

Abstract
LRRK2 is a large and complex protein that possesses kinase and GTPase activities and has emerged as the most relevant player in PD pathogenesis possibly through a toxic gain-of-function mechanism. Kinase activity is a critical component of LRRK2 function and represents a viable target for drug discovery. We now report the development of a mechanism-based TR-FRET assay for the LRRK2 kinase activity using full-length LRRK2. In this assay, PLK-peptide was chosen as the phosphoryl acceptor. Combination of steady state kinetic studies and computer simulations was used to calculate the initial concentrations of ATP and PLK-peptide to generate a steady state situation that favors the identification of ATP noncompetitive inhibitors. The assay was also run in the absence of GTP. Under these conditions, the assay was sensitive to inhibitors that directly interact with the kinase domain and those that modulate the kinase activity by directly interacting with other domains including the GTPase domain. The assay was optimized and used to robustly evaluate our compound library in 384-well format. An inhibitor identified through the screen was further characterized as a noncompetitive inhibitor with both ATP and PLK-peptide and showed similar inhibition against LRRK2 WT and the mutant G2019S.
Keywords: LRRK2 kinase, Assay and analysis
Parkinson's disease (PD) characterized by tremor, rigidity, bradykinesia, and postural instability, is the second most common neurodegenerative disorder after Alzheimer's disease (AD). It affects over 1 million Americans and more than 60,000 patients are newly diagnosed each year [1, 2]. PD is caused by the loss of dopaminergic neurons in the substantia nigra. Normally, these neurons produce dopamine, an essential chemical messenger in the brain. Once damaged, these neurons stop producing dopamine and compromise the brain's ability to control movement. Mutations in several genes have been genetically linked to PD in recent years [3]. Among those genes, the leucine-rich repeat kinase2 (LRRK2) has emerged as the most relevant player in PD pathogenesis and has been associated with typical idiopathic, late-onset PD [4–8]. At least 20 mutations in LRRK2 have been found in the most common familial forms and some sporadic forms of PD. For example, the most common mutant G2019S accounts for approximately 5% of familial cases and 1% of sporadic cases.
LRRK2 is a large and complex protein containing several domains, including a leucine-rich repeat (LRR) domain, a Roc domain followed by its associated COR domain, a kinase domain, and a C-terminal WD40 domain [9, 10]. LRRK2 is unusual in that it encodes two distinct but functionally linked enzymes: a protein kinase and a GTPase. Although a recent study in animals suggests that LRRK2 is involved in regulating dopamine transmission, the physiological function of LRRK2 remains largely unknown [11]. The physiological substrate of LRRK2 is unclear despite the recent studies reporting that ezrine, radixin, and moesin (ERM), proteins which anchor the actin cytoskeleton to the plasma membrane, are efficiently phosphorylated by LRRK2 as potential substrates [12, 13]. Although sequence homology analysis has placed LRRK2 in the tyrosine kinase like family, it functions as a serine/threonine kinase. Recent studies have suggested that LRRK2 is capable of undergoing both autophosphorylation and generic substrate phosphorylation, and the kinase activity is regulated by the GTP domain [10, 14–17]. The elevated kinase activity found in some PD-associated mutations is linked to neurotoxicity in cultured neurons [10, 18, 19]. Although the kinase activity is a critical component of LRRK2 function, its causative role in PD remains debatable due to inconsistent results in literature regarding the mutant effects on the kinase activity. Nonetheless, the most common PD mutation of LRRK2, G2019S, has been consistently reported to increase kinase activity by 2–3 fold.
As a drug discovery center, we initiated a program to identify inhibitors of LRRK2 kinase that could first be used to test the likely role of LRRK2 in the pathogenesis in PD and then, if inhibition of the kinase activity will lead to disease modification. Our strategy is to screen large collections of structurally diverse drug-like small molecules to identify inhibitors of LRRK2 using an assay with full-length LRRK2 that is sensitive to inhibitors interacting directly with the kinase domain and also allosteric inhibitors that modulate kinase activity through interaction with other LRRK2 domains including the GTP binding domain. Here we will discuss the design of a mechanism-based assay upon our understanding of the two enzymatic properties of LRRK2, report the screen performance of this assay, and describe characterization of hits identified through screen.
