

Abstract
A concise synthesis of (±)-pseudotabersonine from commercially available 1-(phenylsulfonyl)-3-indolecarboxaldehyde has been accomplished. This synthesis features the convergent assembly of a key intermediate via a stepwise variant of a Mannich-type multicomponent coupling process, a double ring-closing metathesis, and a one-pot deprotection/cyclization reaction.
Members of the Aspidosperma family of indole alkaloids have long captured the attention of synthetic chemists owing to their complex structural frameworks and their diverse and important biological activites.1 Most of these alkaloids comprise a pentacyclic core with an ethyl group or a functionlized ethyl group appended at the bridgehead carbon atom at C(20), as exemplified by the structure of aspidospermidine (1) (Figure 1). However, there is a small sub-family of Aspidosperma alkaloids related to pandoline (2) having a rearranged skeleton (Figure 1). Pseudotabersonine (3), a member of the pandoline alkaloids, was isolated from Pandaca caducifolia in 1975,2 and two elegant syntheses of this compound were reported in the early 1990s by Kuehne3 and Grieco.4
Figure 1.

Examples of Aspidosperma Alkaloids.
Our laboratory has had a longstanding interest in developing general strategies for the synthesis of natural products. In that context, we pioneered the application of ring-closing metathesis (RCM) to the syntheses of nitrogen heterocycles and a variety of alkaloids as well as other important natural products.5,6,7 Herein, we report a concise total synthesis of (±)-pseudotabersonine that features a stepwise variant of a Mannich-type multicomponent reaction previously developed in our laboratory,8 a double RCM reaction, and a one-pot deprotection/cyclization sequence.
Our retrosynthetic analysis of pseudotabersonine (3) is shown in Scheme 1. It was envisioned that 3 could be assembled from the tetracyclic alcohol 4 via deprotection and cyclization using a protocol developed by Bosch and coworkers.9 Access to 4 would then be achieved by a double RCM of the tetraene 5, followed by a selective reduction of the less substituted double bond. The synthesis of 5 would involve a sequential union of aldehyde 6, allylamine 7, a pentadienyl organometallic reagent, and ethylene oxide.
Scheme 1.

Retrosynthetic Analysis of Pseudotabersonine (3).
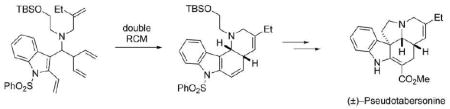
The synthesis of pseudotabersonine commenced with the condensation of commercially available 1-(phenylsulfonyl)-3-indolecarboxaldehyde (6) with 2-ethylallylamine hydrochloride (7)10 to provide the crude imine 8 in virtually quantitative yield (Scheme 2). The next step posed a considerable challenge as it required the selective addition of a pentadienyl organometallic reagent to the imine function to give a branched adduct. Different pentadienyl organometallic reagents (Li, Zn, In and Al) were examined in this reaction. After considerable experimentation, we discovered that the pentadienyl aluminum reagent 9, which was generated in situ by transmetallation of pentadienyllithium, provided the best selectivity for the branched product.7l,11 However, it was essential to allow the reaction to warm to room temperature, because increased amounts of the linear adduct were obtained if the reaction was quenched at −78 °C. In the event, addition of 9 to a solution of crude imine 8 in CH2Cl2 followed by subsequent reaction of the adduct 10 with ethylene oxide in a sealed tube at 60 °C in MeOH afforded a mixure of branched and linear products (branched:linear > 10:1) in 89% combined yield over two steps. After a single recrystallization, the requisite branched product 11 was obtained in 71% overall yield from 6.
Scheme 2.
Synthesis of Triene 11.
