

Abstract
The eubacterial SOS system is a paradigm of cellular DNA damage and repair, and its activation can contribute to antibiotic resistance.1,2,3 Under normal conditions, LexA represses the transcription of many DNA repair proteins by binding to SOS “boxes” in their operators. Under genotoxic stress, accumulating RecA-ssDNA-ATP complexes activate LexA for autocleavage. To address how LexA recognizes its binding sites, we determined three crystal structures of Escherichia coli LexA in complex with SOS boxes. These are the first structures reported of a LexA-DNA complex. The DNA-binding domains of the LexA dimer interact with the DNA in the classical fashion of a winged helix-turn-helix (wHTH) motif. However, the wings of these two DNA binding domains bind to the same minor groove of the DNA. These wing-wing contacts may explain why the spacing between the two half-sites of E. coli SOS boxes is invariant.
The LexA protein contains two domains: an N-terminal wHTH DNA binding domain, and a C-terminal dimerization and latent protease domain (Figure 1, Supplementary Figure 1). The structure of apo-LexA4 shows the two domains connected by a short flexible linker, with the scissile peptide bond lying in a loop just within the C-terminal domain. The C-terminal domains's autoproteolytic activity is activated by interaction with RecA-ssDNA-ATP complexes, and leads to separation of the domains and exposure of a degradation tag on the DNA binding domains5. The protease active site lies in a hydrophobic indentation and includes a nucleophile, Ser 119, and a general base/acid, Lys 156. In the absence of RecA, autocleavage is only appreciable at very high pH, where the lysine would be neutral. Binding to the RecA filament most likely stabilizes a conformation in which the cleavage-site containing loop docks across the active site, shielding Lys156 from solvent, thus by lowering its pKa to make it a more effective catalyst4.
Figure 1.

Overall structure of the E. coli LexA-DNA complex. (a) LexA-DNA complex (in crystal form A22); Individual subunits are colored blue and pink, and the bases comprising the consensus sequence are shown in cyan. The ∼5 residue linker between domains was too poorly ordered to be modeled. The color of each protomer changes from dark to light at the scissile peptide bond (A84-G85). Green balls represent the catalytic residues of the latent protease: S119 (nucleophile) and K156 (general base, here mutated to alanine). (b) Comparison of the DNA-binding domains of bound (magenta), unbound crystal (aqua) (1JHH4), and NMR7 (green) (1LEA) structures. The RMSD values for superimposing the unbound crystal and NMR structures onto A22 are 0.37Å and 0.67Å respectively (CCP4: LSQKAB28). (c) Variable position of the cleavage loop. The catalytic domains are colored as in (a), with the cleavage loops in red. In the A22 dimer both scissile peptide bonds (arrows) are docked in the active sites, in B22, they are displaced from the active sites, and in the apo form (1JHH) one is docked and one is displaced. This suggests that DNA binding does not preferentially induce the loop into one state or the other.
DNA binding by LexA has been studied extensively6,7, but a structural framework for this data has been elusive. The consensus sequence8 for SOS boxes is CTGTN8ACAG, and the length of the 8bp spacer is invariant. Here, we report three crystal structures of a catalytically inactive mutant LexA (K156A) in complex with SOS box-containing DNAs: “A22” and “B22” which differed only in the spacer sequence and formed similar crystals, and “C29” with longer flanking DNA, which led to different crystal packing interactions. The diffraction was anisotropic, with the best data set extending to 3.0, 2.6 and 2.3Å along the 3 principle axes (“A22”, Supplementary Tables 1, 2). These structures offer insights into how the spacer sequence can modulate binding affinity and how LexA measures the length of the SOS box's spacer.