MATERIALS AND METHODS
Materials
ATP, ADP, AMP, GDP, GMP, DTT, magnesium chloride, (HEPES), and bovine serum albumin were purchased from Sigma (St. Louis, MO). GTP was purchased from Bioline (Taunton, MA). ULight-PLK-peptide, Eu-anti-phospho-PLK, and [[α-33P]-GTP were purchased from PerkinElmer (Boston, MA). Full-length LRRK2 (molecular mass of 285 kDa) was purified from BAC-transgenic mouse brain as described by Li [20]. Truncated human LRRK2 WT and the mutant G2019S of residues 970–2527 were purchased from Invitrogen (Carlsbad, California).
Compound Library
The compound library consisted of 63,400 small molecules purchased from Peakdale (High Peak, UK), Maybridge plc. (Cornwall, UK), Bionet Research Ltd. (Cornwall, UK), Chemdiv (San Diego, CA), and Chembridg (San Diego, CA). Compounds were selected from the different vendors by applying a series of filters, including for clogP and predicted solubility. All of the small molecules generally adhere to Lipinski's rules (i.e. molecular weight < 500, H-bond donors ≤ 5, H-bond acceptors ≤ 10 and logP < 5) and contain a low proportion of known toxicophores (i.e. Michael acceptors and alkylating agents) and unwanted functionalities (i.e. imines, thiols, and quaternary amines), and have been optimized for maximization of molecular diversity. All compounds were stored at −20 °C.
High-Throughput Screen—a TR-FRET Assay
A sensitive TR-FRET assay was developed for the LRRK2-catalzyed PLK-peptide phosphorylation, in which the ULight-labeled peptide with a motif of RRRSLLE derived from PLK protein was chosen as the phosphoryl acceptor. In this assay, the formation of phosphorylated PLK-peptide was detected by the binding of antibody Eu-anti-phospho-PLK that allows energy transferring from Eu to the small molecule ULight. Fluorescence intensity generated therefore is proportional to the amount of phosphorylated PLK-peptide produced. The kinase assay for phosphorylation of PLK-peptide was conducted in buffer containing 20 mM HEPES (pH 7.4), 50 mM NaCl, 10 mM MgCl2, 1 mM DTT, BSA 0.5 mg/ml, 1 mM beta-Gly-PO4. Beta-Gly-PO4 is a phosphatase inhibitor and is added to protect against phosphatases. Both enzyme and substrates were prepared in this buffer. The enzyme/compound mixture was incubated (4 nM enzyme and 10 μM compound final concentration) at room temperature for 30 min before PLK-peptide and ATP (50 nM PLK-peptide and 25 μM ATP final concentration) were added to initiate the reaction, which allows the detection of time-dependent inhibitors. The reactions were incubated at room temperature for 6 h and were stopped by the addition of 15 mM EDTA, followed by 2 h incubation with 1 nM Eu-anti-phospho-PLK. The TR-FRET signal was read by an EnVision plate reader (PerkinElmer). In all cases, reaction progress curves were linear for at last 6 h.
Inhibition Studies of LRRK2-Catalyzed LRRKtide Phosphorylation
The inhibition of full-length LRRK2-catalyzed phosphorylation of LRRKtide (RLGRDKYKTLRQIRQ) was conducted in buffer containing 20 mM HEPES (pH 7.4), 50 mM NaCl, 10 mM MgCl2, 1 mM DTT, BSA 0.5 mg/ml, 1 mM beta-Gly-PO4, 50 μM LRRKtide, 100 μM ATP/1 μCi [γ-33P]-ATP, and different concentrations of compound. The reactions were conducted in duplicate, initiated by the addition of 30 nM LRRK2, and incubated at room temperature for 45 min. The reaction was stopped by the addition of 20 mM EDTA, and the mixture was transferred to a multiscreen PH filtration plate (Millipore, Billerica, MA) and washed six times with 75 mM H3PO4. The plate was dried, filters were removed, and the samples were counted with a scintillation counter. Background reaction was conducted in the absence of LRRK2. Reaction progress curve for production of phospho-LRRKtide was linear over at last 60 minutes, and allowed calculation of initial velocities.
