

Abstract
Here we report the discovery of ON044580, an α-benzoyl styryl benzyl sulfide that possesses potent inhibitory activity against two unrelated kinases, JAK2 and BCR-ABL, and exhibits cytotoxicity to human tumor cells derived from chronic myelogenous leukemia (CML) and myelodysplasia (MDS) patients or cells harboring a mutant JAK2 kinase. This novel spectrum of activity is explained by the non–ATP-competitive inhibition of JAK2 and BCR-ABL kinases. ON044580 inhibits mutant JAK2 kinase and the proliferation of JAK2V617F-positive leukemic cells and blocks the IL-3–mediated phosphorylation of JAK2 and STAT5. Interestingly, this compound also directly inhibits the kinase activity of both wild-type and imatinib-resistant (T315I) forms of the BCR-ABL kinase. Finally, ON044580 effectively induces apoptosis of imatinib-resistant CML patient cells. The apparently unrelated JAK2 and BCR-ABL kinases share a common substrate, STAT5, and such substrate competitive inhibitors represent an alternative therapeutic strategy for development of new inhibitors. The novel mechanism of kinase inhibition exhibited by ON044580 renders it effective against mutant forms of kinases such as the BCR-ABLT315I and JAK2V617F. Importantly, ON044580 selectively reduces the number of aneuploid cells in primary bone marrow samples from monosomy 7 MDS patients, suggesting another regulatory cascade amenable to this agent in these aberrant cells. Data presented suggest that this compound could have multiple therapeutic applications including monosomy 7 MDS, imatinib-resistant CML, and myeloproliferative neoplasms that develop resistance to ATP-competitive agents.
Keywords: JAK2, BCR-ABL, CMPD, MDS, CML
Introduction
The Janus Kinase (JAK) family of cytoplasmic protein tyrosine kinases are pivotal mediators of cytokine signaling pathways.1,2 The JAK kinase family consists of 4 members: TYK2, JAK1, JAK2, and JAK3. Activation of JAK kinases1-3 resulting from cytokine/receptor interaction leads to phosphorylation of corresponding interleukin receptors on multiple tyrosine residues, which in turn serve as docking sites for other signal-transducing proteins, the most important of which are the STAT family of transcription factors.1-3 JAK2 is the primary tyrosine kinase activated by erythropoietin (Epo) and is essential for definitive erythropoiesis.1
The importance of JAKs in human cancer has been highlighted by the discovery of genetic alterations in this family of kinases, leading to hyperactivation of the pathways they regulate. These findings include translocations leading to the expression of various forms of JAK2 fusion protein such as TEL/ETV6-JAK2, PCM1-JAK2, BCR-JAK2, RPN1-JAK2, NFE2-JAK2, AML1-JAK2, SSBP2-JAK2, and PAX5-JAK2, which occur in lymphoid/myeloid leukemias and myelodysplasia (MDS).4-15 In addition, amplification of the JAK2 locus has been shown to occur in Hodgkin’s lymphomas,16 and acquired activating mutations in the JAK2 gene have been found in chronic myeloproliferative disorders (CMPD),17-19 acute lymphoblastic leukemias,20-23 and myelogenous leukemias.24-27
A point mutation in the JAK2 kinase has been suggested as the causative molecular event in most patients with polycythemia vera (PV) as well as in half of the cases of essential thrombocythemia (ET) and chronic idiopathic myelofibrosis, all of which are classified as CMPD.28-31 In addition, it has been reported that about half of refractory anemia ringed sideroblasts with thrombocytosis (RARS-T) patients, along with a subset of others with MDS and mixed MDS/CMPD, carry the JAK2 mutation.18,32,33 Remarkably, every sample derived from such patients contained the same amino acid substitution (V617F). Based on the predicted JAK2 structure and atomic level simulations, this substitution is believed to disrupt an autoinhibitory interaction between the pseudokinase (JH2) and kinase (JH1) domains of the protein.4,28,34 Studies using Epo receptor mutants have revealed the need for receptor-dependant dimerization of the mutant kinase for constitutive activation,35 and a recent report provides biochemical evidence for a regulatory role of the FERM domain in hyperactivation of JAK2 with a V617F substitution.36 This mutation has been found to confer Epo-independent growth of the mutant cells in vitro due to deregulation of signaling pathways downstream of JAK2.28 Small interfering RNA-mediated knock-down of JAK2 has also been found to impair EEC formation from PV bone marrow.29 Furthermore, PV patients who lacked the V617F point mutation were found to harbor other activating exon 12 mutations in JAK2,37 making mutations of JAK2 the causative genetic lesion in all cases of this disease.
Activation of the JAK-STAT pathway has also been observed in diseases with signaling defects in proteins upstream of the Janus kinases. One such example is the constitutive activation of JAK238 and STAT139 in cells from monosomy 7 MDS patients, likely due to aberrant cytokine receptor signaling. Monosomy 7 is the second most frequently observed cytogenetic abnormality in MDS, with an incidence of 21%.40 It is the most frequent karyotypic aberration occurring in bone marrow failure patients following immunosuppressive treatment, and it is associated with severe cytopenias and a high propensity for developing acute leukemia.41,42 Patients who develop monosomy 7 AML are difficult to treat and often relapse quickly or die of infection.43 Monosomy 7 is especially common in MDS secondary to exposure to alkylating drugs and in pediatric MDS. Monosomy 7 cells show increases in a differentiation-defective GCSFR isoform (IV) that fails to internalize following GCSF binding as normally occurs for the full-length receptor. It is also defective in facilitating phosphorylation of STAT-3, but its ability to signal phosphorylation of STAT-1 and -5 is unimpaired.39,44 As a result, the cell’s ability to differentiate is limited, whereas its ability to proliferate via JAK-2 remains intact.
These findings open new avenues for diagnosing and classifying patients with these disorders and identify JAK2 as a new molecular target for drug discovery. To date, a number of ATP-competitive JAK-2 inhibitors have been identified.45-49 Here we report the discovery of a new JAK2 inhibitor that is non-ATP competitive and potently inhibits the kinase activity of both wild-type and mutant JAK2 kinase. It readily inhibits the proliferation of JAK2 V617F-positive leukemic cells and blocks the IL-3–mediated phosphorylation of JAK2 and STAT5, a known substrate of JAK2. Importantly, ON044580 selectively inhibits the proliferation of aneuploid cells in bone marrow samples from monosomy 7 MDS patients. Most interestingly, this compound also inhibits both wild-type and imatinib-resistant (T315I) forms of the BCR-ABL kinase and induces apoptosis of imatinib-resistant chronic myelogenous leukemia (CML) patient cells.
Results
The remarkable clinical success of imatinib,50 an inhibitor of the BCR-ABL tyrosine kinase, was followed by the development of a series of kinase inhibitors that have found application in cancer therapy. In the past few years, there has been an increasing realization that tumor cells often develop resistance to ATP-competitive kinase inhibitors as a result of accumulating mutations in the ATP binding site of the kinase. This has been observed in patients undergoing treatment with imatinib.51 Because selection of highly conserved mutable residues in the ATP binding site appears to be relatively common for many kinases, it has been argued that non–ATP-competitive inhibitors might constitute better drug candidates.52 Because there are a limited number of chemotypes that act as non–ATP-competitive inhibitors, we undertook the synthesis and characterization of new chemotypes that are unrelated to ATP or other purine and pyrimidine nucleosides and yet possess kinase inhibitory activity. This effort led to the synthesis and identification of a new class of compounds, α-benzoyl styryl benzyl sulfides, that possess potent kinase inhibitory activity and exhibit cytotoxicity to human tumor cells that express oncogenic kinases.53 Our preliminary screening of these compounds using a high-throughput cell-based assay in combination with kinase assays led to the identification of a compound, ON044580 (Fig. 1a) that is a potent inhibitor of JAK-2. In vitro studies using a recombinant JAK-2 protein produced in insect cells (that is commercially available) showed that this compound inhibits the kinase activity of recombinant JAK2 with an IC50 ranging between 0.9 and 1.2 µM (Figs. 1b and c). Under identical conditions, AG490 was able to inhibit JAK2 kinase activity with an IC50 of 36.4 µM, which is in agreement with published literature.54 Following these observations, we examined the ability of this compound to inhibit the activated V617F mutant form of JAK2 using commercially available recombinant protein produced in insect cells. The results of this study showed that ON044580 inhibited the kinase activity of mutant JAK2 with a similar IC50 (0.8 to 1.1 µM) as that seen with WT JAK2, whereas AG490 had an IC50 of 33.1 µM (Figs. 1b and c). Because the recombinant preparations of wild-type and mutant JAK2 proteins are truncated forms of the kinase, we examined the kinase inhibitory activity of these compounds using JAK2 kinase immunoprecipitated from the Ba/F3:JAK2V617F cell line that expresses the full-length wild-type and mutant forms of JAK2. These studies again showed that ON044580 inhibited the JAK2 kinase activity with an IC50 of approximately 4 µM. Under identical conditions, AG490 inhibited the kinase activity with an IC50 of 145 µM (Figs. 1d and e). The observation that ON44580 inhibits recombinant JAK2 (aa 532-1132) led us to ask whether this compound binds to the catalytic kinase domain (JH1) or the regulatory pseudokinase domain (JH2) of JAK2. To test this, we made use of the commercially available recombinant form of JAK2 containing just the kinase domain (JH1 domain spanning amino acids 808-1132). Interestingly, ON044580 failed to inhibit JAK2 kinase domain (JH1) activity at a concentration of 10 µM, whereas complete inhibition was observed only at 100 µM (Fig. 1f). This significant increase in the inhibitory concentration suggests the ATP-binding kinase domain of JAK2 (JH1) is not the primary site of action of ON044580.
