

Abstract
Experiments were designed to test the hypothesis that endothelin and/or reactive oxygen species contribute to the pressor response induced by acute air jet stress in normotensive Dahl salt-sensitive rats maintained on a normal salt diet (pre-hypertensive). Mean arterial pressure was chronically monitored by telemetry before and after 3-day treatment with the free radical scavenger, 4-hydroxy-2,2,6,6-tetramethyl piperidinoxyl (tempol), or endothelin receptor antagonists, ABT-627 (ETA antagonist) or A-182086 (ETA/B antagonist), supplied in the drinking water. Rats were restrained and subjected to pulsatile air jet stress (3 minutes). Plasma samples at baseline and during acute stress were analyzed for 8-isoprostane (measure of reactive oxygen species production) and endothelin. Neither tempol nor endothelin receptor antagonist treatment had an effect on baseline mean arterial pressure or plasma 8-isoprostane. The pressor response to acute stress was accompanied by significant increases in plasma 8-isoprostane and endothelin. Tempol significantly reduced both the total pressor response (area under the curve) and the stress-mediated increase in plasma 8-isoprostane; conversely, tempol had no effect on the stress-induced increase in plasma endothelin. Combined ETA/B antagonism, but not selective ETA receptor blockade, similarly suppressed the pressor response to stress and stress-mediated rise in 8-isoprostane. Together, these results indicate that reactive oxygen species contribute to the pressor response to acute air jet stress. Furthermore, the increase in reactive oxygen species occurs downstream of ET receptor activation.
Keywords: endothelin, reactive oxygen species, air jet stress, Dahl salt-sensitive rat, blood pressure
INTRODUCTION
Reactive oxygen species (ROS) contribute to the pathogenesis of cardiovascular dysfunction associated with several diseases, including hypertension, chronic heart failure, ischemic heart disease, hyperlipidemia, and diabetes mellitus.1–3 In addition, results from several laboratories suggest that ROS are implicated in normal cardiovascular function. Systemic administration of the free radical scavenger, 4-hydroxyl-2,2,6,6-tetramethylpiperidine-1-oxyl (tempol), leads to significant decreases in mean arterial pressure (MAP), heart rate (HR), and sympathetic nerve activity in normotensive animals,4–6 suggesting an important role of ROS in the regulation of arterial pressure. Studies performed in vitro7, 8 and in vivo9, 10 also demonstrate that ROS are involved in the constrictor or pressor response, respectively, to various agonists.
A growing body of evidence suggests that behavioral stress elicits production of ROS; most of these reports, however, focus on chronic stress paradigms.11, 12. Numerous studies suggest that acute stress may induce cardiovascular dysfunction,13–20 additional studies have shown that cardiovascular hyper-reactivity is strongly associated with future cardiovascular disease.21–28 Proposed as a mechanistic link between acute stress events and chronic disease later in life, the concept of allostatic load suggests that the cumulative effect of repeated challenges over time may lead to disease.29 Thus, we reasoned that delineation of the mechanism(s) mediating the response to a single stress event would aid in understanding the consequences of cumulative effects.
We have utilized a model of acute behavioral stress that combines restraint with pulsatile, unavoidable bursts of air to the head (referred to as air jet stress).30–32 We chose the Dahl salt-sensitive (DS) rat because of its use as a model to evaluate genetically defined risk for salt-sensitive hypertension. Tempol significantly attenuates the high salt-mediated increase in arterial pressure in the DS rat.33, 34 Very little is known, however, to what extent ROS influence pressor responses in DS animals maintained on a normal salt diet or under pre-hypertensive conditions.
ET-1 is an endothelial-derived potent vasoconstrictor peptide. ET-1 is released abluminally and circulating levels are thought to be the result of spillover from elevations in ET-1 production or reduced clearance. ET-1 is also produced in sympathetic nerves35 and renal tubular epithelium.36, 37 Plasma ET-1 levels increase in response to acute mental (mental arithmetic) and physical (cold pressor) stress in human adults38, 39 and in adolescents.38, 40 Treiber et al.38, 40 demonstrated that acute stress-induced elevations in plasma ET-1 correlated with stress-induced increases in blood pressure in pre-hypertensive young adults with verified family histories of cardiovascular disease, suggesting that the stress-induced release of ET-1 may be involved in the acute stress-induced pressor response.
We reasoned that the normotensive DS rat is an appropriate model of pre-hypertensive young adults with a family history of cardiovascular disease. Our focus in this study was to test the hypothesis that ROS contribute to the pressor response to acute air jet stress in pre-hypertensive DS rats. Pre-hypertensive DS rats are more sensitive to the pressor effects of ET-1 than Dahl salt-resistant (DR) counterparts,41 and ET-1 plays a prominent role in the salt-dependent hypertension.42 Furthermore, previous studies have shown that ROS stimulate sympathetic nerve activity5, 6, 43 and promote ET-1 production.44 Therefore, we also tested the hypothesis that the stress-induced increase in ROS in DS rats is downstream of ET-1 receptor activation.
METHODS
Animal model
All experiments used 9- to 12-wk-old male Dahl salt-sensitive (DS) rats (Harlan Laboratories; Indianapolis, IN) fed standard rat chow containing 0.4% NaCl and tap water, ad libitum. One experimental protocol also utilized 9- to 12-wk-old male Dahl salt-resistant (DR) rats. Rats were housed in the animal care facility at the Medical College of Georgia, which is approved by the American Association for the Accreditation of Laboratory Animal Care. All protocols have been approved by the Institutional Animal Care and Use Committee.
Telemetry
Telemetry transmitters (Data Sciences, Inc.) were implanted according to the manufacturer’s specifications, as previously published.45
Air jet stress
DS rats were subjected to 2 sessions of acute air jet stress as previously described45 spaced one week apart, during which the animals were left untreated (week 1) or put on a 3-day regimen of either tempol (1 mM in the drinking water; n=10) or the dual ETA/B receptor antagonist A-182062 (30 mg/kg/day in the food; n=4) (week 2). All animals were previously subjected to at least two 15-minute restraint sessions on days prior to an experiment to reduce the stress associated with the restraint itself. Air jet stress was performed as previously published.45
Determination of plasma concentrations of 8-isoprostane, ET-1, and catecholamines
Rats were anesthetized with ketamine/xylazine (50 mg/kg/10 mg/kg, i.p.), and catheters (Braintree Scientific Inc., Braintree, MA) were inserted into the jugular vein. Catheters were routed subcutaneously, and exteriorized at the back of the neck; catheters were filled with heparin (1000 U/ml). Blood (1 ml) was drawn from restrained animals on two successive days prior to air jet stress to determine baseline (unstressed) plasma levels in the absence or presence of pharmacological treatments, and on the day of stress over the 30–60 sec interval of air jet stress. Blood samples were centrifuged at 10000 × g for 10 min at 4 °C, and plasma was removed, aliquoted, and stored at −80 °C until analyses could be performed. Please see http://hyper.ahajournals.org for detailed methods in the supplemental data section.
