 10.1016/s1534-5807(04)00022-x. [DOI] [PubMed] [Google Scholar]
- 24.Moldrup A, Petersen ED, Nielsen JH. Effects of sex and pregnancy hormones on growth hormone and prolactin receptor gene expression in insulin-producing cells. Endocrinology. 1993;133:1165–1172. doi: 10.1210/endo.133.3.8365359. [DOI] [PubMed] [Google Scholar]
- 25.Leahy JL, Bonner-Weir S, Weir GC. Abnormal glucose regulation of insulin secretion in models of reduced B-cell mass. Diabetes. 1984;33:667–673. doi: 10.2337/diab.33.7.667. [DOI] [PubMed] [Google Scholar]
- 26.Goodner CJ, Koerker DJ, Weigle DS, McCulloch DK. Decreased insulin-and glucagon-pulse amplitude accompanying beta-cell deficiency induced by streptozocin in baboons. Diabetes. 1989;38:925–931. doi: 10.2337/diab.38.7.925. [DOI] [PubMed] [Google Scholar]
- 27.Butler AE, et al. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 28.Mezuk B, Eaton WW, Albrecht S, Golden SH. Depression and type 2 diabetes over the lifespan: a meta-analysis. Diabetes Care. 2008;31:2383–2390. doi: 10.2337/dc08-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kozhimannil KB, Pereira MA, Harlow BL. Association between diabetes and perinatal depression among low-income mothers. JAMA. 2009;301:842–847. doi: 10.1001/jama.2009.201. [DOI] [PubMed] [Google Scholar]
- 30.Gianfrancesco FD, Grogg AL, Mahmoud RA, Wang RH, Nasrallah HA. Differential effects of risperidone, olanzapine, clozapine, and conventional antipsychotics on type 2 diabetes: findings from a large health plan database. J Clin Psychiatry. 2002;63:920–930. doi: 10.4088/jcp.v63n1010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.