

Abstract
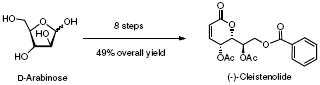
A facile stereoselective total synthesis of cleistenolide (1) from natural chiral template D-arabinose has been achieved in eight steps and 49% overall yield, employing key steps including, Wittig olefination, selective 1,3-trans-acetal formation, and modified Yamaguchi esterification.
The family Annonaceae includes over 2000 species,1 quite a number of which have suffered species extinction before being well investigated. A considerable number of new compounds, having interesting chemical structures and important biological activities, have been isolated from this family.2 In 2007, Nkunya et al.3 discovered two novel constituents, cleistenolide (1) and cleistodienol (2) (Figure 1), from the Annonaceae, Cleistochlamys kirkii Oliver, a plant species found in Tanzania and Mozambique. Extracts made from this plant are used in traditional medicine as a remedy for treatment of wound infections, rheumatism, and tuberculosis.4 Cleistenolide also reportedly exhibits in vitro antibacterial activity against Staphylococcus aureus and Bacillus anthracis, and antifungal activity against Candida albicans.3 Recently, the first total synthesis of cleistenolide 1 was published by Schmidt and co-workers5 in 18% overall yield, by applying a ring-closing metathesis (RCM) protocol to prepare the key building block, an α,β-unsaturated lactone.6 Attracted by the potential pharmacological activity of cleistenolide, a knowledge of its absolute stereochemical configuration, and a shortage of the natural product (only 200 mg of cleistenolide can be extracted from 1 kg of dry plant), we launched a project aimed at the facile synthesis of cleistenolide 1. Herein, we report the stereoselective total synthesis of cleistenolide by taking advantages of the chiral centers present in D-arabinose.
FIGURE 1.

Chemical structures of cleistenolide (1) and cleistodienol (2)
As depicted in the retrosynthetic analysis (Scheme 1), the crucial dihydropyran-2-one 3 was envisioned to be formed from the α,β-unsaturated acid 4 through an intramolecular Yamaguchi esterification.7 Compound 4 would then be prepared from polyhydroxyl intermediate 5 through regio-selective 1,3-trans-acetal formation8 and subsequent ester-acid transformation. Precursor 5 would be constructed from natural chiral template D-arabinose (6) through regioselective silylation and Wittig olefination.9
SCHEME 1. Retrosynthetic Analysis of Cleistenolide (1).

Our synthesis (Schemes 2 and 3) started from D-arabinose. Treatment of D-arabinose 6 with TBDMSCl in pyridine at 0 °C regioselectively afforded the 5-O-silyl aldehyde 7 in a yield of 92%. Wittig olefination of aldehyde 7 with ethyl (triphenylphosphoranylidene)acetate10 in dioxane at 70 °C furnished the α,β-unsaturated ester 5 in 89% isolated yield. The trans configuration (δ 6.16 ppm J = 15.7 Hz, =CHCO2Et) was obtained with no cis product being observed in our reactions. Based on the literature precedent,11 we initially attempted to synthesize 1,3-trans-p-methoxybenzylidene of compound 5 to differentiate the three chiral hydroxyl groups. However, reaction of 5 with 4-MeOPhCH(OMe)2 in the presence of PPTS in CH2Cl2 failed to generate the desired compound 8 in good yield. Fortunately, treatment of ester 5 with 2 equivalents of Me2C(OMe)2 in the presence of a catalytic amount of PPTS at room temperature successfully afforded 1,3-trans-acetal, compound 9, in 87% yield. The correct regioselectivity and relative stereochemistry of compound 9 were confirmed from its 13C NMR spectrum, which showed chemical shifts at 24.74, 25.44, and 102.07 ppm, revealing it to be an anti-1,3-diol acetonide.12
SCHEME 2. Synthesis of 1,3-Diol Protected Fragment 9.

SCHEME 3. Synthesis of (−)-Cleistenolide (1).