Figure 1.
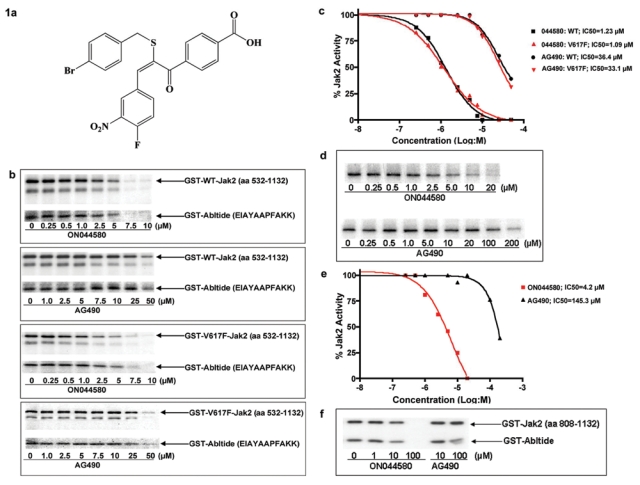
Identification of a novel JAK2 inhibitor. (a) Structure of the JAK2 inhibitor ON044580. (b) JAK2 inhibitory activity of ON044580. Recombinant wild-type or V617F mutant JAK2 (aa 532-1132) was mixed with increasing concentrations of ON044580 (0.25-10 µM) or AG490 (1-50 µM) and kinase assays performed as described in Methods using recombinant GST-Abltide as substrate. The reaction mixtures were subjected to SDS-PAGE and autoradiography. (c) Determination of IC50 values. From the autoradiograms described in b, the values of individual bands corresponding to autophosphorylation of JAK2 were analyzed using MacBas software and plotted as a function of drug concentration using Prism 4 Graphpad software. ON044580 exhibited a 30-fold greater inhibition of wild-type and mutated Jak2. (d) Inhibition of mammalian JAK2 activity by ON044580. Full-length JAK2 was immunoprecipitated from clarified lysates of BaF3:JAK2V617F cells stimulated with 5 ng/mL recombinant IL-3 for 1 hour, as described in the Methods section. The washed immunocomplexes were incubated for 30 minutes in the presence of increasing concentrations of ON044580 and processed for kinase assays as described in b. (e) The samples from d were quantitated, and IC50 curves were plotted as described in c. Although ON044580 showed inhibition with IC50 = 4.2 µM, AG490 had an IC50 value of 145.3 µM. (f) Involvement of the regulatory JH2 domain. Recombinant wild-type catalytic JH1 domain (aa 808-1132) of JAK2 was incubated with increasing concentrations of ON044580 and assayed for kinase activity. No inhibition of JAK2 activity was apparent at an ON044580 concentration of 10 µM in this truncated unregulated form of the kinase. The longer version of the kinase (aa 532-1132) encompassing both the catalytic and regulatory domains was inhibited at 1 µM (1a and b).
ON44580 is a non–ATP-competitive inhibitor
Our observation that the pseudokinase domain is required for the kinase inhibitory activity of ON044580 further led us to postulate that it is not an ATP-competitive JAK2 inhibitor. We directly tested this hypothesis by carrying out kinase inhibition assays either in the presence of increasing amounts of ATP or in the presence of increasing amounts of substrate. The results of this study, shown in Figure 2, demonstrate that increasing the ATP concentration in the kinase reaction mixture did not affect the inhibitory activity of ON044580 (Fig. 2a). On the other hand, increasing the substrate concentration in the reaction mixture resulted in a reduction of the kinase inhibitory activity of ON044580. This was seen to be true for both the autophosphorylation of JAK2 kinase itself (Fig. 2b) and the transphosphorylation of GST-Abltide substrate (Fig. 2c).
Figure 2.
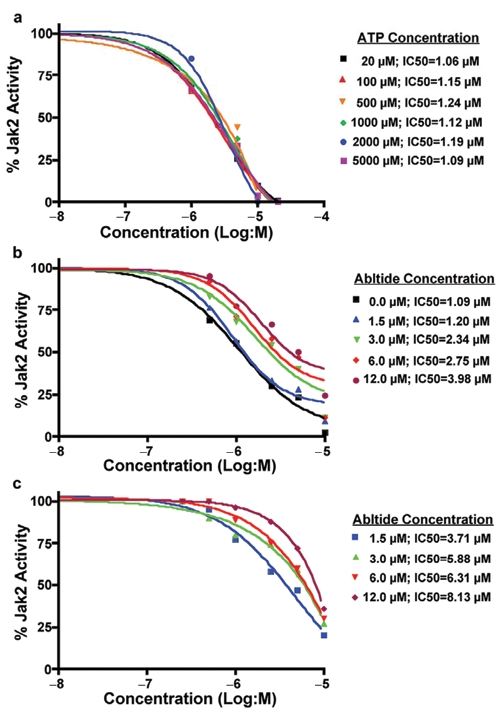
Steady-state kinetic analysis of JAK2 inhibition by ON044580 (a) ATP does not affect the JAK2-V617F inhibitory activity of ON044580. Recombinant JAK2-V617F (aa 532-1124) was mixed with the indicated concentrations of ON044580 and varying concentrations of ATP up to 5 mM. Kinase assays were performed as described in Figure 1b. The values from individual samples were analyzed and plotted as a function of log inhibitor concentration as described in Figure 1c. (b, c) Effect of substrate concentration on the inhibition of V617F-JAK2 autophosphorylation and GST-Abltide phosphorylation. IC50 curves for V617F-JAK2 kinase activity in the presence of varying concentrations of GST-Abltide substrate and ON044580 were generated as described in (a). The upper band, like that seen in Figure 1b was quantitated for autophosphorylation of JAK2-V617F (b) and the lower band for phosphorylation of GST-Abltide (c).
In vivo inhibition of Jak2 autophosphorylation and STAT-5 phosphorylation by ON044580 in Ba/F3:JAK2V617F cells
To test the in vivo kinase inhibitory activity of ON044580, we treated Ba/F3:JAK2V617F cells with increasing concentrations of the compound for 2 hours in the presence of recombinant IL-3 (which enhances the phosphorylation status of JAK2). At the end of the 2-hour incubation period, cells were washed and lysed in detergent containing buffer, and the clarified lysates were subjected to SDS-PAGE followed by Western blotting to detect the phosphorylation status of JAK2. The results of this study (Fig. 3a) showed that ON044580 was able to inhibit the phosphorylation of JAK2 in a concentration-dependent manner. AG490, under identical conditions, did not inhibit JAK2 phosphorylation, which could be due to the high IC50 values seen for full-length JAK2 kinase with this compound. As part of this study, we also examined the time course of inhibition in which we added 10 µM of ON044580 for periods of time ranging from 15 to 60 minutes and examined the phosphorylation status of JAK2 using Western blot analysis. The results of this study presented in Figure 3b showed that in as little as 15 to 30 minutes, the compound was able to inhibit IL-3–mediated JAK2 phosphorylation.