Inhibition Studies of GTP Hydrolysis Catalyzed by LRRK2
The GTPase assay for GTP hydrolysis was conducted in buffer containing 20 mM Tris (pH 7.4), 50 mM NaCl, 10 mM MgCl2, 1 mM DTT, BSA 0.5 mg/ml, 80 μM GTP/1.5 μCi [α-33P]-GTP. The reactions were conducted in triplicate, initiated by the addition of 30 nM LRRK2, and incubated at room temperature for 20 min. The reaction was stopped by the addition of 20 mM EDTA, and the product [α-33P]-GDP was separated from [α-33P]-GTP by PEI-cellulose thin layer chromatography (TLC) (Sigma, ST. Louis, MO) (Sigma, ST. Louis, MO) developed by 0.5 M KH2PO4 (pH3.4) developing buffer and analyzed by scintillation counter. Reaction progress curve for GTP hydrolysis was linear over at last 30 minutes, and allowed calculation of initial velocities.
Data Analysis – Basic Equations
Data were analyzed by nonlinear least-squares, using either Sigma-Plot or Grafit software packages. Simulations were performed using the program Scientist, MicroMath Scientific Software. High-throughput screening data was loading into ActivityBase.
RESULTS AND DISCUSSION
Source of LRRK2 Enzyme
When screening a compound library to identify lead compounds to be used as a starting point for drug development, it would be ideal to use human enzymes. But sometimes practical considerations, such as quantity and purity of protein, override this. In addition, although truncated versions of enzymes expressing multidomains, such as LRRK2, can demonstrate catalytic activity, subtle differences in protein structure and domain composition can have dramatic effects on inhibitor interactions. Thus, it is important, whenever feasible, to work with full-length protein. LRRK2 is expressed in multiple tissues, but the purified LRRK2 from brain has elevated kinase activity compared to enzyme isolated from other sources [20]. The mouse and human LRRK2 share 86% sequence identity and were determined to be kinetically identical (data not shown). Therefore, we chose to use full-length LRRK2 protein from brain in our primary screen, which not only presents the opportunity to identify inhibitors that directly or indirectly interact with the kinase domain, but also allows us to measure the relative affinities of inhibitors under conditions closer to physiological conditions. All the hits identified were subsequently tested against human LRRK2.
Development and Evaluation of the TR-FRET Assay
A TR-FRET assay using PLK-peptide with a motif of RRRSLLE (based on the putative phosphorylation site of PLK protein) as the phosphoryl acceptor was developed. Using this assay, a linear dependence of fluorescence intensity on time was demonstrated, which allowed for calculating the initial velocity by measuring the slope of the linear product versus time. The reaction progress curves were linear for at least 6 h for LRRK2 concentrations ranged from 1– 16 nM (Figure 1A). Over the same range of LRRK2 concentration, FU intensity also showed linear dependence on LRRK2 concentration (Figure 1B). Next, the initial velocities were measured as a function of PLK-peptide at 200 μM ATP and the dependence of v0 on PLK-peptide adhered to the simple Michaelis-Menten equation, yielding steady-state kinetic parameter estimates: (kcat)app = 31870 ± 837 FU/h and (KPLK)app = 592 ± 56 nM (Figure 2). To evaluate this assay, the reaction was run in a radiometric assay under the same condition, and similar kinetics was obtained with steady-state kinetic parameter estimates: (kcat)app = 33.6 ± 3.5 nM/h and (KPLK)app = 434 ± 16 nM. Linear correlation between unit of FU/h and unit of nM/h allowed us to calculate a proportionality factor of 855 ± 108 FU/nM that can be used to convert initial velocities from unit of FU/h to unit of nM/h (Figure 2).
Figure 1.

The reaction progress curves. (A) The reactions were run in the presence of 25 μM ATP and 50 nM PLK-peptide at different enzyme concentrations of 16 (●), 8 (○), 4 (▾), 2 (▿), and 1 (▪) nM. The reaction progress curves were linear for at least 6h. (B) Fluorescence intensity dependence on LRRK2 concentration.
Figure 2.

Comparison of the TR-FRET and radiometric assays for LRRK2-catalyzed PLK-peptide phosphorylation. The initial velocities were measured as a function of [PLK-peptide] at 200 μM ATP, and 24 nM LRRK2 under the same condition in a TR-FRET assay (●) and a radiometric assay (○). Filled circles correspond to the y-axis on the left, while open circles correspond to the y-axis on the right. The data were fit to the simple Michaelis-Menten equation. Best fit parameters are: (kcat)app = 31870 ± 837 FU/h and (KPLK)app = 592 ± 56 nM for the TR-FRET assay; (kcat)app = 33.6 ± 3.5 nM/h and (KPLK)app = 434 ± 16 nM for the radiometric assay. The inset shows the linear correlation of unit FU/h and unit nM/h.