Removal of ester protection from compound 9 with LiOH in THF/H2O afforded the corresponding acid 4 in quantitative yield. Intramolecular esterification of acid 4 under modified Yamaguchi conditions7 afforded key precursor 3 in 90% yield. The formation of 3 could be explained by thermal δ-lactonization through activation of carboxylic acid with 2,4,6-trichlorobenzoyl and subsequent pyridine-assisted addition-elimination. The cis-olefin configuration of 3 was confirmed by the correlated doublet (δ 6.19 ppm, J = 9.8 Hz, =CHCO2Et) and the quartet (δ 6.79 ppm, J = 5.6, 9.8 Hz, RCHCH=) in its 1HNMR spectrum. We were gratified that by using TBAF and Bz2O in THF, the one-pot5 desilylation and benzoylation of 3 proceeded smoothly affording compound 10 in high yield (84%). Removal of isopropylidene group from 10, with bis(acetonitrile)dichloropalladium(II)13,14 at 65 °C, furnished diol 11. Acetylation, with Ac2O in pyridine, completed the synthesis of (−)-cleistenolide (1) in 91% yield over the final two steps. The physical and spectral data of our synthetic sample 1 were in excellent agreement with the literature reports, except for the specific optical rotation. We observed a value of [α]25 D-147 (c 0.4, CHCl3), Schmidt and co-workers5 reported [α]24D -165 (c 0.48, CH2Cl2), while Nkunya et al.,3 reported [α]D -63.5 (c 0.7, CHCl3) for the natural product. Our result, as well as Schmidt's report,5 supports the absolute configuration assigned to natural product as (−)-cleistenolide.
In summary, we have accomplished the concise and stereoselective total synthesis of (−)-cleistenolide (1) in eight steps and 49% overall yield from D-arabinose. Our synthesis is very efficient with high stereoselectivities, indicating the powerful application of carbohydrates as chiral synthons. Key features of our strategy towards practical total synthesis of (−)-cleistenolide are the efficient combination of a Wittig olefination, selective 1,3-trans-acetal formation, and the use of modified Yamaguchi esterification.
Experimental Section
(4R,5S,6R,E)-Ethyl 7-(tert-butyldimethylsilyloxy)-4,5,6-trihydroxyhept-2-enoate (5)
To a solution of D-arabinose (1.50 g, 10.00 mmol) in pyridine (30 mL), tert-butyldimethylchlorosilane (1.65 g, 11.00 mmol) was added in the presence of catalytic amount of DMAP at 0 °C under N2 atmosphere. The mixture was allowed to warm to room temperature and stirred under these conditions for 12 h. The mixture was concentrated and the residue was purified by silica gel column chromatography with ethyl acetate as eluent to afford 7 (2.43 g, 92%) as a colorless syrup. To a solution of 7 (1.00 g, 3.78 mmol) in anhydrous dioxane (15 mL), (ethoxycarbonylmethylene)triphenylphosphorane (1.71 g, 4.92 mmol) was added under a N2 atmosphere at room temperature. The mixture was stirred under these conditions for 2 h and then warmed to 70 °C and stirred for 5 h. The resulting clear solution was concentrated in vacuo, and the residue was poured into saturated aqueous NaCl (20 mL). The aqueous phase was extracted with CH2Cl2 (3 × 20 mL), and the combined organic phase was evaporated under reduced pressure to give a residue, which was subjected to the silica gel column chromatography (1:1 petroleum ether-ethyl acetate) to give 5 (1.12 g, 89%) as a colorless syrup: [α]25D +31 (c 1.2, CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.09 (s, 6H), 0.89 (s, 9H), 1.28 (t, 3H, J = 7.1 Hz), 3.00 (br s, 2H), 3.35 (br s, 1H), 3.62-3.64 (m, 1H), 3.71 (dd, 1H, J = 5.4, 11.9 Hz), 3.76-3.81 (m, 2H), 4.18 (dd, 2H, J = 7.2, 14.3 Hz), 4.60 (s, 1H), 6.16 (d, 1H, J = 15.7 Hz), 7.00 (dd, 1H, J = 4.2, 15.7 Hz); 13C NMR (125 MHz, CDCl3): δ -5.5, 14.2, 18.2, 25.8, 60.5, 64.2, 70.5, 71.4, 73.7, 122.0, 147.4, 166.4. ESI(+)-MS calcd for C15H30O6Si: 334.2 [M]; found 357.1 [M+Na]+. Anal. Calcd for C15H30O6Si: C, 53.86; H, 9.04. Found: C, 53.72; H, 9.15.