Figure 3.
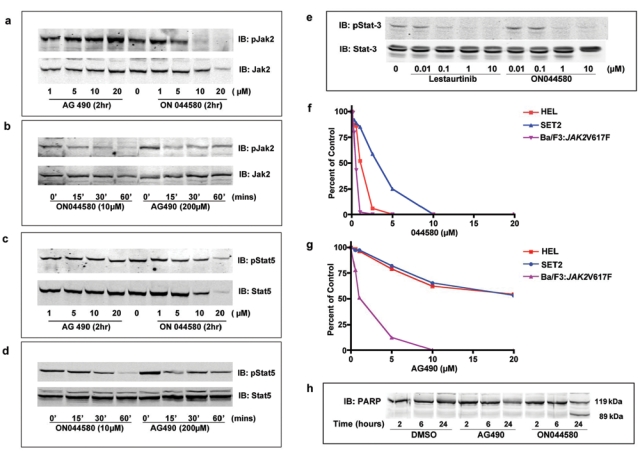
Effects of ON044580 on cellular JAK2-dependent cytokine signaling. (a) Inhibition of JAK2 autophosphorylation by ON044580 in IL-3–stimulated Ba/F3:JAK2V617F cells. Mid-log phase Ba/F3:JAK2V617F cells were treated for 2 hours with indicated concentrations of ON044580 and 1 hour with 5 ng/mL recombinant IL-3. Washed cells were lysed in detergent containing buffer, and the clarified lysates were subjected to SDS-PAGE followed by immunoblotting to detect phosphorylation status of JAK-2. (b) ON044580 inhibits JAK2 autophosphorylation in Ba/F3:JAK2V617F cells within 15 minutes. Mid-log phase Ba/F3:JAK2V617F cells were treated for the indicated times with 10 µM ON044580 or 200 µM AG490 and processed as above. For all time points, the total time for IL-3 stimulation was 60 minutes. (c) Inhibition of STAT-5 phosphorylation by ON044580 in IL-3–stimulated Ba/F3:JAK2V617F cells. Ba/F3:JAK2V617F cells were treated and processed as in (a). Immunoblotting was carried out to assess STAT-5 phosphorylation. (d) ON044580 inhibits STAT-5 phosphorylation in Ba/F3:JAK2V617F cells within 30 minutes. Ba/F3:JAK2V617F cell treatments were carried out as described in (b). Western blot analysis was performed to assay degree of STAT-5 phosphorylation. (e) ON044580 inhibits JAK2-dependent STAT-3 phosphorylation in U266 cells. Exponentially growing U266 cells were treated for 2 hours with increasing concentrations of ON044580. The ATP-competitive JAK2 inhibitor, lestaurtinib, was used as a control. Cells were washed and lysed, and clarified lysates were resolved by SDS-PAGE for immunoblot analysis to assess phosphorylated STAT-3 levels. (f, g) Growth inhibition of JAK2-V617F–expressing cells. Ba/F3:JAK2V617F cells (ectopic expression), HEL cells (homozygous JAK2V617F), and SET-2 cells (hemizygous JAK2V617F) were grown in the presence of varying concentrations of ON044580 (e) or AG490 (f) for 72 hours. Cell viability was measured by Trypan blue exclusion. GI50 values were calculated by plotting the percentage of viable cells as a function of drug concentration. (h) ON044580 induces apoptotic cell death in cells expressing JAK2-V617F. Ba/F3:JAK2V617F cells were treated with 0.5 µM ON044580 or 2 µM AG490 for 2, 6, and 24 hours. Thereafter, the cells were harvested, washed, lysed, and probed with anti-PARP antibody following Western blotting.
Using a similar approach, we also examined the phosphorylation status of STAT-5 (a natural substrate of JAK2) in Ba/F3:JAK2V617F cells treated with increasing concentrations of the compound. The results presented in Figures 3c and d show that ON044580 inhibited STAT-5 phosphorylation in a concentration-dependent and time-dependent manner. Because similar results were seen with JAK2 phosphorylation, these studies suggest that the 2 events are interrelated.
Cellular inhibition of constitutive JAK/STAT signaling by ON044580
The U266 multiple myeloma cell line expresses wild-type JAK2 but has constitutive activation of the IL-6 receptor/JAK2/STAT-3 pathway.55 We treated U266 cells with increasing concentrations of ON044580 for 2 hours to test whether this compound could inhibit aberrant JAK/STAT signaling instigated by mechanisms other than genetic alterations in the JAK2 gene itself. The results of this study showed that such a treatment led to a dose-dependent inhibition of STAT-3 phosphorylation at concentrations comparable to a known ATP-competitive JAK2 inhibitor45 (Fig. 3e).
Growth inhibition of JAK2-V617F–expressing cells
To determine whether ON044580 inhibits the proliferation of JAK2V617F-positive leukemic cells, we studied its effect on the growth and viability of 3 different cell lines that express the mutant form of JAK2. These included the Ba/F3:JAK2V617F cells that were transfected with an expression vector that encodes the mutant JAK2 and two human leukemic cell lines that were derived from leukemic patients who naturally contained this mutation in their JAK2 loci. One of them, HEL, is homozygous for V617F mutation, whereas the second cell line, SET2, is hemizygous for the V617F mutation. The results of this study, presented in Figure 3f, show that ON044580 readily inhibited the proliferation of all three cell lines at nanomolar or low micromolar concentrations. Thus, the GI50 for Ba/F3:JAK2V617F cells was approximately 250 nM, whereas the GI50 for HEL cells was approximately 900 nM. Interestingly, the SET2 cell line that was hemizygous for V617F mutation was more resistant to the cell-killing activity of the compound with a GI50 value of 3.0 µM. In a similar experiment using AG490, the GI50 value for Ba/F3:JAK2V617F cells was 1.0 µM, whereas that for HEL and SET-2 cells was greater than 20 µM (Fig. 3g).
ON044580 exerts its antiproliferative effect by inducing apoptosis in Ba/F3:JAK2V617F cells
After demonstrating effective inhibition of myeloproliferation by ON044580, we examined the mechanisms associated with this cytotoxic effect. Upon microscopic observation of cells treated with ON044580, we did not find evidence for autophagic vacuoles or mitotic arrest. To test if ON044580 activates the apoptotic pathways, we treated Ba/F3:JAK2V617F cells for 2, 6, and 24 hours with ON044580 and AG490 at concentrations twice the GI50 values obtained in the growth inhibition studies (shown in Fig. 3f). DMSO was used as a control. These cells were then harvested and probed for the status of poly-(ADP-ribose) polymerase (PARP), a marker for apoptosis induction.56 The results of this experiment show that treatment with ON044580 did indeed lead to proteolytic cleavage of PARP in 24 hours (Fig. 3h). A similar though less pronounced effect was observed with AG490.
In vitro inhibition of wild-type and T315 mutant forms of BCR-ABL kinase by ON044580
The studies presented above suggest that ON044580 inhibits the JAK2 kinase activity either by binding to the STAT-5 binding domain of JAK2 or by binding to an allosteric site, which results in altered conformation and inhibition of the kinase activity of the protein.57 It is now well established that STAT-5 is also a substrate of the BCR-ABL kinase, and if the two kinases contain a similar substrate-binding structure, it is possible that ON044580 could also inhibit the BCR-ABL kinase. Hence, we evaluated the effect of ON044580 on the in vitro kinase activity of mammalian BCR-ABL proteins immunoprecipitated from cultured cells. Lysates prepared from K562 cells expressing the wild-type BCR-ABL or 32Dcl3 cells expressing the T315I mutant form were incubated with antibodies directed against the BCR-ABL protein. Kinase assays were performed on the washed immunoprecipitates in the presence of different concentrations of ON044580 as described in the Materials and Methods section. Imatinib was used as a control in all of these assays. These studies show that imatinib readily inhibited the kinase activity of WT BCR-ABL but failed to do so with the T315I-BCR-ABL kinase. On the other hand, ON044580 inhibited both WT and T315I mutant forms of the BCR-ABL kinase (Figs. 4a and b), suggesting that mutations that affect the kinase inhibitory activity of imatinib do not affect the inhibitory activity of ON044580. It is interesting to note that ON044580 was more effective in inhibiting the T315I mutant form compared to the wild-type BCR-ABL kinase.
Figure 4.