Steady-state Kinetic Analysis of the LRRK2-catalyzed PLK-peptide Phosphorylation
To determine the kinetic mechanism for the LRRK2-catalyzed PLK-peptide phosphorylation, initial velocities were measured as a function of PLK-peptide concentration (12.5 nM ≤ [PLK-peptide] ≤ 800 nM) at six fixed concentrations of ATP (6.3 μM ≤ [ATP] ≤ 200 μM) (Figure 3). Initial velocities for each substrate pairs were measured in duplicate. The complete data set was subjected to global analysis by nonlinear least-squares fits to the three standard mechanisms, ping-pong, ordered, and random/steady-state ordered, using eqs 1 – 3.
Figure 3.
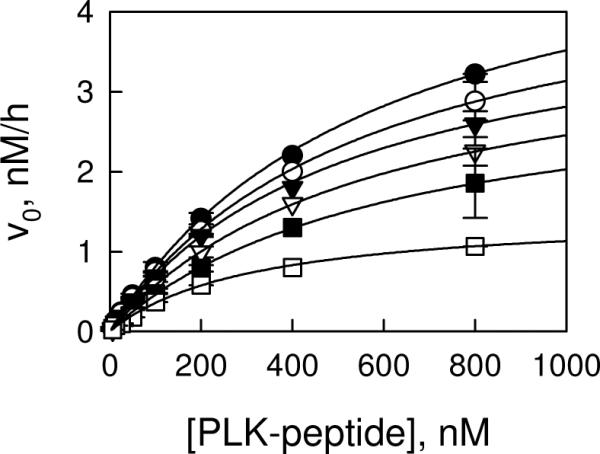
Initial velocity study of LRRK2-catalyzed PLK-peptide phosphorylation. Initial velocities were measured as a function of PLK-peptide concentration at 200 (●), 100 (○), 50 (▾), 25 (▿), 12.5 (▪), and 6.3 μM ATP (□). The complete data set was subjected to global analysis by nonlinear least-squares fits to the random/steady-state ordered mechanism. Each data point is the average of duplicate determinations. Error bars represent the simple deviation from the mean value.
Ping-Pong:
| (1) |
KA and KB are Michaelis constants.
Rapid Equilibrium Ordered:
| (2) |
KA and KB are substrate dissociation constants from EA and EB, respectively.
Rapid Equilibrium Random/Steady-State Ordered:
| (3) |
For rapid equilibrium systems, KA, KB, αKA, and αKB are substrate dissociation constants from EA, EB, and EAB; for steady-state systems, KA is substrate dissociation constant from EA, αKA and αKB are Michaelis constants.
Statistically the random mechanism or steady-state ordered mechanism fits the data the best, yielding the following estimates averaged from two independent experiments: kcat = 0.024 ± 0.002 min−1, KATP = 13 ± 3 μM, KPLK = 0.58 ± 0.1 μM, and α = 1.3 ± 0.2. Inhibition studies using substrate analogue AMP-PNP and LRRKtideA (RLGRDKYKALRQIRQ) and product ADP suggest that the reaction follows a rapid equilibrium random mechanism with either ATP or PLK-peptide binding first to the enzyme (21).
Mechanistic Simulations for an Unbalanced Steady State
The goal of this screening program is to increase the likelihood of identifying ATP noncompetitive inhibitors to have better selectivity without compromising other kinases containing conserved sequence and structure. Adjusting the substrate concentrations can favor one type of inhibitor over another. Having determined the kinetic mechanism and the kinetic parameters for the LRRK2-catalyzed PLK-peptide phosphorylation, we are able to determine [ATP]0 and [PLK-peptide]0 to produce a steady state situation which favors the identification of ATP noncompetitive inhibitors. To calculate concentrations of E, EA, EB, and EAB enzyme species accumulated in the steady state, simulations were performed as a function of [ATP]0 and [PLK-peptide]0 based on the mechanism of Scheme I and [E]total = 4 nM. Examples of these simulations are shown in Figure 4 where the calculated concentrations of enzyme species are plotted as a function of [ATP] under conditions of [PLK-peptide]/Km = 0.1 and [PLK-peptide]/Km close to 1. It can be seen that free E and EA complex are dominant at low [PLK-peptide] for ATP concentrations above its Km. An [ATP] 2.5-fold relative to its Km and [PLK-peptide] 1/10 of its Km were chosen for the screening assay to purposely bias the screen in favor of identifying ATP noncompetitive inhibitors. Under this condition, the major enzyme species are the E:A complex which accounts for 65% of the total enzyme and the free enzyme which accounts for 26% (E:B accounts for 2.6%, and E:A:B central complex 5%). Screening under this condition increases the likelihood of identifying ATP noncompetitive inhibitors which bind to either the free enzyme or the E:A complex and decrease the probability of identifying modest ATP competitive inhibitors, which binds to the free enzyme and the E:B complex.