The LexA-DNA complex structures reveal unexpected interactions between the two DNA binding domains. The individual domains bind DNA in a canonical fashion, with the recognition helix (α3) nestled into the major groove and the wing contacting the phosphodiester backbone across the adjacent minor groove. Unlike many dimeric wHTH proteins such as CAP and MarR family members9,10, the wings point inward and meet in the central minor groove. The wings also contact the α1-α2 loop of the opposite DNA binding domain, forming a small hydrophobic core (Supplementary Table 3). This protein-protein interface buries a relatively small 372Å2 of otherwise solvent-accessible surface area, in agreement with previous results that the DNA binding domain is monomeric in solution (unless otherwise specified, all detailed conclusions are based on the highest-resolution structure, A22)11,12. However, that formation of this interface is synergistic with DNA binding is supported by the results of a genetic screen for LexA variants with enhanced affinity13. All of the selected mutations that map to the DNA binding domain lie at this interface.
Disparate models of the LexA-DNA complex have been proposed. Although most models predicted that the wings of the DNA binding domains would interact with the outer minor grooves14,15, the binding domains were properly oriented in the model of Knegtel et al., which relied the most heavily on experimental data7. The latter also correctly predicted that the DNA would bend towards rather than away from the catalytic domain.
Comparisons of our complex structures and the unbound forms4 yield interesting similarities and differences (Supplementary Figure 2). The dimer formed by the two catalytic domains is essentially the same in all crystal forms, with the exception of the “cleavage loop” containing the scissile peptide bond. The structures of the individual DNA binding domains are also nearly unchanged (Figure 1b). However, the relative position of the N- and C-terminal domains is highly variable. The interdomain linker is short: only ∼5 residues (70-74) are disordered in our structures. However, it is so flexible that it allows an ∼180° re-orientation of the DNA binding domain in the unbound form (Supplementary Figure 2a). We crystallized LexA-DNA complexes in two different crystal forms, and comparison of these structures shows that even with DNA bound the two domains can move freely relative to one another (Supplementary Figure 2b). Such flexibility may allow easier docking to RecA-ssDNA-ATP filaments even while bound to promoter DNA.
The position of the cleavage loop also varies among structures (Figure 1c). In our highest resolution structure, A22, the tip of this loop is ordered in both subunits, with the scissile peptide bond (A84-G85) poised for attack by the active site Ser119. However, in our B22 complex, which differs only in the spacer sequence and formed nearly isomorphous crystals, neither cleavage loop is docked, and the scissile bonds are located 20Å from the cleavage cavity. The Strynadka group observed alternating conformations of this loop in catalytically inactive apo-LexA dimers4. We suggest that, for the catalytically inactive mutants used to obtain these structures, the cleavage loop can dock and undock with an equilibrium close enough to 1 that minor environmental changes can shift its preferred position. However, in the WT case, docking would incur a considerable energetic cost, due to burying Lys156. Presumably the binding energy of RecA filament – LexA contacts is used to pay that price, activating LexA for autocleavage. Given the mismatch in symmetry between LexA dimers and head-to-tail RecA filaments, separate docking events are probably required to cleave both halves of a LexA dimer16. However, loss of one DNA binding domain from a dimer would drastically reduce DNA binding and relieve transcriptional repression.
Recognition of the conserved CTGT motif (C5:G20 to T8:A17 in our numbering scheme) appears to rely heavily on direct readout (Figure 2). The N-terminal portion of the recognition helix, α3, tucks into the major groove and makes direct and/or water-mediated interactions with all four base pairs. C5 donates a hydrogen bond to Glu45, mutation of which (to Val or Ala) results in relaxed specificity at this position.17 T6 forms both a hydrogen bond with Asn41 and a hydrophobic contact to Ala42. A triple mutation that includes Asn41 and Ala42 alters the sequence preference from 5′CTGT to CCGT.11 Asn41 appears to be a key residue, as it also forms direct hydrogen bonds to the next base pair, G7:C18, and a water-mediated one with A17 (paired to T8). Finally, G7 forms a second hydrogen bond to the hydroxyl of Ser39. The only direct contact to the T8:A17 pair is the water-mediated interaction with Asn41, and from our structure, it is unclear how that mediates sequence specificity. However, there may be additional water-mediated contacts, not visible at this resolution, that contribute to specificity. Indirect readout of the DNA's more subtle sequence-dependent conformational preferences may also play a role in sequence recognition here. The protein makes direct contacts to only one base outside the CTGT consensus sequence: the methyl group of T16 (which pairs with A9) lies in a hydrophobic dimple between α3 and the aliphatic portion of R28. However, we found that this makes a relatively small contribution to overall affinity (see below).