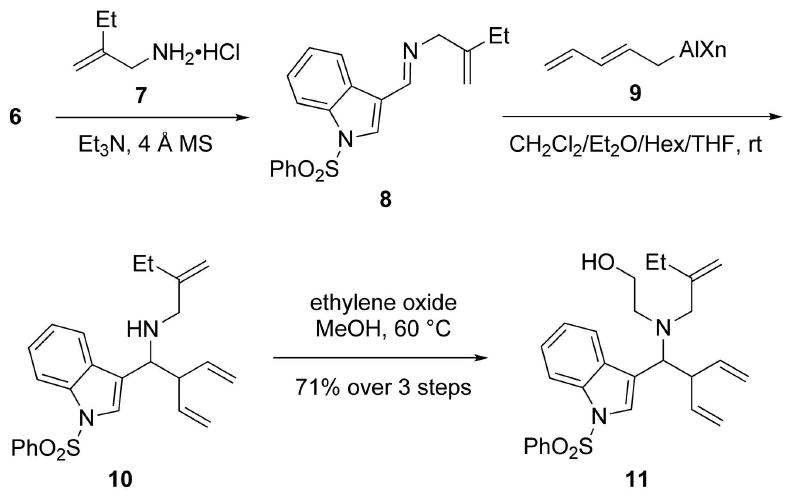
The next stage of the synthesis required the introduction of a vinyl group at C(2) of the indole ring in 11. Toward this end, 11 was first converted to TBS ether 12 in 98% yield (Scheme 3). Deprotonation of 12 at C(2) with LDA followed by the addition of acetaldehyde generated the epimeric alcohols 13 in 78% yield (d.r. ≈ 2.5:1). Dehydration of 13 with Tf2O and Hünig's base furnished the desired tetraene 5 in 92% yield.
Scheme 3.
Synthesis of Tetraene 5.
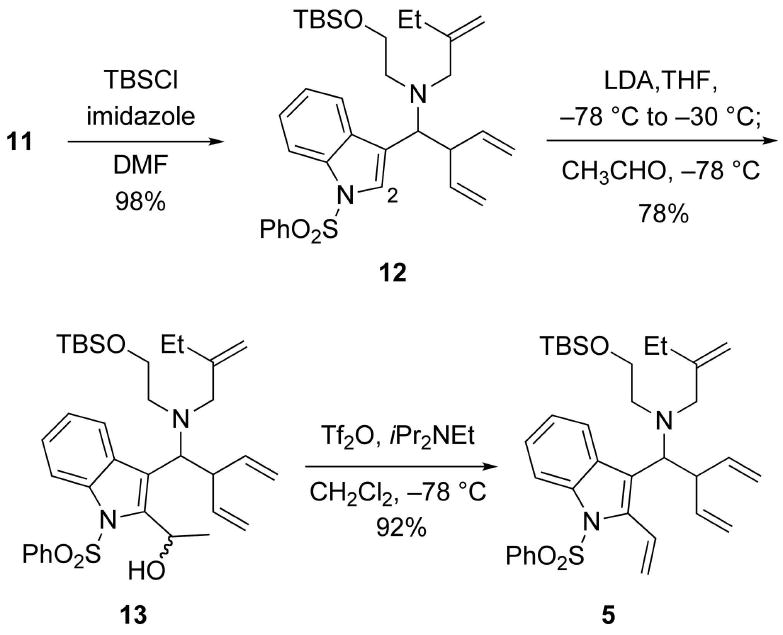
Having the tetraene 5 in hand, we were grateful to find that the key double RCM proceeded smoothly in the presence of 5% of Hoveyda-Grubbs II (H-G II) catalyst at 100 °C (oil bath temp) to afford an inseparable mixture (≈ 7:10) of the D/E cis- and trans-fused tetracycles 14 and 15, respectively (Scheme 4).12 The crude mixture of 14 and 15 was then processed by regioselective reduction of the disubstituted olefinic moiety by catalytic hydrogenation, followed by deprotection of the TBS ether to afford a separable mixture of 4 and 16 in 26% and 44% yields, respectively, from 5 over three steps.
Scheme 4.
Double RCM of Tetraene 5.
It is noteworthy that 14 was unstable to longer reaction times and higher temperatures, apparently fragmenting to give 17 (Scheme 5). For example, when the double RCM of 5 was conducted in toluene under reflux, a mixture (≈ 3:1) of 15 and 17, which presumably arose from 14 via sequential 1,4-elimination and aromatization,13 was obtained in about 65% yield. This deleterious side reaction demanded careful control of the conditions used for the RCM in order to provide optimal quantities of 14. Although conducting the reaction at lower temperatures gave an improved ratio (∼1:1) of 14 and 15, the overall conversions were lower. Accordingly, we decided to continue the synthesis of 3 from 14 and try to epimerize the trans-fused D/E ring system in 15 to the requisite cis-fused ring system at a later stage (vide infra).