Scheme I.

The rapid equilibrium random mechanism of the LRRK2-catalyzed PLK-peptide phosphorylation. A represents ATP and B represents PLK-peptide.
Figure 4.

Steady state concentrations of E, EA, EB, and EAB as a function of [ATP] and [PLK-peptide]. Simulations were performed as a function of [ATP]0 under conditions of [PLK-peptide]0/Km = 0.1 (A) and [PLK-peptide]0/Km close to 1 (B) based on the mechanism of Scheme I and [E]total = 4 nM.
LRRK2 Assay and Its Performance
With the completion of kinetic analyses for the kinase activity of LRRK2, a mechanism-based assay was designed at substrate concentrations of [ATP] = 25 μM and [PLK-peptide] = 50 nM. Next question is whether the screen should be performed in the presence of GTP. It has been hypothesized that the kinase activity of LRRK2 is regulated by the GTPase domain: the binding of GTP stimulates LRRK2 kinase activity [10, 15, 22]. The effect of GTP in a dose-response manner was tested and found not to significantly activate or inhibit the kinase activity [21]. The presence of saturating GTP concentrations does not lead to a stronger signal, and indeed it might decrease the likelihood of identifying compounds which bind to the GTPase domain and mimic the physiological GTPase regulators, such as GTPase-activating proteins (GAP) and guanine nucleotide exchange factors (GEF) [23]. As a result of it, the screen was performed in the absence of GTP.
One challenge of performing screening assays is to conserve the amount of enzyme required. For the LRRK2 assay, an enzyme concentration of 4 nM was chosen, which produced a linear progress curve for at least 6 h. The reaction was allowed to progress for 6 h before being quenched by the addition of 15 mM EDTA. The assay was readily miniaturized to a kinase reaction volume of 8 μL per well in 384-well low volume plates. Conducting the reaction for the longer time period and in the smaller volume significantly reduced the amount of enzyme required, but still resulted in high data quality with a large signal window. The DMSO concentration dependence suggests that the assay could tolerate DMSO up to 2% without significantly decreasing enzyme activity. Therefore, a final DMSO concentration of 1% was chosen for the screen. Also assay performance was tested at different concentrations of antibody Eu-anti-phospho-PLK at a variety of incubation times. An antibody concentration of 1 nM and incubation time of 2 h were chosen to generate a stable and sufficient signal window. The performance of optimized assay was shown in Figure 5, in which the Z'-values in three separate plates are 0.83, 0.87, 0.79. These results suggest that our assay was sufficiently robust to be used for high-throughput screen.
Figure 5.
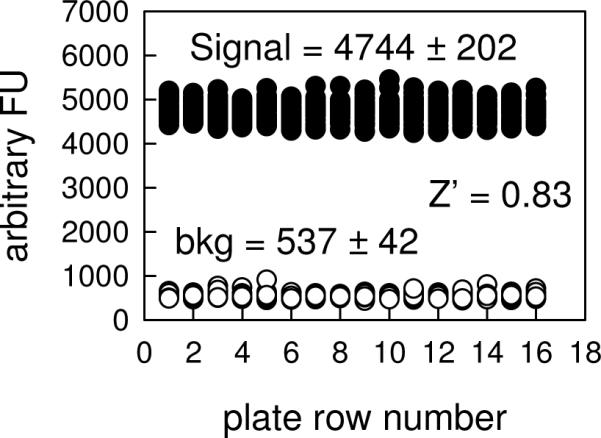
Performance of the high-throughput assay of LRRK2-catalyzed PLK-peptide phosphorylation. The assay was run at 25 μM ATP, 50 nM PLK-peptide, 4 nM LRRK2 in the absence of GTP. The background was run in the absence of LRRK2. The figure describes the intraplate assay performance in which S/N ratio of 9 and Z' values of 0.83 can be calculated.