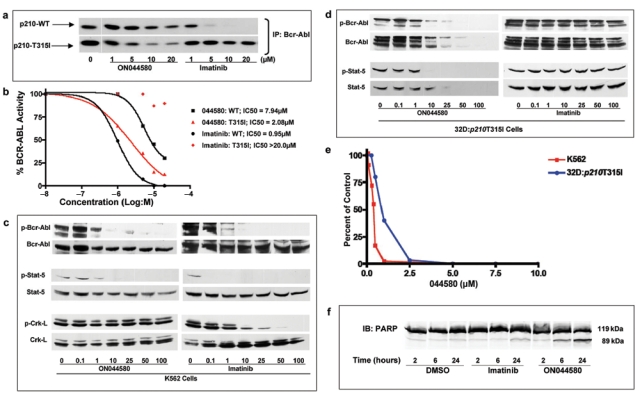
ON044580 is a dual JAK2/BCR-ABL kinase inhibitor. (a) ON044580 inhibits imatinib-sensitive and -resistant forms of BCR-ABL isolated from mammalian cells. BCR-ABL was immunoprecipitated from K562 and 32D:p210T315I cells. The immunocomplexes were incubated with inhibitor for 30 minutes, and radiometric kinase assays were performed. Imatinib was used as a control to inhibit wild-type BCR-ABL. (b) Determination of IC50 values. From the autoradiograms presented in (a), the values of individual bands corresponding to phosphorylatoin of GST-Abltide were analyzed using MacBas software and plotted as a function of drug concentration using Prism 4 Graphpad software. (c) Inhibition of BCR-ABL and STAT-5 phosphorylation by ON044580 in K562 cells. Exponentially growing K562 cells were treated for 2 hours with indicated concentrations of ON044580. Washed cells were lysed in detergent containing buffer, and the clarified lysates were subjected to SDS-PAGE followed by Western blotting and immunodetection with indicated antibodies. (d) Inhibition of BCR-ABL and STAT-5 phosphorylation by ON044580 in imatinib-resistant 32D:p210T315I cells. Exponentially growing 32D:p210T315I cells were treated for 2 hours with indicated concentrations of ON044580. Washed cells were lysed in detergent containing buffer, and the clarified lysates were subjected to SDS-PAGE followed by Western blotting. (e) ON044580 inhibits growth of cells expressing imatinib-sensitive and -resistant forms of BCR-ABL. K562 and 32D:p210T315I cells were grown in the presence of varying concentrations of ON044580 for 72 hours. Cell viability was measured by Trypan blue exclusion. (f) ON044580 induces apoptosis in chronic myelogenous leukemia cells. K562 cells were treated with 1 µM ON044580 or imatinib for 2, 6, and 24 hours. Thereafter, the cells were harvested, washed, lysed, and immunoblotted to ascertain PARP status.
Cellular inhibition of the kinase activity of BCR-ABL
Having demonstrated direct biochemical inhibition of wild-type and imatinib-resistant BCR-ABL kinase activity, we proceeded to evaluate the in vivo inhibition of the BCR-ABL activity by ON044580. We examined the autophosphorylation status of BCR-ABL protein as well as the phosphorylation status of STAT-5 and CrkL in cells treated with increasing concentrations of this compound for 2 hours. Data presented in Figure 4c show that ON044580 inhibited the autophosphorylation of wild-type BCR-ABL protein expressed in K562 cells. This compound also inhibited the phosphorylation of STAT-5 but did not affect the phosphorylation status of CrkL. Imatinib (Gleevec®) was used as a positive control in all of these experiments. These results suggest that ON044580 is selective in its inhibitory activity of BCR-ABL substrates.
Following the establishment of its in vivo activity toward WT BCR-ABL kinase, we next examined the ability of ON044580 to inhibit the autophosphorylation of T315I-BCR-ABL kinase and transphosphorylation of STAT-5. For these studies, we used the 32D:p210T315I cell line that expresses high levels of the T315I-BCR-ABL kinase and is known to be resistant to imatinib. As was done with K562 cells, we treated 32D:p210T315I cells with increasing concentrations of ON044580 for 2 hours followed by Western blot analysis of cell lysates to determine the ability of this compound to inhibit the phosphorylation of BCR-ABL and STAT-5. These studies (Fig. 4d) show that ON044580 was very effective in inhibiting autophosphorylation of T315I-BCR-ABL and STAT-5 phosphorylation in 32D:p210T315I cells, whereas imatinib failed to do so. These results suggest the possibility that ON044580 does not bind to the ATP-binding domain of the BCR-ABL kinase but acts via binding to the substrate-binding domain (that is specific to STAT-5 but not to CrkL) or to an allosteric domain of the BCR-ABL kinase that results in the impairment of its ability to phosphorylate itself and STAT-5. Interestingly, the steady-state levels of BCR-ABL-T315I kinase itself were reduced upon treatment with ON044580, suggesting enhanced degradation of the protein in these murine cells.
In vitro tumor cell–killing activity of ON044580
We next examined the ability of ON044580 to inhibit the proliferation of BCR-ABL–positive myeloid leukemias. For this study, we used K562 cells that express WT BCR-ABL kinase and 32D:p210T315I cells that express an imatinib-resistant form of BCR-ABL. The results presented in Figure 4e show that ON044580 was an effective inducer of myeloid tumor cell death with a GI50 of 300 to 400 nM in cells sensitive to imatinib. Imatinib, in the same assay system, showed a GI50 of 100 to 200 nM (data not shown). Significantly, ON044580 inhibited the proliferation of 32D:p210T315I cells with a GI50 of 500 to 900 nM, whereas imatinib showed a GI50 of 20 to 30 µM.
ON044580 induces apoptosis in K562 cells
After demonstrating effective growth inhibition of cells expressing imatinib-sensitive and imatinib-resistant forms of BCR-ABL by ON044580, we tested whether this effect was mediated by apoptotic cell death. K562 cells were treated with 1 µM ON044580 or imatinib for 2, 6, and 24 hours. DMSO was used as a control. We carried out Western blot analysis of these cell lysates to evaluate the status of PARP. Proteolytic cleavage of PARP upon ON044580 treatment was evident within 6 hours and became more pronounced in 24 hours (Fig. 4f). PARP cleavage was also observed to a lesser extent in K562 cells treated with imatinib for 24 hours.
Short-term ON044580 exposure causes growth inhibition of cells expressing mutated JAK2 and BCR-ABL kinases whereas normal bone marrow cells remain unaffected
Metabolic and/or excretory elimination of drugs after administration is an important pharmacokinetic criterion. It is therefore pertinent that compounds be tested for their ability to effect changes in target cells within short times of exposure. To test these parameters, cells expressing mutated JAK2 (Ba/F3:JAK2V617F and HEL cells) and BCR-ABL (K562 cells) were treated with increasing concentrations of ON044580 for 2 hours, washed extensively, and returned to drug-free growth medium. Normal mouse bone marrow cells were used as a parallel control in all 3 sets of experiments. As can be seen in the results presented in Figure 5a, short-term exposure to ON044580 caused growth inhibition of Ba/F3:JAK2V617F cells in a concentration-dependent manner, whereas mouse bone marrow cells were unaffected. Similar results were obtained for HEL cells that are homogyzous for the JAK2V617F allele and K562 cells expressing the oncogenic BCR-ABL fusion protein. These results are corroborated by those presented in Figure 3b and 3d, where the inhibitory effects of ON044580 on JAK/STAT signaling were elicited within 30 minutes of treatment.
Figure 5.