Whole animal pressor responses
Separate groups of DS animals were either left untreated (tap water alone) or given tempol (1 mM in the drinking water for 3 days) (n=5 for each). Animals were anesthetized with thiobutabarbital (Inactin; 65 mg/kg, i.p.), and the right femoral artery and vein were isolated and cannulated with PE-50 for monitoring MAP and drug infusion, respectively. Peak and steady-state (one minute before introduction of next dose) responses to endothelin peptides and phenylephrine were determined. All measurements were recorded using a Power Lab data acquisition system. Please see http://hyper.ahajournals.org for detailed methods and graphical results in the supplemental data section.
Statistical Analysis
Data are expressed as means ± SE. All baseline MAP and HR values are reported as 24-hour means. Total pressor response refers to the change in MAP during the 3 minutes of air jet stress, and is expressed as the area under the curve (AUC; mmHg × min). Statistical analyses of baseline MAP and HR and of the total pressor response were made by paired t-test. Baseline plasma values of 8-isoprostane, ET-1, and catecholamines represent the average values obtained for the two days before subjecting the animals to air jet stress. Statistical analyses of plasma determinations were made by two-way analysis of variance, followed by Newman-Keuls test for multiple comparisons. Differences are considered significant at p<0.05.
RESULTS
Three days of pretreatment with the free radical scavenger, tempol, had no effect on baseline (24-hour) MAP, but caused a small, yet significant decrease in baseline HR (Table 1). DS animals were restrained and subjected to acute air jet stress. Comparison of the integrated pressor response, calculated as the area under the curve (AUC) indicated that the stress response was significantly lower in tempol-treated rats (15.9±2.5 vs. 27.5±3.8 mmHg × 3 min, tempol vs. untreated, p<0.05) (Fig. 1). MAP was monitored for 20 minutes following the stress period to assess the extent of blood pressure recovery. Post-stress AUC was significantly more negative in the rats given tempol (−109.4±22.9 vs. −4.8±19.6, tempol vs. untreated, p<0.05) (Fig. 1).
Table 1.
Baseline (24-hr) cardiovascular hemodynamics
| Hemodynamic Measurement | Untreated | Tempol |
|---|---|---|
| Mean Arterial Pressure (mmHg) | 111 ± 4 | 109 ± 2 |
| Heart Rate (beats/min) | 402 ± 5 | 387 ± 5* |
| Untreated | A-182086 | |
| Mean Arterial Pressure (mmHg) | 109 ± 4 | 112 ± 9 |
| Heart Rate (beats/min) | 409 ± 3 | 396 ± 6 |
p < 0.05 vs. Untreated
Figure 1.

Summary of integrated pressor response (area under the curve; AUC) to acute air jet stress (left panel) and integrated mean arterial pressure during the 20-minute post-stress period (right panel) in pre-hypertensive Dahl salt-sensitive rats. Animals were either untreated (week 1) or given the free radical scavenger, tempol, (1 mM in the drinking water; week 2) (n=10) for 3 days. AUC was calculated as the sum of the mean arterial pressure data points during or post-air jet stress minus the average MAP obtained over the 3 minutes before the start of air jet stress. *p<0.05
To examine whether acute air jet stress caused an increase in ROS and whether this was affected by tempol, plasma levels of 8-isoprostane were measured as an index of ROS production. DS rats were fitted with venous catheters and blood was drawn at baseline and then again during air jet stress. In untreated animals, air jet stress caused a near doubling in plasma 8-isoprostane; tempol had no effect on the baseline plasma level of 8-isoprostane, but abolished the stress-mediated rise in 8-isoprostane (Fig. 2A).
Figure 2.

Effect of the free radical scavenger, tempol, on plasma concentrations of 8-isoprostane (A) (n=7–12) and endothelin-1 (ET-1) (B) (n=6–11) at baseline (unstressed) and during air jet stress in pre-hypertensive Dahl salt-sensitive rats. Animals were either untreated (week 1) or given tempol (1 mM in the drinking water; week 2) for 3 days. *p<0.05
We were interested in assessing whether the stress-induced rise in arterial pressure and plasma 8-isoprostane in DS rats was related to the hypertensive genetic predisposition of DS rats or was a general phenomenon. Therefore, we measured the effect of tempol on the stress-mediated pressor response and the stress-mediated plasma 8-isoprostane levels in DR rats. Opposite to what was observed in DS rats, tempol enhanced the integrated pressor response in DR rats (27.6±2.0 vs. 17.6±2.3 mmHg × 3 min, tempol vs. untreated, p<0.05). Plasma levels of 8-isoprostane in DR rats were similar before and during air jet stress (18±1 vs. 17±2 pg/ml, baseline vs. stress).
Previous studies have shown that ROS promote ET-1 production.44 Therefore, we evaluated whether tempol suppressed the stress-induced rise in plasma ET-1 levels. Tempol had no effect on the baseline plasma concentrations of ET-1 (Fig. 2B), nor on the plasma ET-1 levels during stress (Fig. 2B).
Other laboratories have demonstrated that ROS stimulate sympathetic nerve activity,5, 6, 43 thus we measured plasma levels of catecholamines as an indirect determinate of sympathetic activity in the presence and absence of tempol treatment. Tempol did not alter the baseline Epi (Fig. 3A), or NE (Fig. 3B) levels. Whereas tempol significantly reduced the stress-induced increase in plasma Epi (368±33 vs. 522±36 pg/ml, tempol vs. untreated, p<0.05) (Fig. 3A), yet tempol augmented the stress-mediated rise in plasma NE concentration (523±96 vs. 303±33 pg/ml, tempol vs. untreated, p<0.05) (Fig. 3B).
Figure 3.