Figure 2.
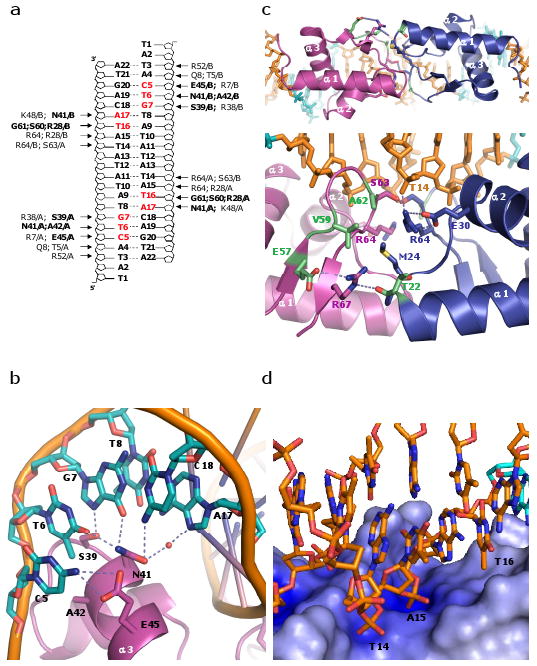
The E. coli LexA-DNA binding. (a) Schematic representation of protein-DNA interactions observed in the LexA-DNA structure. Amino acid residues in bold and bases highlighted in red represent interactions with the bases. Remaining amino acid residues contact the DNA phosphodiester backbone. (b) Detailed of contacts with the consensus sequence. In addition to the hydrogen bonds shown as dotted lines, there is a van der Waals interaction between A42 and the methyl group of T6. (c) Interface between DNA binding domains. Top: viewed from the catalytic domains, and bottom: expanded and rotated ∼90° about a horizontal axis. Residues that appear to make key protein-protein contacts are shown as sticks, and those whose mutations enhanced DNA binding are highlighted in green (specifically, T22I, E57K, V59I, and A62T or V13). (d) Zoomed in view of the electrostatic potential surface map of the wing region. The main chain amide group of S63 and guanidine groups of R64 and R28 interact with the phosphate groups of T14 and A15. The side chains of R28, P40, and E44 form a hydrophobic pocket that stabilizes the methyl group of T16. The electrostatic potential at the protein surface was calculated with APBS, and the color scale ranges from -10kT/e (bright red) to +10kT/e (dark blue)29.
By examining more sites, less stringently conserved bases can be included, and the consensus sequence can be extended to 5′TACTGT(AT)4ACAGTA18,19, which is the sequence used in our highest-resolution “A22” crystal. With the exception of T16, LexA contacts only the phosphate backbones of the additional bases. Therefore its preference for A/T pairs in those positions must reflect indirect rather than direct readout.
The flanking DNA regions curve around the sides of the protein, leading to a global bend of ∼35° in both crystal forms (Supplementary Figure 3). Much of this bend appears to be induced by interactions of the phosphate backbone with a region of positive charge at the N-terminus of α1 (Arg7 and the helix dipole) and the C-terminus of α3 (Arg 52 and Lys 53). Mutation of these residues along α3 reduces binding.11,20 The contacts explain the extended interface predicted from ethylation interference, photocrosslinking and hydroxyl radical cleavage experiments6,21.