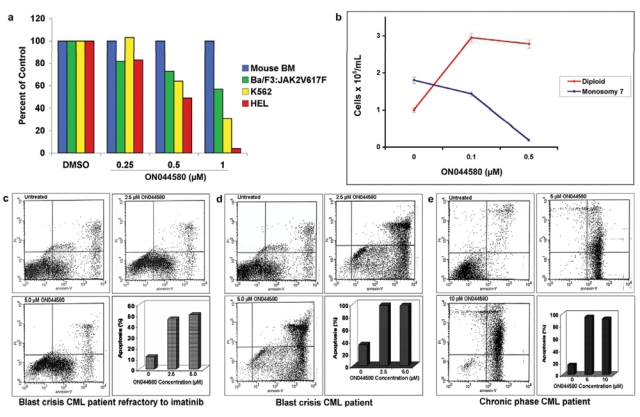
Effects of ON044580 on monosomy 7 MDS and chronic myelogenous leukemia (CML) patient samples. (a) Short-term exposure to ON044580 is cytotoxic to cells expressing oncogenic forms of JAK2 and ABL kinases but not to normal bone marrow cells. Ba/F3:JAK2-V617F cells, HEL cells, K562 cells, and mononuclear cells isolated from normal mouse bone marrow were treated with increasing concentrations of ON044580 for 2 hours. The cells were then washed and allowed to grow in complete medium in the absence of the compound. The data are plotted as percentage viability as compared to DMSO-treated controls. Concentration-dependent growth inhibition was observed for all three oncogenic cell lines, whereas mouse bone marrow cells subjected to identical conditions were unaffected. (b) Ex vivo treatment of monosomy 7 MDS patient bone marrow cells with ON044580 causes favorable cytogenetic changes. MDS patient bone marrow mononuclear cells were cultured in colony-supporting conditions and treated with 0.5 µM and 1.0 µM ON044580. This led to a dose-dependant decrease in the total number of monosomy 7 cells while maintaining/increasing the total number of diploid cells. (c-e) ON044580 induces apoptosis in refractory, chronic phase and blast crisis stages of CML patients. Purified blood samples from a refractory CML patient (c), a CML patient in blast crisis (d), and a chronic phase CML patient (e) were incubated with the indicated concentrations of ON044580, and one untreated sample was kept as a control. After 48 hours, the apoptosis was measured by Annexin-V/IP method. Cells accumulated in the lower right and upper right quadrants indicate the percentage of cells in the early and late stage of apoptosis, respectively.
ON044580 effects favorable cytogenetic changes in primary bone marrow cells from monosomy 7 MDS patients
Monosomy 7 MDS bone marrow mononuclear cells preferentially express the truncated class IV G-CSF receptor, which leads to constitutive signaling through the JAK2 pathway.39 Because ON044580 inhibited the activated IL-6 receptor/JAK2/STAT3 pathway in U266 cells (Fig. 3e) and did not show adverse effects on normal bone marrow cells (Fig. 5a), we were encouraged to test its effects on Monosomy 7 and diploid hematopoietic colony formation from MDS marrow samples. To assess the effect of ON044580 on these cells with constitutive JAK2 activity, bone marrow aspirate mononuclear cells (BMMNCs) derived from patients with monosomy 7 MDS (confirmed by metaphase karyotyping and FISH) were grown in Mylocult media (Stem Cell Technologies, Vancouver, Canada) supplemented with 400 ng/mL G-CSF and growth factor cocktail as previously described.58 Following treatment with ON044580 at 0.1 µM and 0.5 µM, cells harvested were examined for the number of aneuploid and diploid cells by FISH using centromeric probes specific for chromosomes 7 and 8. Results from this study showed that there was a reduction in the number of anueploid cells by 20% and 80%, respectively, compared to vehicle treatment control. The effect of ON044580 appeared to be limited to the aneuploid population as the total number of diploid cells in the treatment groups increased (Fig. 5b). These preliminary results indicate that ON044580 suppresses monosomy 7 bone marrow cell growth while stimulating growth of normal diploid cells, a finding that may translate into a novel targeted therapy for patients with monosomy 7 MDS.
ON044580 induces apoptosis in primary cells from CML patients refractory to imatinib
Because ON044580 was able to induce the apoptotic death of 32D:p210T315I cells, it was of interest to examine its effects on primary tumor cells derived from patients who were refractory to imatinib treatment. For this study, we used cells derived from three different patients: a blast crisis CML patient refractory to imatinib treatment, (Fig. 5c), a CML patient in blast crisis (Fig. 5d), and a CML patient in chronic phase (Fig. 5e). Cells were maintained for 48 hours in the absence of cytokines, to enhance the level of BCR-ABL+ cells in the blood cell population, and treated for an additional 48 hours with various doses of the drug. Our results show that the blast crisis and chronic phase cells were highly sensitive to apoptosis induction by ON044580, which suggests that ON044580 may be useful to treat the unusually resistant blast crisis patients.
Discussion
Chronic myeloproliferative disorders (CMPD) are clonal malignancies characterized by overproduction of one or more hematopoietic lineages with relatively normal differentiation. The molecular pathogenesis of several CMPD has been well characterized and is frequently attributable to mutations that result in constitutive activation of a protein tyrosine kinase. The classic CMPD are subdivided into chronic myeloid leukemia (Ph+ CML) and the Ph-negative CMPD, that is, polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF).59
Although Ph+ CML is characterized at the molecular level by the expression of the BCR-ABL fusion protein with deregulated kinase activity, a novel gain of function mutation in the JAK2 tyrosine kinase (JAK2V617F) has been observed in about 95% of patients with PV and 50% of those with either ET or PMF. This mutation has also been found in patients with nonclassic CMPD such as RARS-T, chronic neutrophilic leukemia, atypical CML, and chronic myelomonocytic leukemia at 50%, 20%, 20%, and 3% incidence, respectively.60 Both the BCR-ABL and JAK2-V617F proteins have been found to confer cytokine-independent growth of the mutant cells in vitro due to deregulation of signaling pathways downstream of JAK2. Expression of either of these mutant proteins were found to result in the constitutive activation of downstream effector proteins of the JAK signaling pathway, the STAT family of transcription factors. These findings have opened new avenues for diagnosing and classifying patients with these disorders and identify JAK2 as a new molecular target for the therapy of myeloproliferative disorders.
Selection of highly conserved mutable residues in the ATP binding site appears to be relatively common for many kinases. It has hence been argued that substrate-competitive inhibitors might constitute better drug candidates.53 In addition, many of the kinase inhibitors that are ATP mimetics inhibit their enzyme targets at nanomolar concentrations while requiring micromolar concentration for tumor cell growth inhibition, and this discrepancy may be related to the need to overcome millimolar concentrations of ATP known to exist inside the cell.53 Mutations occur at residues directly implicated in imatinib binding or, more commonly, at residues important for the ability of the kinase to adopt the specific closed (inactive) conformation to which imatinib binds.61 Because of the frequency of mutations, efforts are now focused on the identification of novel inhibitors that are active against imatinib-resistant mutants of BCR-ABL.
In this report, we describe the development of a novel small-molecule inhibitor of JAK2 and BCR-ABL that appears to target regions outside the ATP-binding site of its target and offers the potential to be unaffected by mutations in the kinase domain that make tumor cells resistant to ATP-competitive inhibitors. The results presented in this study show that this novel pharmacophore,53 which inhibits recombinant JAK2 kinase (both wild-type and mutant forms) at a concentration of 0.9 to 1.2 µM, is noncompetitive with ATP (Fig. 2a). However, the addition of increasing concentrations of the substrate peptide to the reaction mixture results in an increase in the IC50 of the compound, suggesting that this could be a substrate-competitive inhibitor (Figs. 2b and c). It is interesting to note that both autophosphorylation and substrate phosphorylation are affected by increasing concentrations of the substrate peptide, suggesting that the binding of the compound to JAK2 might affect both these activities. Our studies also suggest that at least a portion of the regulatory JH2 domain is essential for the inhibitory activity of ON044580 (Fig. 1f).
ON044580 is also able to actively down regulate IL-3 signaling by inhibiting the phosphorylation of JAK2 and STAT5 proteins in cells harboring mutant JAK2 kinase within 30 minutes following the addition of the compound. This, in turn, appears to result in growth arrest and apoptosis of human and mouse leukemic cells expressing the JAK2V617F protein (Figs. 3f and h). Because both BCR-ABL and JAK2-V617F proteins confer cytokine-independent growth of mutant cells in vitro due to deregulation of signaling pathways downstream of JAK2 and share several of the substrates associated with cytokine signaling (such as STAT5), it was of interest to see if ON044580 exhibits any cross-reactivity with BCR-ABL kinase activity. Initially, we carried out experiments aimed at determining the ability of ON044580 to inhibit the kinase activity of recombinant BCR-ABL kinase produced in insect cells. Our results presented in Supplementary Figure S1 show that this compound failed to inhibit the kinase activity of recombinant BCR-ABL even at a concentration of 20 µM. It has been known that some of the non–ATP-competitive inhibitors of BCR-ABL do not exhibit in vitro kinase inhibitory activity with recombinant enzymes.57 An explanation for the lack of in vitro activity against recombinant protein could be the absence of posttranslational modifications that might affect the conformation of the protein as well as the binding of the inhibitor to the protein.