(E)-Ethyl 3-{(4R,5S,6R)-6-[(tert-butyldimethylsilyloxy) methyl]-5-hydroxy-2,2-dimethyl-1,3-dioxan-4-yl}acrylate (9)
To a mixture of 5 (245 mg, 0.73 mmol) and 2,2′-dimethoxypropane (0.18 mL, 1.46 mmol) in anhydrous CH2Cl2 (10 mL) at 0 °C PPTS (18.4 mg, 0.073 mmol) was added. The mixture was stirred at room temperature for 10 h under N2 atmosphere. Triethylamine (0.3 mL) was then added and the reaction mixture was concentrated to dryness. Purification of the remaining syrup by flash chromatography (4:1 petroleum ether-ethyl acetate) gave the acetonide 9 (238 mg, 87%) as a colorless syrup: [α]25D +97 (c 0.9, CHCl3); 1H NMR (600 MHz, CDCl3): δ 0.08 (s, 3H), 0.09 (s, 3H), 0.90 (s, 9 H), 1.29 (t, 3H, J = 7.2 Hz), 1.34 (s, 3H), 1.44 (s, 3H), 2.04 (d, 1H, J = 5.4 Hz), 3.64-3.65 (m, 1H), 3.72 (dd, 1H, J = 6.0, 10.1 Hz), 3.84 (dd, 1H, J = 5.4, 10.2 Hz), 3.94-3.95 (m, 1H), 4.18-4.21 (m, 2H), 4.55 (s, 1H), 6.18 (d, 1H, J = 15.7 Hz), 7.00 (dd, 1H, J = 4.0, 15.7 Hz); 13C NMR (150 MHz, CDCl3): δ -4.62, -4.60, 15.0, 19.0, 24.7, 25.4, 26.6, 61.2, 65.3, 71.4, 72.9, 75.2, 102.1, 123.1, 138.9, 143.8, 166.9. ESI(+)-MS calcd for C18H34O6Si: 374.2 [M]; found 397.1 [M+Na]+. Anal. Calcd for C18H34O6Si: C, 57.72; H, 9.15. Found: C, 57.57; H, 9.28.
(4R,4aS,8aR)-4-[(tert-Butyldimethylsilyloxy)methyl]-2,2-dimethyl-4,4a-dihydropyrano[3,2-d][1,3]dioxin-6(8aH)-one (3)
To a solution of 9 (150 mg, 0.40 mmol) in THF (3 mL), 2 M aqueous LiOH (3 mL) was added dropwise and the reaction was stirred for 5 h at room temperature. Amberlite IR-120 (H+) was then added to neutralize the solution and the mixture was poured into water (10 mL) and extracted with CH2Cl2 (3 × 10 mL). Evaporation of the combined organic phase followed by flash chromatography with (1:2 petroleum ether-ethyl acetate) gave acid 4 as colorless syrup: 1H NMR (500 MHz, CDCl3): δ 0.08 (s, 6H), 0.90 (s, 9H), 1.39 (s, 3H), 1.40 (s, 3H), 3.60 (t, 1H, J = 6.9 Hz), 3.73 (dd, 1H, J = 6.0, 11.2 Hz), 3.77-3.81 (m, 2H), 4.48 (s, 1H), 6.05, (d, 1H, J = 15.5 Hz), 6.85 (dd, 1H, J = 4.4, 15.5 Hz). To a solution of 4 (67.5 mg, 0.19 mmol) in pyridine (2 mL) a solution of 2,4,6-trichlorobenzoyl chloride (0.052 g, 0.21 mmol) in CH2Cl2 (0.2 mL) at 0 °C was added and the reaction mixture was stirred at room temperature for 2 h. The reaction mixture was concentrated in vacuo to give a residue, which was subjected to the silica gel column chromatography (6:1 petroleum ether-ethyl acetate) to afford 3 (57.6 mg, 90% for two steps) as a colorless syrup: [α]25D -55 (c 0.5, CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.08 (s, 6H), 0.90 (s, 9H), 1.41 (s, 3H), 1.43 (s, 3H), 3.83-3.89 (m, 3H), 4.28 (t, 1H, J = 5.2 Hz), 4.65 (t, 1H, J = 5.0 Hz), 6.19 (d, 1H, J = 9.8 Hz), 6.79 (dd, 1H, J = 5.6, 9.8 Hz); 13C NMR (125 MHz, CDCl3): δ -5.3, -5.2, 18.3, 23.6, 24.7, 30.9, 59.2, 62.7, 72.2, 75.3, 101.9, 124.5, 139.8, 161.9. ESI(+)-MS calcd for C16H28O5Si: 328.2 [M]; found 351.2 [M+Na]+. Anal. Calcd for C16H28O5Si: C, 58.50; H, 8.59. Found: C, 58.32; H, 8.71.