Effect of the free radical scavenger, tempol, on plasma concentration of epinephrine (Epi) (A) and norepinephrine (NE) (B) (n=7 each) at baseline (unstressed) and during air jet stress Dahl salt-sensitive rats. Animals were either untreated (week 1) or given tempol (1 mM in the drinking water; week 2) for 3 days. *p<0.05
Because ET-1 has been demonstrated to mediate an increase in ROS,46, 47 we tested the effects of endothelin receptor blockade on plasma 8-isoprostane levels to determine whether ROS production was downstream of ET-1 receptor activation. We previously demonstrated that selective ETA receptor blockade does not affect the pressor response to air jet stress or the stress-mediated increase in catecholamines in pre-hypertensive DS rats.45 Nevertheless, we examined whether ETA receptor blockade altered the production of ROS. Treatment of DS rats with the ETA receptor antagonist, ABT-627, had no effect on plasma 8-isoprostane either at baseline or during stress (Fig. 4A). We next examined the effect of dual ETA/B receptor inhibition with A-182086. Pretreatment with A-182086 had no effect on baseline (24-hour) MAP or HR (Table 1). A-182086 also had no effect on baseline plasma 8-isoprostane, but blocked the stress-induced increase in plasma 8-isoprostane (Fig. 4B).
Figure 4.

Effect of the selective endothelin A receptor antagonist ABT-627 (A) (n=15–18) and the dual endothelin A/B receptor antagonist A-182086 (B) (n=13) on plasma concentrations of 8-isoprostane at baseline (unstressed) and during air jet stress in pre-hypertensive Dahl salt-sensitive rats. Animals were either untreated (week 1), or given ABT-627 (5 mg/kg/day in the drinking water; week 2) or A-182086 (30 mg/kg/day in the food; week 2) for 3 days. *p<0.05
Similar to the results obtained with tempol, combined ETA/B receptor antagonism significantly reduced the integrated pressor response to acute stress (6.9±6.7 vs. 43.8±12.5 mmHg × 3 min, A-182086 vs. untreated, p<0.05) (Fig. 5, left panel); post-stress recovery of MAP appeared to be greater, but this difference was not statistically significant (p=0.11) (Fig. 5, right panel). Dual ETA/B receptor antagonism did not affect the baseline plasma catecholamines (Epi: 147±14 vs. 145±15 pg/ml, A-182086 vs. untreated; NE: 406±37 vs. 357±43 pg/ml, A-182086 vs. untreated) or stress-mediated elevation in catecholamines (Epi: 231±34 vs. 282±26 pg/ml, A-182086 vs. untreated; NE: 562±123 vs. 482±55 pg/ml, A-182086 vs. untreated). Treatment with A-182086 increased baseline plasma ET-1 levels (22.65±1.42 pg/ml vs. 0.73±0.14 pg/ml, A-182086 vs. untreated, p<0.0001). DS rats treated with A-182086 during stress did not demonstrate a stress-induced increase in plasma ET-1 (22.65±1.42 pg/ml vs. 20.36±2.74 pg/ml, baseline vs. stress).
Figure 5.

Summary of integrated pressor response (area under the curve; AUC) to acute air jet stress (left panel) and integrated mean arterial pressure during the 20-minute post-stress period (right panel) in pre-hypertensive Dahl salt-sensitive rats. Animals were either untreated (week 1) or given the dual endothelin A/B receptor antagonist, A-182086 (30 mg/kg/day in the food; week 2) (n=4) for 3 days. AUC was calculated as the sum of the mean arterial pressure data points during or post-air jet stress minus the average MAP obtained over the 3 minutes before the start of air jet stress. *p<0.05
ROS have been shown to partially mediate the constrictor response to various agonists, including ET-1.9 The whole animal pressor response to ET-1 and S6c, selective ETB receptor agonist, in anesthetized DS rats with and without tempol treatment was determined. Peak and steady-state pressor responses to exogenous ET-1 (Fig. S3) or S6c (Fig S4) were unaffected by treatment with tempol. We examined the whole animal pressor response to exogenous phenylephrine in anesthetized animals to determine whether there is reduced responsiveness of the vascular smooth muscle to α1 adrenergic stimulation. Experiments were performed in both the absence and presence of autonomic ganglion blockade with chlorisondamine. Chlorisondamine produced comparable decreases in MAP in untreated and tempol-treated animals (Fig S5). Tempol had no effect on the phenylephrine-mediated pressor response in the absence and presence of chlorisondamine (Fig. S6).
DISCUSSION
The principal finding of this study is that the blood pressure responsiveness during acute air jet stress in normotensive DS animals is dependent on ET mediated increases in ROS. Specifically, the free radical scavenger, tempol, significantly lowered the pressor response to air jet stress and abolished the stress-mediated rise in ROS. Similar responses were obtained with combined ETA/B, but not selective ETA receptor blockade. Neither tempol nor combined ETA/B receptor blockade had any effect on 24 hour baseline MAP.
Several studies have shown that ROS can increase ET-1 production in cultured endothelial and vascular smooth muscle cells.48–50 Moreover, under various experimental or pathologic conditions, tempol reduces ET-1 generation in vivo.44, 51, 52 Tempol had no effect on basal or stress-mediated increases in circulating ET-1, suggesting that ROS, per se, is not the stimulus for ET-1 release during acute stress. Conversely, ET-1 can stimulate O2·− generation in aortic rings,8 setting up a potential feed-forward mechanism for further production of ET-1 and ROS. In the present study, ET receptor antagonism prevented the stress-mediated increases in plasma 8-isoprostane and arterial pressure without changes in baseline values. These data indicate a causal relationship between ROS and the pressor response to acute stress, and that the increase in ROS occurs downstream of ET receptor activation. Specifically, dual ETA/B receptor antagonism prevented the stress-mediated rise in ROS, whereas selective ETA receptor blockade had no effect. These data indicate that the increase in ROS most likely occurs in response to ETB receptor stimulation. Our experimental approach used the comparison of a dual ETA/B antagonist and a selective ETA antagonist to discern the effects of ET receptors in response to stress. We used this approach because treatment with an ETB selective antagonist will produce large increases in arterial pressure, vascular resistance, and activate ETA receptor activity, which will make interpretation of results especially difficult. We recognize that the inability to directly probe the ETB receptor is a limitation of our study. A role for both ETA and ETB receptors in stimulating ROS in the vasculature and sympathetic nervous system is supported by previously published studies.34–35, 53, 54 Alternatively, the two receptor subtypes may functionally interact to produce an increase in ROS.8
Tempol is a redox-cycling nitroxide that promotes the metabolism of many reactive oxygen species and is utilized as a free radical scavenger.70 Since tempol treatment blunted the air jet stress pressor response, we concluded that acute stress induces ROS in DS rats. Plasma isoprostane measurements are routinely used as a biomarker of in vivo ROS production or oxidative stress and lipid peroxidation.55 In the vast majority of studies, changes in plasma isoprostane are examined under the context of a chronic disease state or following a more prolonged pathological insult. In this regard, our results are indeed novel in that we reproducibly detect changes after the start of air jet stress in DS rats. We found that DR rats did not display a similar stress-induced increase in plasma isoprostane, thus we reasoned that the increased isoprostane levels in DS rats is relevant. Isoprostanes are also widely recognized to mediate increases in DNA synthesis, cellular proliferation, and collagen synthesis.56 Thus, isoprostanes are biomarkers of ROS production and have direct biological effects in the vasculature. Possibly, these isoprostane-specific biological effects may play a role in the vascular pathologies observed in chronic repetitive stress paradigms.