Hits Confirmation and Characterization
A screen of 63,400 compounds at a single concentration of 10 μM produced 63 hits using a 50% cutoff (0.1% hit rate). Close to 50% hits was confirmed in 5-dose response test. Among those, 21 compounds were prioritized in a 12-dose response test and showed IC50 ≤ 10 μM in the TR-FRET assay and were reproduced in a radiometric assay using LRRKtide (RLGRDKYKTLRQIRQ) as a phosphoryl acceptor to rule out fluorescence or other artifacts. All the compounds showed similar IC50 in the radiometric assay, and were further tested against human LRRK2 WT and the mutant G2019S. One inhibitor from the screen, LDN-22684, was selected as an example to illustrate compound characterization. First, the reversibility of LDN-22684 inhibition was tested by the method of rapid and large dilution. After half hour preincubation of 400 nM LRRK2 with 100 μM LDN-22684 (100- and 10-fold, respectively, over the screen assay concentrations), this mixture was diluted 100-fold into reaction buffer containing the substrate to initiate the reaction. The enzyme activity was rescued after the rapid dilution thus demonstrating that LDN-22684 is a reversible inhibitor. Then, LDN-22684 inhibition as a function of time was evaluated as shown in Figure 6. The progress curve in the presence of LDN-22684 was not linear and showed time-dependence inhibition, with inhibition being quite minimal early in the progress curve and increasing with reaction time, demonstrating that LDN-22684 is a slow-binding inhibitor. Next, different concentrations of LDN-22684 were preincubated with the enzyme for 3h at room temperature before the initiation of the reaction by the addition of the substrate. The dependence of initial velocity on inhibitor concentration adhered to the simple inhibition expression of general form: vinhib = vcontrol/(1 + [I]/Ki,app) with an IC50 of 5.4 ± 0.8 μM. There is no sign of partial inhibition and no need to include the higher-order terms to attain good data fitting. Similar IC50 of 6.4 ± 0.5 μM was obtained in the radiometric assay using LRRKtide as a phosphoryl acceptor. The compound LDN-22684 was also tested in the LRRK2-catazlyzed GTP hydrolysis reaction and no significant activation or inhibition was observed (data not shown). It suggests that the compound LDN-22684 modulates the kinase activity of LRRK2 without affecting the GTPase activity. Finally, the compound LDN-22684 was tested against commercially available human LRRK2 WT and the mutant G2019S. The studies were performed using a truncated version of LRRK2 of residues 970–2527. The inhibition of LDN-22684 for the mutant G2019S and the truncated wild-type still showed time-dependence. The initial velocities were measured as a function of inhibitor concentration after a 3h preincubation of the inhibitor with the enzyme. The dependence of initial velocity on inhibitor concentration adhered to the simple inhibition expression of general form: vinhib = vcontrol/(1 + [I]/Ki,app) with IC50's of 6.9 ± 1.0 μM and 6.1 ± 0.8 μM for the mutant G2019S and wild-type, respectively (Figure 7). There was no significant difference in the IC50 values between the mutant G2019S and wild-type, and between the mouse and human proteins.
Figure 6.

Product progress curves for LRRK2-catalyzed PLK-peptide phosphorylation in the presence (●) and absence (○) of LDN-22684. The reaction was carried out at 25 μM ATP, 50 nM PLK-peptide, and 4 nM LRRK2.
Figure 7.

Inhibition of the human LRRK2 WT- and the mutant G2019S-catalyzed PLK-peptide phosphorylation by LDN-22684. The LRRK2 WT (●) and the mutant G2019S (○) were preincubated with LDN-22684 for 3h prior to the reaction initiation by the addition of substrates. The initial velocity of LRRK2-catalyzed phosphorylation of PLK-peptide were measured as a function of concentration of LDN-22684 at ATP = 25 μM and PLK = 50 nM. Each data point is the average of triplicate determinations. The data was fit to the simple inhibition expression of general form: vinhib = vcontrol/(1 + [I]/Ki,app).
Mechanism of Inhibition of LDN-22684
The mechanism of inhibition of the compound LDN-22684 was studied using full-length LRRK2 and data were analyzed using the methods of replots as described previously [21, 24]. First we determined (i) at a single [PLK-peptide], the dependence of v0 on [ATP] at several concentrations of [LDN-22684] (Figure 8A) and (ii) at a single [ATP], the dependence of v0 on [PLK-peptide] at several concentrations of [LDN-22684] (Figure 8D). Next, for (i), we analyzed the dependence of vo on [ATP] at each [LDN-22684] to calculate apparent values (kcat)ATP and (kcat/Km)ATP. For (ii), we analyzed the dependence of vo on [PLK] at each [LDN-22684] to calculate apparent (kcat)PLK and (kcat/Km)PLK. Replots of apparent values of (kcat)X (X = ATP or PLK-peptide) vs [LDN-22684] and apparent values of (kcat/Km)X vs [LDN-22684] were then constructed, which shows a specific, mechanism-based pattern [21, 24].