At the center of the complex, where the minor groove faces the protein, LexA's two wings cross, forming the protein-protein interface described above (Figure 2c, d). The wingtips bind across the minor groove, and in conjunction with R28 and the N-terminus of helix 2, form positively charged binding pockets for the phosphate groups of T14 and A15. The minor groove is unusually narrow in this region, presumably to fit into this network of interactions (Supplementary Graph 1a).
Narrow minor grooves are often associated with A/T rich regions of DNA, in agreement with LexA's preference for an A/T spacer22. However, many functional SOS boxes do include G:C pairs within the spacer22. To investigate how a G/C-rich spacer interacts with LexA, we changed the central base pairs from ATAT to GCGC, resulting in structure “B22”. The minor groove width is essentially unchanged (Supplementary Graph 1). Although B22 is a lower resolution structure (3.6/3.1/2.9 Å), the density for the DNA backbone is strong enough to define the overall groove width. Thus, the protein-protein interface between DNA binding domains appears strong enough to compress even a G/C rich minor groove. The alternating pyrmidine-purine nature of both spacer sequences may also facilitate such compression, since 5′Y-R steps are particularly flexible.
To further assess the role of the spacer, we characterized binding with various DNA substrates (Table 1), including those sequences used in our crystallizations (the duplex ends were extended to prevent hairpinning at low concentrations). As expected, the site carrying the consensus spacer sequence, (AT)422, yielded the tightest binding, with an apparent Kd of 0.8nM under our conditions. The AT rich consensus sequence and narrow minor groove suggest that GC base pairs would be disfavored, as in the cases of bacteriophage 434 and P22 repressors23,24. Surprisingly, the four G:C base pairs found in the B22 structure's spacer decreased affinity by only two fold. This was similar to the effect of the two G:C pairs found in the symmetrized RecA SOS box used in C29. Changing the entire spacer from alternating As and Ts to a more rigid A-tract decreased the affinity ∼5 fold. This relatively minor decrease in affinity may reflect the fact that only in the center of the structure is the minor groove as narrow as an A-tract's (Supplementary Graph 1). It could also reflect some other advantage of the alternating sequence's flexibility, such as the ability to adopt the alternating high/low twist seen in the structure.
Table 1.
Affinities of E. coli LexA for various DNA substrates.
| Duplex | DNA sequencea | Kdb (nM) |
|---|---|---|
| RecA SOS boxc | GATGCCTGCGGATA CTGT ATGATCAT ACAG TATCAATTCTGGCT | 1.67 ± 0.26 |
| CTACGGACGCCTAT GACA TACTAGTA TGTC ATAGTTAAGACCGA | ||
| AT–repeat spacer | GATGCCTGCGGATA CTGT ATATATAT ACAG TATCAATTCTGGCT | 0.80 ± 0.27 |
| CTACGGACGCCTAT GACA TATATATA TGTC ATAGTTAAGACCGA | ||
| AT–GC spacer | GATGCCTGCGGATA CTGT ATGCGCAT ACAG TATCAATTCTGGCT | 1.59 ± 0.30 |
| CTACGGACGCCTAT GACA TACGCGTA TGTC ATAGTTAAGACCGA | ||
| U spacer | GATGCCTGCGGATA CTGT ATGCGCAU ACAG TATCAATTCTGGCT | 8.16 ± 1.75 |
| CTACGGACGCCTAT GACA UACGCGTA TGTC ATAGTTAAGACCGA | ||
| A–tract spacer | GATGCCTGCGGATA CTGT AAAAAAAA ACAG TATCAATTCTGGCT | 5.64 ± 0.78 |
| CTACGGACGCCTAT GACA TTTTTTTT TGTC ATAGTTAAGACCGA | ||
| GC–repeat spacer | GATGCCTGCGGATA CTGT GCGCGCGC ACAG TATCAATTCTGGCT | ∼100d |
| CTACGGACGCCTAT GACA CGCGCGCG TGTC ATAGTTAAGACCGA | ||
| TGC spacer | GATGCCTGCGGATA CTGT ACGCGCGT ACAG TATCAATTCTGGCT | 2.40 ± 0.50 |
| CTACGGACGCCTAT GACA TGCGCGCA TGTC ATAGTTAAGACCGA | ||
| −1 spacer | GATGCCTGCGGATA CTGT ATATATT ACAG TATCAATTCTGGCT | ∼250d |
| CTACGGACGCCTAT GACA TATATAA TGTC ATAGTTAAGACCGA | ||
| +1 spacer | GATGCCTGCGGATA CTGT ATATATAAT ACAG TATCAATTCTGGCT | ∼200d |
| CTACGGACGCCTAT GACA TATATATTA TGTC ATAGTTAAGACCGA | ||
Note: the central 29 bp of the “RecA SOS box” duplex match those used in crystal form C29, and the central 22bp of the “AT-repeat spacer” and “AT-GC” spacer duplexes match those in crystal forms A22 and B22 respectively. The extended duplexes were designed to prevent hairpinning at low concentrations.