It stands to reason that reduced sympathetic nerve activity resulting from a decrease in neuronal ROS may contribute to the effect seen with tempol. Bolus intravenous administration of tempol has been shown to lower renal sympathetic nerve activity in both normotensive and hypertensive rats.5, 6, 57 Dai et al.58 demonstrated that ET-1 activation of ROS in celiac ganglia isolated from DOCA-salt hypertensive rats was sensitive to ETB but not ETA receptor blockade. ETB receptor stimulation caused a similar increase in ganglionic O2·− levels in normotensive rats.54 From these studies, one would predict that reducing ROS with tempol or with ETB receptor blockade would lower the stress-mediated rise in plasma catecholamines. While tempol did reduce the stress-mediated increase in epinephrine, we found that there was a paradoxical increase in plasma norepinephrine. Moreover, dual ETA/B receptor blockade had no effect on the stress-mediated rise of either catecholamine. Li et al.59 reported that chronic ETB receptor activation by S6c, selective ETB agonist, induced hypertension that was ameloriated by tempol and decreased superoxide levels in ganglia. Furthermore, these investigators found that although plasma norepinephrine levels were not increased in S6c hypertension, surgical ablation of the celiac ganglion plexus, which provides most of the sympathetic innervation to the splanchnic organs, significantly attenuated the development of S6c-induced hypertension. These results are relevant to our study by demonstrating that ETB receptor activation does involve sympathetic pathways, without changes in plasma catecholamines. Plasma levels of catecholamines are an indirect surrogate measurement of sympathetic activation and should be interpreted accordingly. Further investigation with direct sympathetic nerve recording is necessary to fully elucidate the role of ROS and ET receptor activation in the stress-induced pressor response.
Considerable evidence links ROS to cardiovascular disease in various animal models, particularly hypertension, yet ROS also contribute to normal cardiovascular function.60 Bolus intravenous administration of tempol has been shown to acutely lower blood pressure in normotensive rats.6, 61–63 Using an oral dosing paradigm, however, we did not observe any effect on baseline blood pressure over 3 days in pre-hypertensive DS rats; a similar lack of effect on pressure was obtained in studies by Schnackenberg et al61, 64 with chronic dosing of Wistar Kyoto rats. A possible explanation of the acute versus long-term effects of tempol in normotensive rats may be explained by the dose and dosing route. Consistent with no change in baseline pressure, we found no effect of tempol on basal plasma 8-isoprostane levels. Finally, tempol can cross the blood-brain barrier,65, 66 and so we cannot rule out the possibility that reductions in ROS within the central nervous system contribute to the tempol-mediated attenuation of the pressor response to acute air jet stress.
ROS also partially mediate the response to contractile agonists.7, 9, 10 We reasoned that in the absence of an effect of tempol on the stress-mediated increase in plasma ET-1, tempol may blunt the constrictor response to ET-1. We found, however, that tempol had no effect on the anesthesized whole animal pressor responses to exogenous ET-1 or S6c. Given the role of the sympathetic nervous system in the response to an acute stress, we also examined the effect of tempol on the pressor response to α adrenergic stimulation in anesthesized DS rats. Tempol similarly had no effect on the PE-mediated pressor response. This can be explained by previous findings showing that α adrenergic stimulation does not elicit an increase in ROS.7 It is plausible that a bolus injection of PE in anesthesized preparations is not an accurate model for acute stress-mediated adrenergic activation in conscious rats. Therefore, future studies are necessary to fully examine the interaction of the adrenergic, ET, and ROS pathways in acute stress induced pressor responses in normotensive DS rats.
The results in the present study in conjunction with various reports in the literature, have led us to propose a causal chain of events that, in part, mediate the acute stress-induced pressor response in DS rats. However, the cellular mechanisms by which this occurs remain to be elucidated. Mayorov et al67 have shown that bilateral injection of tempol into the rostral ventrolateral medulla significantly attenuates the pressor response to air jet stress, suggesting that ROS mediate at least in part the cardiovascular response to acute stress. Also, acute increases in blood pressure or increased vascular pressure lead to increased ROS production.68, 69 Since both ET receptor blockade and tempol reduced the stress-induced pressor response, we concluded that the rise in blood pressure is most likely not the stimulus for the increased ET-1 or ROS but that ROS activate the pressor response. A link between the increase in ROS and increased blood pressure was not revealed in our study. Tempol is a redox-cycling nitroxide that promotes the metabolism of ROS and improves nitric oxide bioavailability in vivo,70 thus we speculate that increased ROS may lead to a loss of NO bioavailability mediating the increase in blood pressure. Vascular NOS activity in DS rats compared to DR rats is very low (Pollock, et al; unpublished observations), so it is possible that the NO buffering capacity is greatly reduced in this animal model. Future experiments are necessary to elucidate the mechanism(s) of ROS-mediated increase in blood pressure in pre-hypertensive DS rats. Figure 6 shows our hypothetical scheme that air jet stress stimulates the sympathetic nervous system followed by increased ET-1 and ET receptor activation leading to the production of ROS and finally increased blood pressure. These data indicate a causal relationship between ROS and the pressor response to acute stress, and that the increase in ROS occurs downstream of ET receptor activation.
Figure 6.