{(4R,4aS,8aR)-2,2-Dimethyl-6-oxo-4,4a,6,8a-tetrahydro pyrano[3,2-d][1,3]dioxin-4-yl}methyl benzoate (10)
To a solution of 3 (152 mg, 0.46 mmol) in anhydrous THF (50 mL) TBAF (158 mg, 0.50 mmol) was added. The mixture was stirred at room temperature for 30 min, then benzoic anhydride (416 mg, 1.80 mmol) was added and the reaction was stirred for an additional 8 h. The reaction was quenched by addition of water, and the aqueous layer was extracted with EtOAc (3 × 30 mL). The combined organic phase was subsequently washed with aqueous NH4Cl (50 mL) and saturated aqueous NaCl (50 mL), then dried over anhydrous Na2SO4, and concentrated in vacuo. Purification by column chromatography (5:1 petroleum ether-ethyl acetate) gave 10 (123 mg, 84%) as a colorless syrup: [α]25D -15 (c 0.7, CHCl3); 1H NMR (500 MHz, CDCl3): δ 1.44 (s, 3H), 1.49 (s, 3H), 4.23 (ddd, 1H, J = 3.0, 5.5, 7.5 Hz), 4.40 (t, 1H, J = 4.5 Hz), 4.55 (dd, 1H, J = 6.0, 12.0 Hz), 4.59 (dd, 1H, J = 3.0, 12.0 Hz), 4.66 (dd, 1H, J = 4.5, 7.5 Hz), 6.22 (dd, 1H, J = 1.0, 10.0 Hz), 6.80 (dd, 1H, J = 5.5, 9.5 Hz), 7.45 (t, 2H, J = 8.0 Hz), 7.57 (t, 1H, J = 7.5 Hz), 8.04 (d, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3): δ 23.6, 24.5, 30.9, 59.1, 63.8, 69.8, 76.0, 102.4, 124.6, 128.4, 129.7, 130.2, 133.2, 133.6, 139.5, 161.2, 166.1; ESI(+)-MS calcd for C17H18O6: 318.1 [M]; found 341.1 [M+Na]+. Anal. Calcd for C17H18O6: C, 64.14; H, 5.70. Found: C, 63.87; H, 5.88.
Cleistenolide (1)
A solution of 10 (55 mg, 0.17 mmol) in acetonitrile/water (30 mL, v/v 1:1) was heated at 65 °C in the presence of PdCl2(CH3CN)2 (50 mg, 0.19 mmol) for 24 h. The mixture was cooled to room temperature and filtered through Celite. The filtrate was concentrated in vacuo to afford 11 as a syrup: 1H NMR (500 MHz, CDCl3): δ 2.92 (br s, 1H), 3.56 (br s, 1H), 4.36-4.40 (m, 2H), 4.54 (d, 1H, J = 4.4 Hz), 4.59 (dd, 1H, J = 4.8, 12.3 Hz), 4.83 (dd, 1H, J = 1.2, 11.9 Hz), 6.18 (d, 1H, J = 9.7 Hz), 7.03 (dd, 1H, J = 5.9, 9.7 Hz), 7.46 (t, 2H, J = 7.6 Hz), 7.59 (t, 1H, J = 7.6 Hz), 8.05 (d, 2H. J = 7.9 Hz). ESI(+)-MS calcd for C14H14O6: 278.1 [M]; found 301.2 [M+Na]+. Compound 11 was directly dissolved into a solution of acetic anhydride (1 mL) and pyridine (2 mL). After TLC indicated that all of the starting material was consumed, the solution was concentrated in vacuo, and the residue was subjected to the silica gel column chromatography (3:1 petroleum ether-ethyl acetate) to give 1 (56 mg, 91% for two steps) as a white crystal: mp 130-133 °C; [α]25D -147 (c 0.4, CHCl3); 1H NMR (600 MHz, CDCl3): δ 2.04 (s, 3H), 2.09 (s, 3H), 4.53 (dd, 1H, J = 12.5, 4.4 Hz), 4.80 (dd, 1H, J = 9.6, 2.5 Hz), 4.93 (dd, 1H, J = 12.5, 2.0 Hz), 5.42 (dd, 1H, J = 6.0, 2.5 Hz), 5.52 (ddd, 1H, J = 9.5, 4.0, 2.3 Hz), 6.29 (d, 1H, J = 9.7 Hz), 7.00 (dd, 1H, J = 9.6, 6.1 Hz), 7.45 (t, 2H, J = 7.6 Hz), 7.57 (t, 1H, J = 7.4 Hz), 8.02 (d, 2H. J = 7.7 Hz); 13C NMR (150 MHz, CDCl3): δ 20.5, 20.7, 59.7, 62.0, 67.7, 75.5, 125.4, 128.5, 129.6, 129.7, 129.7, 133.3, 139.7, 161.1, 166.0, 169.5, 169.9. HR-ESI(+)-MS calcd for C18H18O8: 362.1002 [M]; found: 385.0896 [M+Na]+, 401.0636 [M+K]+.