Figure 8.

Inhibition of the LRRK2-catalyzed phosphorylation of PLK-peptide by the compound LDN-22684. (A) Plot of initial velocities vs [ATP] at 25 (●), 12.5 (○), 6.3 (▾), 3.1 (▿), 1.6 (▪), and 0 μM LDN-22684 (□), all at a fixed PLK-peptide concentration of 200 nM. (B and C) LDN-22684 concentration dependencies of apparent values of (kcat)ATP and (kcat/Km)ATP derived from analysis of the data in panel A. (D) Plot of initial velocities vs [PLK-peptide] at 25 (●), 12.5 (○), 6.3 (▾), 3.1 (▿), 1.6 (▪), and 0 μM LDN-22684 (□), all at a fixed ATP concentration of 50 μM. (E and F) LDN-22684 concentration dependencies of apparent values of (kcat)PLK and (kcat/Km)PLK derived from analysis of the data in panel D.
When ATP was the variable substrate, we found that apparent values of (kcat)ATP and (kcat/Km)ATP both depend on [LDN-22684] (Figures 8B and 8C) according to a simple inhibition expression of general form: vinhib = vcontrol/(1 + [I]/Ki,app), yielding similar Ki,app's values of 8.2 ± 1.2 μM and 7.2 ± 1.4 μM at saturating and low ATP concentrations, respectively. These patterns indicate that the compound LDN-22684 is noncompetitive with ATP. As a comparison, (kcat)ATP should be independent of a competitive inhibitor. When PLK-peptide was the variable substrate, both apparent values of (kcat)PLK and (kcat/Km)PLK titrate with the compound LDN-22684 (Figures 8E and 8F), suggesting that the compound LDN-22684 is also a noncompetitive inhibitor of PLK-peptide and can bind to all the enzyme forms accumulated in the steady-state as shown in Scheme I. This sort of complex mechanism of inhibition that we observed for LDN-22684 has been reported for N4-(6-aminopryimidin-4-yl)-sulfanilamide (APS) in the study for cyclin-dependent kinase 5 [24]. The kinetic data suggested that APS is noncompetitive with both ATP and the peptidic substrate and can bind to all four enzyme forms. The superposition of structures of ATP bound enzyme and APS bound enzyme clearly showed simultaneous binding of ATP and APS to the same site on the enzyme. Future crystal structure of the kinase domain of LRRK2 would help us elucidate the binding site of LDN-22684.
In conclusion, a mechanism-based HTS assay for LRRK2 was developed and utilized to identify LRRK2 inhibitors. The assay was also used to robustly evaluate the potency of hits and to further characterize hits for their inhibitory efficacy between LRRK2 WT and the mutant G2019S. Identification of LRRK2 kinase inhibitors using the screening protocol described herein can provide an important first-step in the development of new therapeutics for the treatment of PD and also provide useful probes to further investigate the physiological functions of LRRK2 in normal cellular biology and in pathological conditions.
Table 1.