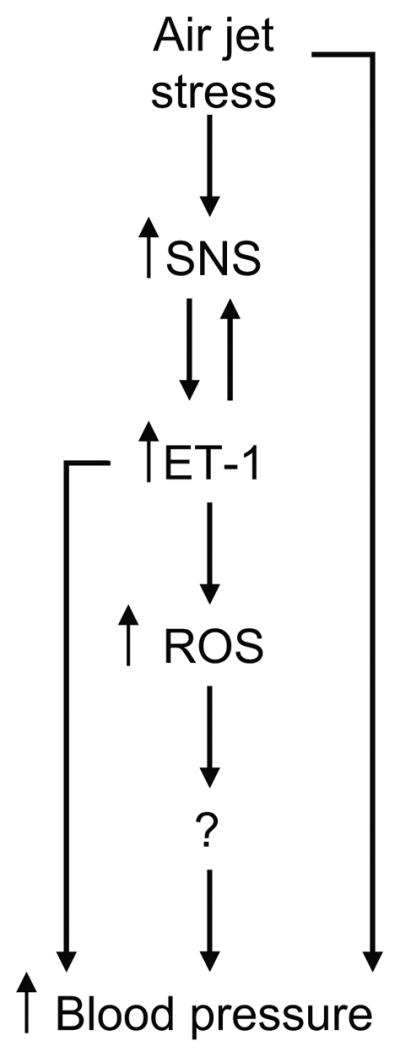
Scheme depicting the causal relationship of the ET pathway and ROS production in the acute stress-mediated rise in blood pressure in pre-hypertensive DS rats.
Perspectives
There is a growing body of evidence suggesting that exaggerated cardiovascular responses to acute stress can identify individuals at increased risk of cardiovascular disease later in life.21–28 The concept of allostatic load suggests, however, that disease results from the cumulative effects of multiple exaggerated responses to stress over time.29 Because the vascular dysfunction that occurs with aging is associated with increased ROS,71 it is therefore plausible that repeated responses to stress that invoke a rise in ROS may further contribute to the pathogenesis of cardiovascular disease.
Our group previously demonstrated that acute stress-induced elevations in plasma ET-1 correlated with stress-induced increases in blood pressure in pre-hypertensive adolescents and young adults with verified family histories of cardiovascular disease.38,40 Our current study in an animal model, the pre-hypertensive DS rat, determined a mechanism of the stress-induced pressor response is via ET-1 activation of the ROS pathway. The pre-hypertensive DS rat is a model of pre-hypertensive young adults with family histories of cardiovascular disease; thus we predict that behavioral stress in the young adults activates an ET-dependent ROS pathway. Future translational studies will explore these hypotheses.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the excellent technical assistance of Hiram Ocasio, Amy Dukes, Carolyn Rhoden, Christopher Middleton, and Paul Wach. We would also like to thank Dr. Jennifer Sullivan for expert editorial suggestions.
FUNDING
This study was supported by grants from the National Heart, Lung, and Blood Institute (D.M. Pollock: HL-64776; J.S. Pollock: HL-69999) and from the American Heart Association (G. D’Angelo: Scientist Development Grant 0530361N; D.M. Pollock: Established Investigator 0340443N; J.S. Pollock: Established Investigator 0440073N).
Footnotes
DISCLOSURES
None
References
- 1.Schnackenberg CG. Physiological and pathophysiological roles of oxygen radicals in the renal microvasculature. Am J Physiol Regul Integr Comp Physiol. 2002;282:R335–342. doi: 10.1152/ajpregu.00605.2001. [DOI] [PubMed] [Google Scholar]
- 2.Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension. 2004;44:248–252. doi: 10.1161/01.HYP.0000138070.47616.9d. [DOI] [PubMed] [Google Scholar]
- 3.Taniyama Y, Griendling KK. Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension. 2003;42:1075–1081. doi: 10.1161/01.HYP.0000100443.09293.4F. [DOI] [PubMed] [Google Scholar]
- 4.Xu H, Fink GD, Chen A, Watts S, Galligan JJ. Nitric oxide-independent effects of tempol on sympathetic nerve activity and blood pressure in normotensive rats. Am J Physiol Heart Circ Physiol. 2001;281:H975–980. doi: 10.1152/ajpheart.2001.281.2.H975. [DOI] [PubMed] [Google Scholar]
- 5.Shokoji T, Fujisawa Y, Kimura S, Rahman M, Kiyamoto H, Matsubara K, Moriwaki K, Aki Y, Miyatake A, Kohno M, Abe Y, Nishiyama A. Effects of local administration of tempol and diethyldithio-carbamic on peripheral nerve activity. Hypertension. 2004;44:236–243. doi: 10.1161/01.HYP.0000136393.26777.63. [DOI] [PubMed] [Google Scholar]
- 6.Shokoji T, Nishiyama A, Fujisawa Y, Hitomi H, Kiyamoto H, Takahashi N, Kimura S, Kohno M, Abe Y. Renal sympathetic nerve responses to tempol in spontaneously hypertensive rats. Hypertension. 2003;41:266–273. doi: 10.1161/01.hyp.0000049621.85474.cf. [DOI] [PubMed] [Google Scholar]
- 7.Chen Y, Pearlman A, Luo Z, Wilcox CS. Hydrogen peroxide mediates a transient vasorelaxation with tempol during oxidative stress. Am J Physiol Heart Circ Physiol. 2007;293:H2085–2092. doi: 10.1152/ajpheart.00968.2006. [DOI] [PubMed] [Google Scholar]
- 8.Loomis ED, Sullivan JC, Osmond DA, Pollock DM, Pollock JS. Endothelin mediates superoxide production and vasoconstriction through activation of NADPH oxidase and uncoupled nitric-oxide synthase in the rat aorta. J Pharmacol Exp Ther. 2005;315:1058–1064. doi: 10.1124/jpet.105.091728. [DOI] [PubMed] [Google Scholar]
- 9.Just A, Whitten CL, Arendshorst WJ. Reactive oxygen species participate in acute renal vasoconstrictor responses induced by ETA and ETB receptors. Am J Physiol Renal Physiol. 2008;294:F719–728. doi: 10.1152/ajprenal.00506.2007. [DOI] [PubMed] [Google Scholar]
- 10.Just A, Olson AJ, Whitten CL, Arendshorst WJ. Superoxide mediates acute renal vasoconstriction produced by angiotensin II and catecholamines by a mechanism independent of nitric oxide. Am J Physiol Heart Circ Physiol. 2007;292:H83–92. doi: 10.1152/ajpheart.00715.2006. [DOI] [PubMed] [Google Scholar]
- 11.Lee KW, Kim JB, Seo JS, Kim TK, Im JY, Baek IS, Kim KS, Lee JK, Han PL. Behavioral stress accelerates plaque pathogenesis in the brain of Tg2576 mice via generation of metabolic oxidative stress. J Neurochem. 2009;108:165–175. doi: 10.1111/j.1471-4159.2008.05769.x. [DOI] [PubMed] [Google Scholar]