When we carried out the in vitro kinase inhibition assays with BCR-ABL protein immunoprecipitated from K562 cells or 32D:p210T315I cells, we could detect direct inhibition of both the wild-type and mutant T315I forms of BCR-ABL kinase by ON044580 (Figs. 4a and b). Notably, ON044580 exhibited a better inhibitory activity against the mutant kinase (IC50 = 2.08 µM) as compared to the wild type (IC50 = 7.94 µM). It is known that the T315I mutation confers imatinib resistance partly by phosphorylation of endogenous BCR, suggesting aberrant substrate activation by BCR/ABL harboring the T315I mutation.62 It is conceivable that ON044580, being a substrate competitive inhibitor, has an additional site to effect its activity against the T315I version of BCR-ABL as opposed to the wild-type protein, which does not display such aberrant substrate activation. Alternatively, it is possible that the conformation adapted by the mutant BCR-ABL protein allows a tighter binding of ON044580.
We also examined whether ON044580 inhibits the in vivo autophosphorylation and substrate (STAT5) phosphorylation of BCR-ABL kinase. These studies showed that incubation of K562 cells with ON044580 resulted in an inhibition of cellular phosphorylation of the BCR-ABL kinase in a dose-dependent manner. In 2 hours, autophosphorylation levels started to decrease at 0.3 µM and were undetectable at a concentration of 10 µM. In the same assay, imatinib exhibited similar inhibitory activity with respect to BCR-ABL autophosphorylation (Fig. 4c). Notably, such inhibition required lower concentrations of ON044580 compared to the in vitro inhibition of immunoprecipitated BCR-ABL (IC50 = 8 µM). This could be due to interference by the antibody used to immunoprecipitate the BCR-ABL protein or subtle changes that the protein undergoes during immunoprecipitation resulting in a poorer binding of the compound to the kinase.
Most interestingly, ON044580 could bring about a similar reduction in the autophosphorylation status of BCR-ABL-T315I kinase (Fig. 4d), suggesting that mutations in the kinase domain of BCR-ABL do not adversely affect the inhibitory activity of ON044580. As can be expected, in a similar assay, imatinib failed to affect the autophosphorylation status of BCR-ABL-T315I kinase even at a concentration of 100 µM. Interestingly, ON044580 could readily inhibit the phosphorylation status of STAT5 in cells expressing the WT or the T315I mutant forms of BCR-ABL but failed to inhibit CrkL phosphorylation. These results suggest that substrate-competitive inhibitors may not inhibit phosphorylation of all substrates and thus differ from ATP-competitive inhibitors such as imatinib. We also observed enhanced degradation of BCR-ABL-T315I kinase in ON044580-treated cells (Fig. 4d). A similar effect is seen on the levels of JAK2 and STAT5 in lysates from murine myeloid cells treated with 20 µM ON044580 (Figs. 3a and c). It has been previously shown that inhibition of JAK2 kinase activity in CML cells by AG490, its analogs, or JAK2-directed siRNA results in the destabilization of the large BCR-ABL-JAK2 multiprotein network leading to the degradation of BCR-ABL kinase.63-66 Our preliminary studies show that ON044580 brings about similar destabilization of the large BCR-ABL-JAK2 multiprotein complex (which contains STAT5) and could account for its degradation.
To further substantiate the usefulness of this compound in CML therapy, we examined the effects of this compound on freshly isolated human leukemic cells from patients who were refractory to imatinib. In this study, we used cells derived from three different patients, a blast crisis CML patient refractory to imatinib treatment (Fig. 5c), a CML patient in blast crisis (Fig. 5d), and a CML patient in chronic phase (Fig. 5e), for treatment with various doses of the drug. Our results show that the blast crisis and chronic phase cells were highly sensitive to apoptosis induction by ON044580, which suggests that this compound may be useful to treat the unusually resistant blast crisis patients.
The observation that ON044580 inhibits the wild-type JAK2 (Fig. 1b and c) led us to test its effect in U266 multiple myeloma cells that have constitutively activated IL-6 recpetor/JAK2/STAT-3 pathway. As would be expected, we found ON044580 inhibited the phosphorylation of STAT-3 at nanomolar concentrations (Fig. 3e). We further tested the usefulness of ON044580 in shutting down aberrant JAK/STAT signaling in bone marrow samples from monosomy 7 MDS patients. Monosomy 7 MDS is characterized by constitutive activation of JAK2 and STAT1. Treatment with ON044580 specifically decreased the monosomy 7 population whereas normal diploid cells continued to proliferate (Fig. 5b). These results along with the induction of apoptosis in cells expressing the activated V617F mutant form of JAK2 (Figs. 3f and h) suggest that ON044580 also holds promise for use as a therapeutic against both myelodysplastic syndromes and myeloproliferative neoplasms where JAK2 is aberrantly activated.
In the past few years, several cases have been reported in which both the BCR-ABL translocation and JAK2-V617F mutation have been observed concomitantly in bone marrow samples from CMPD patients.67-70 These studies have revealed that JAK2-V617F mutation-associated CMPD develops predominantly after selective treatment of Ph+ CML with imatinib. Furthermore, the emergence of the BCR-ABL translocation on the background of JAK2V617F CMPD seems to be unrelated to prior myelosuppressive treatment (standard treatment for CMPD). Finally, JAK2V617F mutation seems to precede the acquisition of the Philadelphia chromosome.71 Importantly, there has been recent evidence showing that the kinase activity of JAK2 is required for the stability of BCR-ABL protein and thus maintenance of the oncogenic signal. In this context, JAK2 has been shown to be activated in Bcr-Abl+ hematopoietic cells and CML cell lines.58,64,72,73 In these cell lines, activated JAK2 appears to be important for stimulating the PI-3-kinase/Akt pathway. Jak2 is also involved in elevating SET levels,58 which inhibits the PP2A/Shp1 pathway leading to the maintenance of high levels of pTyr Bcr-Abl.74
It is significant that ON044580 is the first reported dual JAK2/BCR-ABL inhibitor and holds the unique clinical promise to eliminate all clones that have both or either of these 2 genetic lesions. In addition, ON044580 can potentially reduce the outgrowth of JAK2V617F clones from Ph+ cells and prevent the development of resistance.
In conclusion, we have identified ON044580, a substrate-competitive kinase inhibitor active against two oncogenic kinases, BCR-ABL and activated JAK2 that appear to play a central role in CMPD. This inhibitor is active against mutant versions of these kinases and causes apoptotic cell death of imatinib-resistant CML clones on one hand while selectively inhibiting the proliferation of monosomy 7 MDS clones on the other. Being a dual JAK2/BCR-ABL inhibitor, ON044580 could possibly become the first in a class of inhibitors effective in treating a variety of myeloproliferative disorders such as CML, Ph-negative CMPD, and MDS typified by aberrant JAK/STAT signaling.
Materials and Methods
Kinase assays and IC50 determination
Ten nanograms of GST-JAK2 (aa 808-1132; Invitrogen PV4210), 200 ng of GST-JAK2-WT (532-1132; Invitrogen PV4393), 200 ng of GST-JAK2-V617F (532-1132; Invitrogen PV4336), or 100 ng affinity purified GST-p210-WT kinase (baculovirally expressed in Sf21 cells; Invitrogen B821-01) was diluted into kinase buffer (20 mM Tris pH 7.5, 10 mM MgCl2, 0.01% NP-40, 1 mM EGTA, 2 mM DTT) and incubated with the indicated concentration of inhibitor at room temperature for 30 minutes. The kinase reactions were initiated by the addition of 1 µg (1.5 µM) recombinant GST-Abltide (Upstate 12-525), 20 µM ATP, and 20 µCi γ-32P-ATP. The reactions were incubated at 30°C for 20 minutes, terminated by the addition of 2x Laemmli sample buffer, boiled for 2 minutes, resolved by 12% acrylamide SDS-PAGE, and subjected to autoradiography. For Src (Invitrogen P3044), LynB (Invitrogen P2907), Plk-1 (Invitrogen PV3501), and Cdk-1/cyclinB (Upstate 14-450) kinase assays, 10 ng of recombinant kinase was used with 1 µg GST-Sam68 (aa 331-443; Santa Cruz Biotechnologies sc-4249), dephosphorylated α-Casein (Sigma C8032), or Histone H1 (Roche Diagnostics 223549) as protein substrate, respectively. Full-length BCR-ABL (WT and T315I) and JAK2-V617F were immunoprecipitated from cultured cells and the immune-complexes processed as above to assay kinase activity. The autoradiograms were scanned, and the band corresponding to autophosphorylation of JAK2 or phosphorylation of GST-Abltide was quantitated using MacBas software. The densitometric values obtained were plotted as a function of log drug concentration using Prism 4 Graphpad software and IC50 values determined by plotting sigmoidal nonlinear regression curves with variable slope.