Kinetic parameters of LRRK2 kinase activity
| kcat, min−1 | 0.024 ± 0.001 |
| KATP, μM | 8.3 ± 1.4 |
| KPLK, μM | 0.57 ± 0.09 |
| α | 1.5 ± 0.2 |
ABBREVIATIONS
- PD
Parkinson's disease
- LRRK2
leucine-rich repeat kinase2
- PLK-peptide
PLK-derived peptide with a motif of RRRSLLE
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- [2].Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68:384–386. doi: 10.1212/01.wnl.0000247740.47667.03. [DOI] [PubMed] [Google Scholar]
- [3].Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu. Rev. Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- [4].Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, López de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Martí-Massó JF, Pérez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- [5].Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Müller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- [6].Berg D, Schweitzer K, Leitner P, Zimprich A, Lichtner P, Belcredi P, Brussel T, Schulte C, Maass S, Nagele T. Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson's disease. Brain. 2005;128:3000–3011. doi: 10.1093/brain/awh666. [DOI] [PubMed] [Google Scholar]
- [7].Khan NL, Jain S, Lynch JM, Pavese N, Abou-Sleiman P, Holton JL, Healy DG, Gilks WP, Sweeney MG, Ganguly M, Gibbons V, Gandhi S, Vaughan J, Eunson LH, Katzenschlager R, Gayton J, Lennox G, Revesz T, Nicholl D, Bhatia KP, Quinn N, Brooks D, Lees AJ, Davis MB, Piccini P, Singleton AB, Wood NW. Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson's disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain. 2005;128:2786–2796. doi: 10.1093/brain/awh667. [DOI] [PubMed] [Google Scholar]
- [8].Mata IF, Kachergus JM, Taylor JP, Lincoln S, Aasly J, Lynch T, Hulihan MM, Cobb SA, Wu RM, Lu CS, Lahoz C, Wszolek ZK, Farrer MJ. Lrrk2 pathogenic substitutions in Parkinson's disease. Neurogenetics. 2005;6:171–177. doi: 10.1007/s10048-005-0005-1. [DOI] [PubMed] [Google Scholar]
- [9].Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, López de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Martí-Massó JF, Pérez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- [10].Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- [11].Li X, Patel JC, Wang J, Avshalumov MV, Nicholson C, Buxbaum JD, Elder GA, Rice ME, Yue Z. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. J Neurosci. 2010;30:1788–1797. doi: 10.1523/JNEUROSCI.5604-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, Alessi DR. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. 2007;405:307–317. doi: 10.1042/BJ20070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Parisiadou L, Xie C, Cho HJ, Lin X, Gu XL, Long CX, Lobbestael E, Baekelandt V, Taymans JM, Sun L, Cai H. Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J Neurosci. 2009;29:13971–13980. doi: 10.1523/JNEUROSCI.3799-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Greggio E, Zambrano I, Kaganovich A, Beilina A, Taymans JM, Daniëls V, Lewis P, Jain S, Ding J, Syed A, Thomas KJ, Baekelandt V, Cookson MR. The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J Biol Chem. 2008;283:16906–16914. doi: 10.1074/jbc.M708718200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Guo L, Gandhi PN, Wang W, Petersen RB, Wilson-Delfosse AL, Chen SG. The Parkinson's disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp Cell Res. 2007;313:3658–3670. doi: 10.1016/j.yexcr.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ito G, Okai T, Fujino G, Takeda K, Ichijo H, Katada T, Iwatsubo T. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial parkinsons's disease. Biochemistry. 2007;46:1380–1388. doi: 10.1021/bi061960m. [DOI] [PubMed] [Google Scholar]
- [17].West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, Dawson VL, Dawson TM, Ross CA. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci. 2005;102:18676–18681. doi: 10.1073/pnas.0508052102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- [20].Li X, Tan YC, Poulose S, Olanow CW, Huang XY, Yue Z. Leucine- rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson's disease R1441C/G mutants. J Neurochem. 2007;103:238–247. doi: 10.1111/j.1471-4159.2007.04743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu M, Dobson B, Glicksman MA, Yue Z, Stein RL. Kinetic Mechanistic Studies of WT Leucine-Rich Repeat Kinase2: Characterization of the Kinase and GTPase Activities. Biochemistry. 2010;49:2008–2017. doi: 10.1021/bi901851y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Manning G, Whyte DB, Martinex R, Hunter T, Sudarsanam S. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemsitry. 2002;46:1380–1388. doi: 10.1021/bi061960m. [DOI] [PubMed] [Google Scholar]
- [23].Anand VS, Reichling LJ, Lipinski K, Stochaj W, Duan W, Kelleher K, Pungaliya P, Brown EL, Reinhart PH, Somberg R, Hirst WD, Riddle SM, Braithwaite SP. Investigation of leucine-rich repeat kinase 2 : enzymological properties and novel assays. FEBS J. 2009;276:466–478. doi: 10.1111/j.1742-4658.2008.06789.x. [DOI] [PubMed] [Google Scholar]
- [24].Liu M, Choi S, Cuny GD, Ding K, Dobson BC, Glicksman MA, Auerbach K, Stein RL. Kinetic studies of Cdk5/p25 kinase: phosphorylation of tau and complex inhibition by two prototype inhibitors. Biochemistry. 2008;47:8367–8377. doi: 10.1021/bi800732v. [DOI] [PubMed] [Google Scholar]