- 12.Song L, Zheng J, Li H, Jia N, Suo Z, Cai Q, Bai Z, Cheng D, Zhu Z. Prenatal stress causes oxidative damage to mitochondrial DNA in hippocampus of offspring rats. Neurochem Res. 2009;34:739–745. doi: 10.1007/s11064-008-9838-y. [DOI] [PubMed] [Google Scholar]
- 13.Brodsky MA, Sato DA, Iseri LT, Wolff LJ, Allen BJ. Ventricular tachyarrhythmia associated with psychological stress: the role of the sympathetic nervous system. JAMA. 1987;257:2064–2067. [PubMed] [Google Scholar]
- 14.Jern C, Eriksson E, Tengborn L, Risberg B, Wadenvik H, Jern S. Changes of plasma coagulation and fibrinolysis in response to mental stress. Thromb Haemost. 1989;62:767–771. [PubMed] [Google Scholar]
- 15.Jiang W, Babyak M, Krantz DS, Waugh RA, Coleman RE, Hanson MM, Frid DJ, McNulty S, Morris JJ, O’Connor CM, Blumenthal JA. Mental stress-induced myocardial ischemia and cardiac events. JAMA. 1996;275:1651–1656. doi: 10.1001/jama.275.21.1651. [DOI] [PubMed] [Google Scholar]
- 16.Levine SP, Towell BL, Suarez AM, Kniereim LK, Harris MM, George JN. Platelet activation and secretion associated with emotional stress. Circulation. 1985;71:1129–1134. doi: 10.1161/01.cir.71.6.1129. [DOI] [PubMed] [Google Scholar]
- 17.Pettersson K, Bejne B, Bjork H, Strawn WB, Bondjers G. Experimental sympathetic activation causes endothelial injury in the rabbit thoracic aorta via beta 1-adrenoceptor activation. Circ Res. 1990;67:1027–1034. doi: 10.1161/01.res.67.4.1027. [DOI] [PubMed] [Google Scholar]
- 18.Reich P, DaSilva RA, Lown B, Murawski BJ. Acute psychological disturbances preceding life-threatening ventricular arrhythmias. JAMA. 1981;246:233–235. [PubMed] [Google Scholar]
- 19.Rozanski A, Bairey CN, Krantz DS, Friedman J, Resser KJ, Morell M, Hilton-Chalfen S, Hestrin L, Bietendorf J, Berman DS. Mental stress and the induction of silent myocardial ischemia in patients with coronary artery disease. N Engl J Med. 1988;318:1005–1012. doi: 10.1056/NEJM198804213181601. [DOI] [PubMed] [Google Scholar]
- 20.Skantze HB, Kaplan J, Pettersson K, Manuck SB, Blomqvist N, Kyes R, Williams K, Bondjers G. Psychosocial stress causes endothelial injury in cynomolgus monkeys via B1-adrenoceptor activation. Atherosclerosis. 1998;136:153–161. doi: 10.1016/s0021-9150(97)00202-5. [DOI] [PubMed] [Google Scholar]
- 21.Chida Y, Steptoe A. Greater cardiovascular responses to laboratory mental stress are associated with poor subsequent cardiovascular risk status: a meta-analysis of prospective evidence. Hypertension. 2010;55:1026–1032. doi: 10.1161/HYPERTENSIONAHA.109.146621. [DOI] [PubMed] [Google Scholar]
- 22.Carroll D, Smith GD, Shipley MJ, Steptoe A, Brunner EJ, Marmot MG. Blood pressure reactions to acute psychological stress and future blood pressure status: a 10-year follow-up of men in the Whitehall II study. Psychosom Med. 2001;63:737–743. doi: 10.1097/00006842-200109000-00006. [DOI] [PubMed] [Google Scholar]
- 23.Matthews KA, Woodall KL, Allen MT. Cardiovascular reactivity to stress predicts future blood pressure status. Hypertension. 1993;22:479–485. doi: 10.1161/01.hyp.22.4.479. [DOI] [PubMed] [Google Scholar]
- 24.Matthews KA, Katholi CR, McCreath H, Whooley MA, Williams DR, Zhu S, Markovitz JH. Blood pressure reactivity to psychological stress predicts hypertension in the CARDIA study. Circulation. 2004;110:74–78. doi: 10.1161/01.CIR.0000133415.37578.E4. [DOI] [PubMed] [Google Scholar]
- 25.Flaa A, Eide IK, Kjeldsen SE, Rostrup M. Sympathoadrenal stress reactivity is a predictor of future blood pressure: an 18-year follow-up study. Hypertension. 2008;52:336–341. doi: 10.1161/HYPERTENSIONAHA.108.111625. [DOI] [PubMed] [Google Scholar]
- 26.Kasagi F, Akahoshi M, Shimaoka K. Relation between cold pressor test and development of hypertension based on 28-year follow-up. Hypertension. 1995;25:71–76. doi: 10.1161/01.hyp.25.1.71. [DOI] [PubMed] [Google Scholar]
- 27.Light KC, Girdler SS, Sherwood A, Bragdon EE, Brownley KA, West SG, Hinderliter AL. High stress responsivity predicts later blood pressure only in combination with positive family history and high life stress. Hypertension. 1999;33:1458–1464. doi: 10.1161/01.hyp.33.6.1458. [DOI] [PubMed] [Google Scholar]
- 28.Menkes MS, Matthews KA, Krantz DS, Lundber U, Mead LA, Qaqish B, Liang K-Y, Thomas CB, Pearson TA. Cardiovascular reactivity to the cold pressor test as a predictor of hypertension. Hypertension. 1989;14:524–530. doi: 10.1161/01.hyp.14.5.524. [DOI] [PubMed] [Google Scholar]
- 29.McEwen BS, Seeman TE. Protective and damaging effects of mediators of stress: elaborating and testing the concepts of allostasis and allostatic load. Ann NY Acad Sci. 1999;896:30–47. doi: 10.1111/j.1749-6632.1999.tb08103.x. [DOI] [PubMed] [Google Scholar]
- 30.Koepke JP, Jones S, DiBona GF. Stress increases renal nerve activity and decreases sodium excretion in Dahl rats. Hypertension. 1988;11:334–338. doi: 10.1161/01.hyp.11.4.334. [DOI] [PubMed] [Google Scholar]
- 31.Koepke JP, DiBona GF. High sodium intake enhances renal and antinatriuretic responses to stress in spontaneously hypertensive rats. Hypertension. 1985;7:357–363. [PubMed] [Google Scholar]
- 32.DiBona GF, Jones SY. Analysis of renal sympathetic nerve responses to stress. Hypertension. 1995;25:531–538. doi: 10.1161/01.hyp.25.4.531. [DOI] [PubMed] [Google Scholar]