Cell culture
Ba/F3:JAK2V617F cells were maintained in RPMI medium 1640 supplemented with 10% FBS, 1 U/mL penicillin-streptomycin, and 1% WEHI-3B conditioned medium as a source of IL-3. 32D:p210WT and 32D:p210T315I cells were maintained in Iscove’s modified Dulbecco’s medium (IMDM) supplemented with 10% FBS, 1 U/mL penicillin-streptomycin. K562 (DSMZ no. ACC 10) cells were maintained in RPMI medium 1640 with 10% FBS and 1 U/mL penicillin-streptomycin. U266 (DSMZ no. ACC 9), HEL (DSMZ no. ACC 11), and SET-2 (DSMZ no. ACC 608) cells were maintained in RPMI medium 1640 containing 10%, 20%, and 10% heat-inactivated FBS, respectively. Patients’ samples were diluted with PBS in a 1:1 ratio and were separated by Histopaque-1077 (Sigma-Aldrich). After separation of the whole blood, the cells were washed with PBS, suspended in RPMI-1640 supplemented with 10% FBS, and treated with various doses of ON044580 for 48 hours. Subsequently, the cells were assayed for apoptosis by Annexin-V/IP method involving flow cytometry.
Western blot analysis and immunoprecipitation
Mid-log phase Ba/F3:JAK2V617F cells were treated for 0 to 120 minutes with indicated concentrations of inhibitor and 1 hour with 5 ng/mL recombinant mouse IL-3 (R&D Systems 403-ML). Washed cells were lysed in detergent containing buffer, and the 50 µg of the clarified lysates were resolved by SDS-PAGE and analyzed by Western blotting using anti-phopho-STAT5A/B (Tyr694/699) clone 8-5-2 (Upstate 05-495) and anti-phosho-JAK2 (Tyr1007/1008; Upstate 07-606) antibodies and the corresponding HRP-conjugated secondary antibodies (GE Healthcare). Subsequently, the blots were stripped and reprobed with anti-STAT5A/B (Upstate 06-588) and anti-JAK2 (Upstate 06-255) antibodies. In apoptosis studies, cells were treated with compounds (or DMSO) for 2, 6, and 24 hours. The harvested cells were washed in PBS and lysed as above. Fifty micrograms of clarified lysates resolved by SDS-PAGE were analyzed by Western blotting using anti-PARP antibody (Cell Signaling Technology 9542). For immunoprecipitation, cells grown to mid-log phase were stimulated with 5 ng/mL recombinant IL-3 for 1 hour, washed with PBS, lysed in detergent containing buffer (25 mM HEPES pH 7.5, 0.1% Triton X-100, 150 mM NaCl, 1 mM DTT, 1 mM EGTA, 1.5 mM MgCl2, 20 mM β-glycerophosphate, 0.2 mM Na3VO4, and 1x protease inhibitor mixture [Roche Diagnostics]), and 500 µg of the clarified lysate was incubated with 5 µL anti-JAK2 antibody and 25 µL (50% slurry) of Protein A Sepharose CL-4B beads (GE Healthcare 17-0780-01) for 2 hours at 4°C. The immune-complexes were washed 3 times in PBS and used for in vitro kinase assays or Western blot analysis. Exponentially growing K562 cells and 32D:p210T315I cells were treated for 2 hours with indicated concentrations of inhibitor. Cells were washed with PBS, and 50 µg of the clarified lysate was used for Western blot analysis. BCR-ABL was immunoprecipitated from 500 µg of the clarified lysates using 5 µL of anti-Bcr (N-20) antibody (Santa Cruz Biotechnologies sc-885) and processed as above. For kinase assays, untreated 32D:p210WT and 32D:p210T315I cells were used for immunoprecipitation. Western blot analysis was carried out with antiphosphotyrosine antibody (Santa Cruz Biotechnologies sc-885) to check the levels of p210 autophosphorylation. The blot was stripped and reprobed for levels of p210 protein with anti-Bcr (N-20) antibody. Similarly, anti-phospho-CrkL (Tyr207; Cell Signaling Technologies 3181S) and anti-CrkL (32H4; Cell Signaling Technology 182) antibodies were used to assay levels of CrkL phosphorylation.
Growth inhibition and GI50 determination
Ba/F3:JAK2V617F cells, HEL cells, SET-2 cells, K562 cells, and 32D:p210T315I cells were grown in the presence of varying concentrations of ON044580 for 72 hours. Cell viability was measured by Trypan blue exclusion. GI50 values were calculated by plotting percentage viable cells as a function of drug concentration using Prism 4 Graphpad software.
Short-term exposure wash-out experiments
Ba/F3:JAK2V617F cells, HEL cells, and K562 cells were treated with increasing concentrations of ON044580 or vehicle for 2 hours (in duplicate), washed 3 times in growth medium, and plated in 12-well dishes. The total number of viable cells was determined 96 hours later by Trypan blue exclusion. Bone marrow was harvested from femur and tibia of CD-1 mice and treated with increasing concentrations of ON044580 or vehicle (in duplicate) for 2 hours. The cells were washed 3 times in IMDM medium and cultured in methylcellulose medium supplemented with 50 ng/mL rmStem Cell Factor, 10 ng/mL rmIL-3, 10 ng/mL rhIL-6, 200 µg/mL human Transfeffin, and 3 U/mL rhErythropoietin (Stem Cell Technologies). Cultures were seeded in duplicates using 35-mm plastic petri dishes, and colony-forming units were determined after 1 week. Histograms of percentage viable cells/colony-forming units (compared with DMSO-treated controls) as a function of drug concentration were plotted using Excel software.
Treatment of primary cells from CML patients
Blood samples were received from 3 CML patients through our approved lab protocol. One was from a blast crisis CML patient refractory to imatinib treatment. The other was from a CML patient in blast crisis. The 3rd was a sample from a CML patient in chronic phase. White blood cells were maintained for 48 hours in the absence of cytokines, to enhance the level of BCR-ABL+ cells in the blood cell population. Cells were treated for an additional 48 hours with various doses of the inhibitor. Cells were assayed for apoptosis by the annexinV/PI method involving flow cytometry. Apoptotic cells accumulate in the lower right (quadrant 3) and upper right quadrants (quadrant 4).
Treatment of bone marrow samples from monosomy 7 MDS patients
BMMNCs were obtained from patients with monosomy 7 MDS confirmed by metaphase karyotyping and FISH after informed consent according to protocols approved by the Institutional Review Board of the National Heart, Lung, and Blood Institute. BMMNCs were aspirated from the posterior iliac crest into syringes containing RPMI media supplemented 1:10 with heparin (O’Neill and Feldman, St. Louis, MO) and prepared by density gradient centrifugation using lymphocyte separation medium (Organon, Durham, NC). Cryopreserved BMMNCs were grown in Mylocult media (Stem Cell Technologies) supplemented with 400 ng/mL G-CSF and growth factor cocktail as previously described.75
FISH studies
FISH studies on cryopreserved BMMNCs were performed as described previously75 using centromeric probes for chromosome 7 and 8 (Vysis, Downers Grove, IL). Percentage positive staining was based on counting of 400 cells. Three different observers, who were blinded with respect to sample identity, scored 3 different sets of slides. Scores were averaged, and the mean of the 3 was recorded. The total number of aneuploid and diploid cells was calculated by multiplying the percentage aneuploidy by the number of viable cells in each sample.
Supplementary Material
Acknowledgments
The authors thank Dr. Hajop Kantarjian, Dr. Jorge Cortes, and Dr. Xiaoping Sun for providing them with the patient material used in this study, and they thank Dr. Richard A. Van Etten for providing the Ba/F3:JAK2V617F cell line.