Scheme 5.

Double RCM of Tetraene 5 at Higher Temperature.
Conversion of 4 to the pentacyclic intermediate 18 was achieved by an intriguing one-pot process, which involved a sequential N-deprotection/O-sulfonylation and cyclization process, that had been reported by Bosch and coworkers (Scheme 6).9 In the event, addition of a solution of KOtBu in THF to a solution of 4 in DME afforded 18 in 66% yield. Finally, following a procedure developed by Rawal and coworkers,14 18 was deprotonated with LDA, and the intermediate metallo enamine was selectively acylated on carbon using Mander's reagent to furnish (±)-pseudotabersonine (3) in 61% yield with only trace amounts of the corresponding N-acylated product being detected. The synthetic pseudotabersonine thus obtained gave 1H and 13C NMR spectra that are consistent with the assigned structure of 3 and with those reported and provided by Kuehne.3,15
Scheme 6.
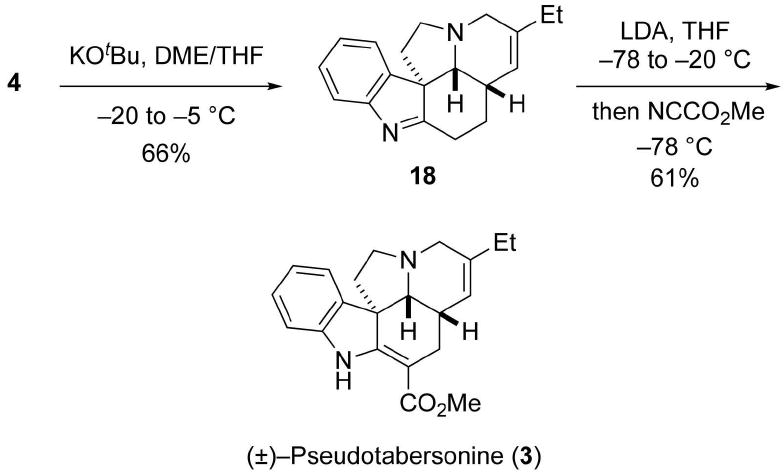
Completion of Total Synthesis of (±)-Pseudotabersonine (3)
The tetracycle 16 was then transformed to (±)-14-epi pseudotabersoine (20) by a series of reactions analogous to those used to convert 4 into 3 (Scheme 7). Namely, treatment of 16 with KOtBu afforded the D/E trans pentacycle 19 in 75% yield. Deprotonation of 19 followed by trapping with Mander's reagent afforded 20 in 46% yield together with the N-acylation product 21 in 26% yield. The significant difference in the regioselectivity in the acylations of 18 and 19 using Mander's reagent is both noteworthy and unexpected.
Scheme 7.
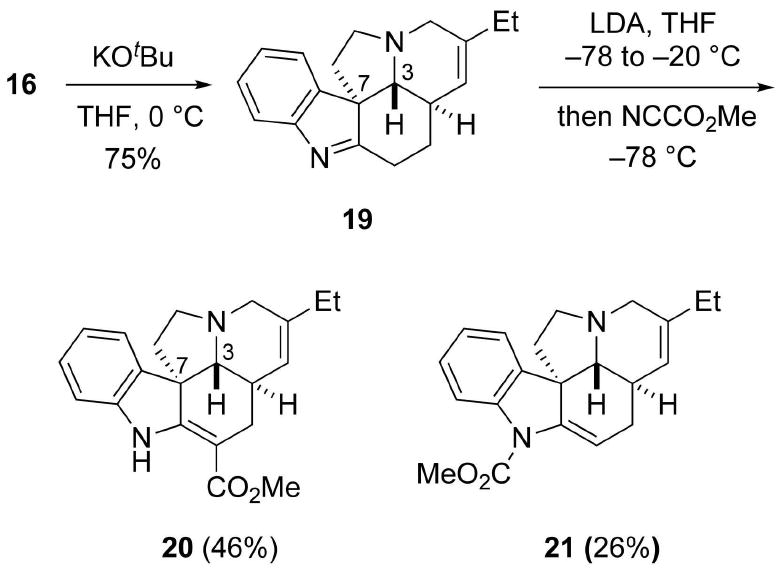
Synthesis of (±)-14-epi-Pseudotabersonine (20).