Coding: underline, LexA consensus sequence; bold, spacers.
Apparent Kd.
Nucleotide number 23 was changed from the original G to T to make the DNA self-complementary for crystallization.
No distinct product bands are observed. Rather the gels showed smearing indicating that the complexes fall apart during the course of the EMSA,and unbound DNA bands that decreased with increasing protein concentration (data not shown). These binding affinities were estimated by eye.
When the outer region of the spacer sequences was changed, differences were much more apparent. When the GC repeat was extended to cover the entire spacer region, the apparent Kd weakened by ∼100-fold (Table 1, AT-GC spacer vs. GC- repeat spacer). However, restoring the outermost base pair to A:T in both half-sites increased binding by ∼40 fold (Table 1, GC-repeat vs. TGC spacer). Our structure shows that the methyl group of this T, T16, is docked in a hydrophobic pocket (Figure 2d). We tested the importance of this methyl group by replacing the thymine with uracil. Although this should leave the hydrophobic pocket unfavorably open to solvent, it lowered the affinity by only ∼5 fold (Table 1, AT-GC vs. U spacer). The remaining observed differences lay in the thymine to cytosine substitution. It is unclear why thymine is favored over cytosine.
LexA is less tolerant of spacer length modifications: extending or shortening it by one base pair, decreased affinity by 200-250 fold (Table 1, +1 and -1 spacers). Nevertheless, a small amount of complex was still observed as smears on EMSA (data not shown). The altered spacers would disrupt the synergistic protein-protein and protein-DNA interactions between the two binding domains and their respective half-sites.
Crystal structures of winged HTH motif have revealed versatile wing functions in DNA binding, i.e. establishing protein-protein interactions like HSF-DNA25 complex or intercalating into the minor groove like the AhrC26 repressor. While LexA shares the canonical wHTH motif, it utilizes its wing not only to stabilize DNA binding but also to fulfill spacer length requirement of its SOS operators.
Methods Summary
Briefly, a catalytically inactive mutant of E. coli LexA (K156A) was crystallized in complex with its SOS operator sequences in two space groups, A22 (5′-TATACTGTATATATATACAGTA) and B22 (5′-TATACTGTATGCGCATACAGTA) in P21212; C29 (5′-GTTGATACTGTA*TGATCATACAGTATCAA in which A* denotes the position of a nick in the sequence) in P212121. The anisotropic resolutions for A22 and B22 are 3.0, 2.6, 2.3 and 3.6, 3.1, 2.9; R and Rfree are 26.6/30.6 for A22 and 26.6/28.7 for B22. C29 was not refined due to its low-resolution nature (4.0, 3.2, 4.0). Supplementary Figure 3 shows the anomalous difference Fourier electron density map illustrating the location of the Br-peaks for A22. Figures were generated with Pymol27.