- 33.Hoagland KM, Maier KG, Roman RJ. Contributions of 20-HETE to the antihypertensive effects of tempol in Dahl salt-sensitive rats. Hypertension. 2003;41:697–702. doi: 10.1161/01.HYP.0000047881.15426.DC. [DOI] [PubMed] [Google Scholar]
- 34.Meng S, Cason GW, Gannon AW, Racusen LC, Manning RD., Jr Oxidative stress in Dahl salt-sensitive hypertension. Hypertension. 2003;41:1346–1352. doi: 10.1161/01.HYP.0000070028.99408.E8. [DOI] [PubMed] [Google Scholar]
- 35.Milner P, Loesch A, Burnstock G. Endothelin immunoreactivity and mRNA expression in sensory and sympathetic neurones following selective denervation. Int J Dev Neurosci. 2000;18:727–734. doi: 10.1016/s0736-5748(00)00054-x. [DOI] [PubMed] [Google Scholar]
- 36.Chen M, Todd-Turla K, Wang WH, Cao X, Smart A, Brosius FC, Killen PD, Keiser JA, Briggs JP, Schnermann J. Endothelin-1 mRNA in glomerular and epithelial cells of kidney. Am J Physiol Renal Physiol. 1993;265:F542–550. doi: 10.1152/ajprenal.1993.265.4.F542. [DOI] [PubMed] [Google Scholar]
- 37.Kohan DE, Padilla E. Endothelin-1 production by rat inner medullary collecting duct: effect of nitric oxide, cGMP, and immune cytokines. Am J Physiol Renal Physiol. 1994;266:F291–F297. doi: 10.1152/ajprenal.1994.266.2.F291. [DOI] [PubMed] [Google Scholar]
- 38.Treiber FA, Musante L, Braden D, Arensman F, Strong WB, Levy M, Leverett S. Racial differences in hemodynamic responses to the cold face stimulus in children and adults. Psychosom Med. 1990;52:286–296. doi: 10.1097/00006842-199005000-00003. [DOI] [PubMed] [Google Scholar]
- 39.Mangiafico RA, Malatino LS, Attinà T, Messina R, Fiore CE. Exaggerated endothelin release in response to acute mental stress in patients with intermittent claudication. Angiology. 2002;53:383–390. doi: 10.1177/000331970205300403. [DOI] [PubMed] [Google Scholar]
- 40.Treiber FA, Jackson RW, Davis H, Pollock JS, Kapuku G, Mensah GA, Pollock DM. Racial differences in endothelin-1 at rest and in response to acute stress in adolescent males. Hypertension. 2000;35:722–725. doi: 10.1161/01.hyp.35.3.722. [DOI] [PubMed] [Google Scholar]
- 41.Ikeda T, Ohta H, Okada M, Kawai N, Nakao R, Siegl PKS, Kobayashi T, Maeda S, Miyauchi T, Nishikibe M. Pathophysiological roles of endothelin-1 in Dahl salt-sensitive hypertension. Hypertension. 1999;34:514–519. doi: 10.1161/01.hyp.34.3.514. [DOI] [PubMed] [Google Scholar]
- 42.Schiffrin EL. Role of endothelin-1 in hypertension. Hypertension. 1999;34:876–881. doi: 10.1161/01.hyp.34.4.876. [DOI] [PubMed] [Google Scholar]
- 43.Campese VM, Shaohua Y, Huiquin Z. Oxidative stress mediates angiotensin II-dependent stimulation of sympathetic nerve activity. Hypertension. 2005;46:533–539. doi: 10.1161/01.HYP.0000179088.57586.26. [DOI] [PubMed] [Google Scholar]
- 44.Ortiz MC, Manriquez MC, Romero JC, Juncos LA. Antioxidants block angiotensin II-induced increases in blood pressure and endothelin. Hypertension. 2001;38:655–659. doi: 10.1161/01.hyp.38.3.655. [DOI] [PubMed] [Google Scholar]
- 45.D’Angelo G, Pollock JS, Pollock DM. In vivo evidence for endothelin-1-mediated attenuation of α1-adrenergic stimulation. Am J Physiol Heart Circ Physiol. 2006;290:H1251–H1258. doi: 10.1152/ajpheart.00203.2005. [DOI] [PubMed] [Google Scholar]
- 46.Callera GE, Touyz RM, Teixeira SA, Muscara MN, Carvalho MHC, Fortes ZB, Nigro D, Schiffrin EL, Tostes RC. ETA receptor blockade decreases vascular superoxide generation in DOCA-salt hypertension. Hypertension. 2003;42:811–817. doi: 10.1161/01.HYP.0000088363.65943.6C. [DOI] [PubMed] [Google Scholar]
- 47.Callera GE, Tostes RC, Yogi A, Montezano ACI, Touyz RM. Endothelin-1-induced oxidative stress in DOCA-salt hypertension involves NADPH-oxidase-independent mechanisms. Clin Sci. 2006;110:246–253. doi: 10.1042/CS20050307. [DOI] [PubMed] [Google Scholar]
- 48.Yura T, Fukunaga M, Khan R, Nassar GN, Badr KF, Montero A. Free-radical-generated F2-isoprostane stimulates cell proliferation and endothelin-1 expression on endothelial cells. Kidney Int. 1999;56:471–478. doi: 10.1046/j.1523-1755.1999.00596.x. [DOI] [PubMed] [Google Scholar]
- 49.Kahler J, Ewart A, Weckmuller J, Stobbe S, Mittmann C, Koster R, Paul M, Meinertz T, Munzel T. Oxidative stress increases endothelin-1 synthesis in human coronary artery smooth muscle cells. J Cardiovasc Pharmacol. 2001;38:49–57. doi: 10.1097/00005344-200107000-00006. [DOI] [PubMed] [Google Scholar]
- 50.Kaehler J, Sill B, Koester R, Mittmann C, Orzechowski HD, Muenzel T, Meinertz T. Endothelin-1 mRNA and protein in vascular wall cells is increased by reactive oxygen species. Clin Sci. 2002;103:176S–178S. doi: 10.1042/CS103S176S. [DOI] [PubMed] [Google Scholar]
- 51.Fujii T, Takaoka M, Ohkita M, Matsumura Y. Tempol protects against ischemic acute renal failure by inhibiting renal noradrenaline overflow and endothelin-1 overproduction. Biol Pharm Bull. 2005;28:641–645. doi: 10.1248/bpb.28.641. [DOI] [PubMed] [Google Scholar]
- 52.Bell D, Zhao Y, McCoy FP, Devine AB, McDermott BJ. Differential effects of an anti-oxidant intervention on cardiomyocyte expression of adrenomedullin and intermedin and their receptor components in chronic nitric oxide deficiency. Cell Physiol Biochem. 2007;20:269–282. doi: 10.1159/000107513. [DOI] [PubMed] [Google Scholar]