We briefly examined the possibility of epimerizing at C(3) and C(7) of both 19 and 20 via a reversible retro-Mannich/Mannich process, which is precedented for related compounds having a cis-fused D/E ring system.16 Stork has also observed the trans- to cis-isomerization of the D/E ring subunit of an intermediate during the synthesis of aspidospermine.17 However, all efforts to convert either 19 or 20 into either 18 or 3 under a variety of acidic conditions (TsOH, AcOH, TFA, TMSOTf, BF3·Et2O, HCl and Cu(OTf)2) led only to recovery of starting material or decomposition. Our failure to effect this equilibration was disappointing, but not wholly unexpected as Kuehne was also unable to epimerize a similar D/E trans-fused pentacycle.18 There is thus a significant, and heretofore underappreciated, difference in the propensity of cis- and trans-fused D/E ring derivatives having the Aspidosperma skeleton to undergo reversible retro-Mannich/Mannich reactions.
In conclusion, we have developed a concise entry to the pentacyclic core of Aspidosperma alkaloids via a sequence that featured a stepwise variant of a Mannich-type coupling process to generate a highly functionalized intermediate that was elaborated by a double ring-closing metathesis and a one-pot deprotection/cyclization reaction. This strategy was applied to a total synthesis of (±)-pseudotabersonine that required a total of 14 steps in which the longest linear sequence starting from commercially available 6 was 11 steps. The application of this strategy to the syntheses of other Aspidosperma alkaloids having an ethyl group at the D/E bridgehead are currently under investigation in our laboratory.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM 25439), the Robert A. Welch Foundation (F-652) for their generous support of this research. We are also grateful to Dr. Richard Pederson (Materia, Inc.) for catalysts support. Finally, we would like to thank Prof. Martin E. Kuehne (The University of Vermont) for providing copies of 1H and 13C NMR spectra of pseudotabersonine.
Footnotes
Supporting Information Available Experimental procedures, spectral data and copies of 1H and 13C NMR spectra for all new compounds and 7. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Cordell GA. In: The Alkaloids. Manske RHF, Rodrigo RGA, editors. Vol. 17. Academic Press; New York: 1979. pp. 199–384. [Google Scholar]; (b) Saxton JE. In: The Alkaloids. Cordell GA, editor. Vol. 50. Academic Press; New York: 1998. pp. 343–376. [Google Scholar]; (c) Saxton JE. In: The Alkaloids. Cordell GA, editor. Vol. 51. Academic Press; New York: 1998. pp. 2–197. [Google Scholar]
- 2.Zeches M, Debray MM, Ledouble G, Le Men-Olivier L, Le Men J. Phytochemistry. 1975;14:1122–1124. [Google Scholar]
- 3.Bornmann WG, Kuehne ME. J Org Chem. 1992;57:1752–1760. [Google Scholar]
- 4.Carroll WA, Grieco PA. J Am Chem Soc. 1993;115:1164–1165. [Google Scholar]
- 5.For a general review of olefin metathesis, see: Grubbs RH, editor. Handbook of Metathesis. 1-3. Wiley-VCH; 2003.
- 6.For reviews of applications of RCM to the synthesis of complex organic molecules and natural products, see: Deiters A, Martin SF. Chem Rev. 2004;104:2199–2238. doi: 10.1021/cr0200872.Nicolaou KC, Bulger PG, Sarlah D. Angew Chem Int Ed. 2005;44:4490–4527. doi: 10.1002/anie.200500369.