Supplementary Material
Acknowledgments
This work was supported by NNSF of China (Projects 20621703, 20872172, and 20732001), Project 2009ZX09501-011, and the US NIH AI065786.
Footnotes
Supporting Information Available: Spectral data for compounds 1, 3, 4, 5, 7, 9, 10, and 11 are available free of charge via Internet at http://pubs.acs.org.
Contributor Information
Yuguo Du, Email: duyuguo@rcees.ac.cn.
Robert J. Linhardt, Email: linhar@rpi.edu.
References
- 1.Alali FQ, Liu XX, McLaughlin JL. J Nat Prod. 1999;62:504–540. doi: 10.1021/np980406d. [DOI] [PubMed] [Google Scholar]
- 2.For natural product isolated from Annonaceae species: Nkunya M, Weenen H, Koyi N, Thijs L, Zwanenburg B. Phytochemistry. 1987;26:2563–2565.Weenen H, Nkunya M, Mgani Q, Achenbach H, Posthumus M, Waibel R. J Org Chem. 1991;56:5865–5867.Kamperdick C, Van NH, Sung TV. Phytochemistry. 2002;61:991–994. doi: 10.1016/s0031-9422(02)00374-6.Li C, Lee D, Graf TN, Phifer SS, Nakanishi Y, Riswan S, Wani MC, Oberlies NH. J Nat Prod. 2009;72:1949–1953. doi: 10.1021/np900572g.
- 3.Samwel S, Mdachi SJM, Nkunya MHH, Irungu BN, Moshi MJ, Moulton B, Luisi BS. Nat Prod Commun. 2007;2:737–741. [Google Scholar]
- 4.Nkunya MHH. Pure Appl Chem. 2005;77:1943–1955. [Google Scholar]
- 5.Schmidt B, Kunz O, Biernat A. J Org Chem. 2010;75:2389–2394. doi: 10.1021/jo1002642. [DOI] [PubMed] [Google Scholar]
- 6.For earlier synthesis of pyran-2-one derivatives through RCM reaction: De Fatima A, Pilli RA. Tetrahedron Lett. 2003;44:8721–8724.Kumar P, Naidu SV. J Org Chem. 2006;71:3935–3941. doi: 10.1021/jo0603781.Curran DP, Moura-Letts G, Pohlman M. Angew Chem Int Ed. 2006;45:2423–2426. doi: 10.1002/anie.200600041.G Subhash, R Ch Nageswara. Tetrahedron Lett. 2010;51:2052–2054.
- 7.Ono M, Zhao XY, Shida Y, Akita H. Tetrahedron. 2007;63:10140–10148. [Google Scholar]
- 8.Marco JA, Carda M, Murga J, Falomir E. Tetrahedron. 2007;63:2929–2958. [Google Scholar]
- 9.Wadsworth WS, Jr, Emmons WD. J Am Chem Soc. 1961;83:1733–1738. [Google Scholar]
- 10.Liu SL, Shi XX, Xu YL, Xu W, Dong J. Tetrahedron: Asymmetry. 2009;20:78–83. [Google Scholar]
- 11.Takebuchi K, Hamada Y, Shioiri T. Tetrahedron Lett. 1994;35:5239–5242. [Google Scholar]
- 12.Rychnovsky SD, Rogers B, Yang G. J Org Chem. 1993;58:3511–3515. [Google Scholar]
- 13.Schmeck C, Hegedus LS. J Am Chem Soc. 1994;116:9927–9934. [Google Scholar]
- 14.Lipshutz BH, Pollart D, Monforte J, Kotsuki H. Tetrahedron Lett. 1985;26:705–708. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.