Supplementary Material
Acknowledgments
We thank Elizabeth Harris and Olajumoke Adekeye for help with crystal growth and optimization, Yuen-Ling Chan Leung for advice on DNA binding assays, and Dr. Sherwin Montaño and other members of the Rice laboratory for help and discussions; the staffs at beamlines BioCARS 14, SBC 19-ID and 19-BM, and LS-CAT 21-ID-F at the Advanced Photon Source, Argonne National Laboratory, for helping with the synchrotron X-ray data collection. The research is supported in part by NIH GM058827.
Footnotes
Full methods accompany this paper.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions
Adrianna Zhang grew the crystals, determined the structure, and carried out the affinity assays. Ying Pigli assisted greatly with cloning, protein purification, and crystal screening and optimization. Phoebe Rice designed and directed the project and assisted in crystallographic data collection and structure determination. AZ and PAR wrote the manuscript.
Atomic coordinates and structure factor files have been deposited with the Protein Data Bank under the accession codes 3JSO (A22), 3JSP (B22), and 3K3R (C29). Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Walker GC. Inducible DNA repair systems. Annu Rev Biochem. 1985;54:425–457. doi: 10.1146/annurev.bi.54.070185.002233. [DOI] [PubMed] [Google Scholar]
- 2.Shinagawa H. SOS response as an adaptive response to DNA damage in prokaryotes. EXS. 1996;77:221–235. doi: 10.1007/978-3-0348-9088-5_14. [DOI] [PubMed] [Google Scholar]
- 3.Erill I, Campoy S, Barbe J. Aeons of distress: an evolutionary perspective on the bacterial SOS response. FEMS Microbiol Rev. 2007;31:637–656. doi: 10.1111/j.1574-6976.2007.00082.x. [DOI] [PubMed] [Google Scholar]
- 4.Luo Y, et al. Crystal structure of LexA: a conformational switch for regulation of self-cleavage. Cell. 2001;106:585–594. doi: 10.1016/s0092-8674(01)00479-2. [DOI] [PubMed] [Google Scholar]
- 5.Neher SB, Flynn JM, Sauer RT, Baker TA. Latent ClpX-recognition signals ensure LexA destruction after DNA damage. Genes Dev. 2003;17:1084–1089. doi: 10.1101/gad.1078003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hurstel S, Granger-Schnarr M, Schnarr M. Contacts between the LexA repressor--or its DNA-binding domain--and the backbone of the recA operator DNA. EMBO J. 1988;7:269–275. doi: 10.1002/j.1460-2075.1988.tb02809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knegtel RM, et al. A model for the LexA repressor DNA complex. Proteins. 1995;21:226–236. doi: 10.1002/prot.340210305. [DOI] [PubMed] [Google Scholar]
- 8.Butala M, Zgur-Bertok D, Busby SJ. The bacterial LexA transcriptional repressor. Cell Mol Life Sci. 2009;66:82–93. doi: 10.1007/s00018-008-8378-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schultz SC, Shields GC, Steitz TA. Crystal structure of a CAP-DNA complex: the DNA is bent by 90 degrees. Science. 1991;253:1001–1007. doi: 10.1126/science.1653449. [DOI] [PubMed] [Google Scholar]
- 10.Hong M, Fuangthong M, Helmann JD, Brennan RG. Structure of an OhrR-ohrA operator complex reveals the DNA binding mechanism of the MarR family. Mol Cell. 2005;20:131–141. doi: 10.1016/j.molcel.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 11.Thliveris AT, Little JW, Mount DW. Repression of the E coli recA gene requires at least two LexA protein monomers. Biochimie. 1991;73:449–456. doi: 10.1016/0300-9084(91)90112-e. [DOI] [PubMed] [Google Scholar]
- 12.Mohana-Borges R, et al. LexA repressor forms stable dimers in solution. The role of specific dna in tightening protein-protein interactions. J Biol Chem. 2000;275:4708–4712. doi: 10.1074/jbc.275.7.4708. [DOI] [PubMed] [Google Scholar]