- 53.Li M, Dai X, Watts S, Kreulen D, Fink G. Increased superoxide levels in ganglia and sympathoexcitation are involved in sarafotoxin 6c-induced hypertension. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1546–1554. doi: 10.1152/ajpregu.00783.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lau YE, Galligan JJ, Kreulen DL, Fink GD. Activation of ETB receptors increases superoxide levels in sympathetic ganglia in vivo. Am J Physiol Regul Integr Comp Physiol. 2006;290:R90–R95. doi: 10.1152/ajpregu.00505.2005. [DOI] [PubMed] [Google Scholar]
- 55.Montuschi P, Barnes PJ, Roberts LJ., II Isoprostanes: markers and mediators of oxidative stress. FASEB J. 2004;18:1791–1800. doi: 10.1096/fj.04-2330rev. [DOI] [PubMed] [Google Scholar]
- 56.Comporti M, Signorini C, Arezzini B, Vecchio D, Monaco B, Gardi C. F2-isoprostanes are not just markers of oxidative stress. Free Radic Biol Med. 2008;44:247–256. doi: 10.1016/j.freeradbiomed.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 57.Xu H, Fink GD, Galligan JJ. Tempol lowers blood pressure and sympathetic nerve activity but not vascular O2·− in DOCA-salt rats. Hypertension. 2004;43:329–334. doi: 10.1161/01.HYP.0000112304.26158.5c. [DOI] [PubMed] [Google Scholar]
- 58.Dai X, Galligan JJ, Watts SW, Fink GD, Kreulen DL. Increased O2·− production and upregulation of ETB receptors by sympathetic neurons in DOCA-salt hypertensive rats. Hypertension. 2004;43:1048–1054. doi: 10.1161/01.HYP.0000126068.27125.42. [DOI] [PubMed] [Google Scholar]
- 59.Li L, Fink GD, Watts SW, Northcott CA, Galligan JJ, Pagano PJ, Chen AF. Endothelin-1 Increases Vascular Superoxide via EndothelinA-NADPH Oxidase Pathway in Low-Renin Hypertension. Circulation. 2003;107:1053–1058. doi: 10.1161/01.cir.0000051459.74466.46. [DOI] [PubMed] [Google Scholar]
- 60.Wilcox CS, Pearlman A. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol Rev. 2008;60:418–469. doi: 10.1124/pr.108.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schnackenberg CG, Welch WJ, Wilcox CS. Normalization of blood pressure and renal vascular resistance in SHR with a membrane-permeable superoxide dismutase mimetic: role of nitric oxide. Hypertension. 1998;32:59–64. doi: 10.1161/01.hyp.32.1.59. [DOI] [PubMed] [Google Scholar]
- 62.Xu H, Fink GD, Chen A, Watts S, Galligan JJ. Nitric oxide-independent effects of tempol on sympathetic nerve activity and blood pressure in normotensive rats. Am J Physiol Heart Circ Physiol. 2001;281:H975–980. doi: 10.1152/ajpheart.2001.281.2.H975. [DOI] [PubMed] [Google Scholar]
- 63.Xu H, Fink GD, Chen A, Watts S, Galligan JJ. Nitric oxide-dependent effects of tempol on sympathetic nerve activity and blood pressure in normotensive rats. Am J Physiol Heart Circ Physiol. 2002;281:H975–H980. doi: 10.1152/ajpheart.2001.281.2.H975. [DOI] [PubMed] [Google Scholar]
- 64.Schnackenberg CG, Wilcox CS. Two-week administration of tempol attenuates both hypertension and renal excretion of 8-Iso prostaglandin F2α. Hypertension. 1999;33:424–428. doi: 10.1161/01.hyp.33.1.424. [DOI] [PubMed] [Google Scholar]
- 65.Matsumoto S, Mori N, Tsuchihashi N, Ogata T, Lin Y, Yokoyama H, Ishida S-I. Enhancement of nitroxide-reducing activity in rats after chronic administration of vitamin E, vitamin C, and idebenone examined by an in Vivo electron spin resonance technique. Magn Reson Med. 1998;40:330–333. doi: 10.1002/mrm.1910400219. [DOI] [PubMed] [Google Scholar]
- 66.Behringer W, Safar P, Kentner R, Wu X, Kagan VE, Radovsky A, Clark RSB, Kochanek PM, Subramanian M, Tyurin VA, Tyurina YY, Tisherman SA. Antioxidant tempol enhances hypothermic cerebral preservation during prolonged cardiac arrest in dogs. J Cereb Blood Flow Metab. 2002;22:105–117. doi: 10.1097/00004647-200201000-00013. [DOI] [PubMed] [Google Scholar]
- 67.Mayorov DN, Head GA, De Matteo R. Tempol attenuates excitatory actions of angiotensin II in the rostral ventrolateral medulla during emotional stress. Hypertension. 2004;44:101–106. doi: 10.1161/01.HYP.0000131290.12255.04. [DOI] [PubMed] [Google Scholar]
- 68.Jacobson GM, Dourron HM, Liu J, Carretero OA, Reddy DJ, Andrzejewski T, Pagano PJ. Novel NAD(P)H oxidase inhibitor suppresses angioplasty-induced superoxide and neointimal hyperplasia of rat carotid artery. Circ Res. 2003;92:637–643. doi: 10.1161/01.RES.0000063423.94645.8A. [DOI] [PubMed] [Google Scholar]
- 69.Ungvari Z, Csiszar A, Huang A, Kaminski PM, Wolin MS, Koller A. High pressure induces superoxide production in isolated arteries via protein kinase C-dependent activation of NAD(P)H oxidase. Circulation. 2003;108:1253–1258. doi: 10.1161/01.CIR.0000079165.84309.4D. [DOI] [PubMed] [Google Scholar]
- 70.Wilcox CS. Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol Ther. 2010;26:119–145. doi: 10.1016/j.pharmthera.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hamilton CA, Brosnan MJ, McIntyre M, Graham D, Dominiczak AF. Superoxide excess in hypertension and aging: a common cause of endothelial dysfunction. Hypertension. 2001;37:529–534. doi: 10.1161/01.hyp.37.2.529. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.