- 7.For selected examples, see: Martin SF, Liao Y, Wong Y, Rein T. Tetrahedron Lett. 1994;35:691–694.Martin SF, Chen HJ, Courtney AK, Liao Y, Pätzel M, Ramser MN, Wagman AS. Tetrahedron. 1996;52:7251–7164.Fellows IM, Kaelin DE, Jr, Martin SF. J Am Chem Soc. 2000;122:10781–10787.Kirkland TA, Colucci J, Geraci LS, Marx MA, Schneider M, Kaelin DE, Jr, Martin SF. J Am Chem Soc. 2001;123:12432–12433. doi: 10.1021/ja011867f.Humphrey JM, Liao Y, Ali A, Rein T, Wong YL, Chen HJ, Courtney AK, Martin SF. J Am Chem Soc. 2002;124:8584–8592. doi: 10.1021/ja0202964.Washburn DG, Heidebrecht RW, Jr, Martin SF. Org Lett. 2003;5:3523–3525. doi: 10.1021/ol0354066.Neipp C, Martin SF. J Org Chem. 2003;68:8867–8878. doi: 10.1021/jo0349936.Brenneman JB, Machauer R, Martin SF. Tetrahedron. 2004;60:7301–7314.Andrade RB, Martin SF. Org Lett. 2005;7:5733–5735. doi: 10.1021/ol0525009.Kummer DA, Brenneman JB, Martin SF. Tetrahedron. 2006;62:11437–11449.Simila STM, Martin SF. J Org Chem. 2007;72:5342–5349. doi: 10.1021/jo070732a.Deck JA, Martin SF. Org Lett. 2010;12:2610–2613. doi: 10.1021/ol100819f.
- 8.(a) Sunderhaus JD, Dockendorff C, Martin SF. Org Lett. 2007;9:4223–4226. doi: 10.1021/ol7018357. [DOI] [PubMed] [Google Scholar]; (b) Sunderhaus JD, Dockendorff C, Martin SF. Tetrahedron. 2009;65:6454–6469. doi: 10.1016/j.tet.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rubiralta M, Diez A, Bosch J, Solans X. J Org Chem. 1989;54:5591–5597. [Google Scholar]
- 10.See supporting information for the synthesis of 7.
- 11.For some related references of 1,2-additions of pentadienyl organometallic agents, see: Gerard F, Miginiac P. Bull Soc Chim Fr. 1974;11(Pt. 2):2527–2533.Courtois G, Mesnard D, Mahoungou JR, Miginiac L. Bull Soc Chim Fr. 1986:449–453.Alvaro G, Grepioni F, Grilli S, Maini L, Martelli G, Savoia D. Synthesis. 2000:581–587.Nishigaichi Y, Ishihara M, Fushitani S, Uenaga K, Takuwa A. Chem Lett. 2004;33:108–109.
- 12.The ratio of 14 and 15 was determined by integrating the signal for H(3) in the 1H NMR of each in the crude double RCM reaction mixture: for 14, H(3) (δ 4.03, d, J = 7.2 Hz); for 15, H(3) (δ 4.10, d, J = 16.4 Hz).
- 13.For similar aromatizations, see: Magnus P, Gallagher T, Brown P, Huffman JC. J Am Chem Soc. 1984;106:2105–2114.Biolatto B, Kneeteman M, Mancini P. Tetrahedron Lett. 1999;40:3343–3346.Gagnon D, Spino C. J Org Chem. 2009;74:6035–6041. doi: 10.1021/jo9009497.
- 14.Kozmin S, Iwama T, Huang Y, Rawal VH. J Am Chem Soc. 2002;124:4628–4641. doi: 10.1021/ja017863s. [DOI] [PubMed] [Google Scholar]
- 15.Kuehne reported a multiplet for three protons in the range δ 2.01-2.20 and a peak for one proton at δ 1.61 in the 1H NMR spectrum of 3. Examination of our spectra, including 2D spectra, shows that there are four protons in the range of δ 2.03-2.20, and although there is sometimes a proton at δ 1.60, that has been shown to be water. Given the difference in field strength of our spectra and those of Kuehne, the spectra appear consistent, although there are no integrations on the spectrum of the authentic sample. There are no significant differences in the 13C NMR spectra.
- 16.For examples, see: Smith GF, Wróbel JT. J Chem Soc. 1960:792–795.Kutney JP, Piers E, Brown RT. J Am Chem Soc. 1970;92:1700–1704. doi: 10.1021/ja00709a048.Lewin G, Poisson J. Tetrahedron Lett. 1984;23:3813–3814.Pilarčík T, Havlíček J, Hájíček J. Tetrahedron Lett. 2005;46:7909–7911.Tóth F, Oláh J, Kalaus G, Greiner I, Szőllősy Á, Gömöry Á, Hazai L, Szántay C. Tetrahedron. 2008;64:7949–7955.
- 17.Stork G. Pure Appl Chem. 1964;9:131–144. [Google Scholar]
- 18.Kuehne ME, Zebovitz TC. J Org Chem. 1987;52:4331–4339. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.