- 13.Oertel-Buchheit P, Porte D, Schnarr M, Granger-Schnarr M. Isolation and characterization of LexA mutant repressors with enhanced DNA binding affinity. J Mol Biol. 1992;225:609–620. doi: 10.1016/0022-2836(92)90389-2. [DOI] [PubMed] [Google Scholar]
- 14.Groban ES, et al. Binding of the Bacillus subtilis LexA protein to the SOS operator. Nucleic Acids Res. 2005;33:6287–6295. doi: 10.1093/nar/gki939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butala M, Hodoscek M, Anderluh G, Podlesek Z, Zgur-Bertok D. Intradomain LexA rotation is a prerequisite for DNA binding specificity. FEBS Lett. 2007;581:4816–4820. doi: 10.1016/j.febslet.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 16.Giese KC, Michalowski CB, Little JW. RecA-dependent cleavage of LexA dimers. J Mol Biol. 2008;377:148–161. doi: 10.1016/j.jmb.2007.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thliveris AT, Mount DW. Genetic identification of the DNA binding domain of Escherichia coli LexA protein. Proc Natl Acad Sci U S A. 1992;89:4500–4504. doi: 10.1073/pnas.89.10.4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernandez De Henestrosa AR, et al. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol Microbiol. 2000;35:1560–1572. doi: 10.1046/j.1365-2958.2000.01826.x. [DOI] [PubMed] [Google Scholar]
- 19.Wade JT, Reppas NB, Church GM, Struhl K. Genomic analysis of LexA binding reveals the permissive nature of the Escherichia coli genome and identifies unconventional target sites. Genes Dev. 2005;19:2619–2630. doi: 10.1101/gad.1355605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oertel-Buchheit P, Lamerichs RM, Schnarr M, Granger-Schnarr M. Genetic analysis of the LexA repressor: isolation and characterization of LexA(Def) mutant proteins. Mol Gen Genet. 1990;223:40–48. doi: 10.1007/BF00315795. [DOI] [PubMed] [Google Scholar]
- 21.Dumoulin P, Oertel-Buchheit P, Granger-Schnarr M, Schnarr M. Orientation of the LexA DNA-binding motif on operator DNA as inferred from cysteine-mediated phenyl azide crosslinking. Proc Natl Acad Sci U S A. 1993;90:2030–2034. doi: 10.1073/pnas.90.5.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis LK, Harlow GR, Gregg-Jolly LA, Mount DW. Identification of high affinity binding sites for LexA which define new DNA damage-inducible genes in Escherichia coli. J Mol Biol. 1994;241:507–523. doi: 10.1006/jmbi.1994.1528. [DOI] [PubMed] [Google Scholar]
- 23.Koudelka GB, Carlson P. DNA twisting and the effects of non-contacted bases on affinity of 434 operator for 434 repressor. Nature. 1992;355:89–91. doi: 10.1038/355089a0. [DOI] [PubMed] [Google Scholar]
- 24.Wu L, Vertino A, Koudelka GB. Non-contacted bases affect the affinity of synthetic P22 operators for P22 repressor. J Biol Chem. 1992;267:9134–9139. [PubMed] [Google Scholar]
- 25.Littlefield O, Nelson HC. A new use for the ‘wing’ of the ‘winged’ helix-turn-helix motif in the HSF-DNA cocrystal. Nat Struct Biol. 1999;6:464–470. doi: 10.1038/8269. [DOI] [PubMed] [Google Scholar]
- 26.Garnett JA, Marincs F, Baumberg S, Stockley PG, Phillips SE. Structure and function of the arginine repressor-operator complex from Bacillus subtilis. J Mol Biol. 2008;379:284–298. doi: 10.1016/j.jmb.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 27.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 28.Kabsch W. A solution for the best rotation to relate two sets of vectors. Acta Crystallogr A. 1976;32:922–923. [Google Scholar]
- 29.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.