Footnotes
Dr. E.P. Reddy is a stockholder, board member, and consultant for Onconova Therapeutics, Inc. Dr. M.V. Ramana Reddy is a stockholder and consultant for Onconova Therapeutics, Inc. Drs. Stephen C. Consenza and Stacey J. Baker are consultants for Onconova Therapeutics, Inc. Drs. Jatiani, Ha, Samanta, Olnes, Pfannes, Sloand, and Arlinghaus declare no potential conflicts of interest.
This work was supported by grants from the Department of Defense (W81XWH-06-1-0267), the National Heart, Lung, and Blood Institute (HL080666), and Onconova Therapeutics, Inc.
Supplementary material for this article is available on the Genes & Cancer website at http://ganc.sagepub.com/supplemental.
References
- 1. Rane SG, Reddy EP. JAKs, STATs and Src kinases in hematopoiesis. Oncogene. 2002;21:3334-58 [DOI] [PubMed] [Google Scholar]
- 2. O’Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109:S121-31 [DOI] [PubMed] [Google Scholar]
- 3. Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93:385-95 [DOI] [PubMed] [Google Scholar]
- 4. Schwaller J, Frantsve J, Aster J, Williams IR, Tomasson MH, Ross TS, et al. Transformation of hematopoietic cell lines to growth-factor independence and induction of a fatal myelo- and lymphoproliferative disease in mice by retrovirally transduced TEL/JAK2 fusion genes. EMBO J. 1998;17:5321-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffé M, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278:1309-12 [DOI] [PubMed] [Google Scholar]
- 6. Peeters P, Raynaud SD, Cools J, Wlodarska I, Grosgeorge J, Philip P, et al. Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood. 1997;90:2535-40 [PubMed] [Google Scholar]
- 7. Aringer M, Cheng A, Nelson JW, Chen M, Sudarshan C, Zhou YJ, et al. Janus kinases and their role in growth and disease. Life Sci. 1999;64:2173-86 [DOI] [PubMed] [Google Scholar]
- 8. Reiter A, Walz C, Watmore A, Schoch C, Blau I, Schlegelberger B, et al. The t(8;9)(p22;p24) is a recurrent abnormality in chronic and acute leukemia that fuses PCM1 to JAK2. Cancer Res. 2005;65:2662-7 [DOI] [PubMed] [Google Scholar]
- 9. Griesinger F, Hennig H, Hillmer F, Podleschny M, Steffens R, Pies A, et al. A BCR-JAK2 fusion gene as the result of a t(9;22)(p24;q11.2) translocation in a patient with a clinically typical chronic myeloid leukemia. Genes Chromosomes Cancer. 2005;44:329-33 [DOI] [PubMed] [Google Scholar]
- 10. Cirmena G, Aliano S, Fugazza G, Bruzzone R, Garuti A, Bocciardi R, et al. A BCR-JAK2 fusion gene as the result of a t(9;22)(p24;q11) in a patient with acute myeloid leukemia. Cancer Genet Cytogenet. 2008;183:105-8 [DOI] [PubMed] [Google Scholar]
- 11. Murati A, Gelsi-Boyer V, Adélaïde J, Perot C, Talmant P, Giraudier S, et al. PCM1-JAK2 fusion in myeloproliferative disorders and acute erythroid leukemia with t(8;9) translocation. Leukemia. 2005;19:1692-6 [DOI] [PubMed] [Google Scholar]
- 12. Mark HF, Sotomayor EA, Nelson M, Chaves F, Sanger WG, Kaleem Z, et al. Chronic idiopathic myelofibrosis (CIMF) resulting from a unique 3;9 translocation disrupting the janus kinase 2 (JAK2) gene. Exp Mol Pathol. 2006;81:217-23 [DOI] [PubMed] [Google Scholar]
- 13. Najfeld V, Cozza A, Berkofsy-Fessler W, Prchal J, Scalise A. Numerical gain and structural rearrangements of JAK2, identified by FISH, characterize both JAK2617V>F-positive and -negative patients with Ph-negative MPD, myelodysplasia, and B-lymphoid neoplasms. Exp Hematol. 2007;35:1668-76 [DOI] [PubMed] [Google Scholar]
- 14. Poitras JL, Dal Cin P, Aster JC, Deangelo DJ, Morton CC. Novel SSBP2-JAK2 fusion gene resulting from a t(5;9)(q14.1;p24.1) in pre-B acute lymphocytic leukemia. Genes Chromosomes Cancer. 2008;47:884-9 [DOI] [PubMed] [Google Scholar]
- 15. Nebral K, Denk D, Attarbaschi A, König M, Mann G, Haas OA, et al. Incidence and diversity of PAX5 fusion genes in childhood acute lymphoblastic leukemia. Leukemia. 2009;23:134-43 [DOI] [PubMed] [Google Scholar]
- 16. Joos S, Küpper M, Ohl S, von Bonin F, Mechtersheimer G, Bentz M, et al. Genomic imbalances including amplification of the tyrosine kinase gene JAK2 in CD30+ Hodgkin cells. Cancer Res. 2000;60:549-52 [PubMed] [Google Scholar]
- 17. Shannon K, Van Etten RA. JAKing up hematopoietic proliferation. Cancer Cell. 2005;7:291-3 [DOI] [PubMed] [Google Scholar]
- 18. Szpurka H, Tiu R, Murugesan G, Aboudola S, Hsi ED, Theil KS, et al. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis (RARS-T), another myeloproliferative condition characterized by JAK2 V617F mutation. Blood. 2006;108:2173-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ma W, Kantarjian H, Zhang X, Yeh CH, Zhang ZJ, Verstovsek S, et al. Mutation profile of JAK2 transcripts in patients with chronic myeloproliferative neoplasias. J Mol Diagn. 2009;11:49-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bercovich D, Ganmore I, Scott LM, Wainreb G, Birger Y, Elimelech A, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet. 2008;372:1484-92 [DOI] [PubMed] [Google Scholar]
- 21. Kearney L, Gonzalez De Castro D, Yeung J, Procter J, Horsley SW, Eguchi-Ishimae M, et al. Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood. 2009;113:646-8 [DOI] [PubMed] [Google Scholar]
- 22. Mullighan CG, Zhang J, Harvey RC, Collins-Underwood JR, Schulman BA, Phillips LA, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2009;106:9414-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gaikwad A, Rye CL, Devidas M, Heerema NA, Carroll AJ, Izraeli S, et al. Prevalence and clinical correlates of JAK2 mutations in Down syndrome acute lymphoblastic leukaemia. Br J Haematol. 2009;144:930-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tono C, Xu G, Toki T, Takahashi Y, Sasaki S, Terui K, et al. JAK2 Val617Phe activating tyrosine kinase mutation in juvenile myelomonocytic leukemia. Leukemia. 2005;19:1843-4 [DOI] [PubMed] [Google Scholar]
- 25. Jelinek J, Oki Y, Gharibyan V, Bueso-Ramos C, Prchal JT, Verstovsek S, et al. JAK2 mutation 1849G>T is rare in acute leukemias but can be found in CMML, Philadelphia chromosome-negative CML, and megakaryocytic leukemia. Blood. 2005;106:3370-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Levine RL, Loriaux M, Huntly BJ, Loh ML, Beran M, Stoffregen E, et al. The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood. 2005;106:3377-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee JW, Kim YG, Soung YH, Han KJ, Kim SY, Rhim HS, et al. The JAK2 V617F mutation in de novo acute myelogenous leukemias. Oncogene. 2006;25:1434-6 [DOI] [PubMed] [Google Scholar]
- 28. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Cancer Genome Project. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054-61 [DOI] [PubMed] [Google Scholar]
- 29. James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144-8 [DOI] [PubMed] [Google Scholar]
- 30. Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779-90 [DOI] [PubMed] [Google Scholar]
- 31. Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387-97 [DOI] [PubMed] [Google Scholar]
- 32. Hellström-Lindberg E, Cazzola M. The role of JAK2 mutations in RARS and other MDS. Hematol Am Soc Hematol Educ Program. 2008:52-9 [DOI] [PubMed] [Google Scholar]
- 33. Remacha AF, Nomdedéu JF, Puget G, Estivill C, Sarda MP, Canals C, et al. Occurrence of the JAK2 V617F mutation in the WHO provisional entity: myelodysplastic/myeloproliferative disease, unclassifiable-refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Haematologica. 2006;91:719-20 [PubMed] [Google Scholar]
- 34. Lee TS, Ma W, Zhang X, Giles F, Kantarjian H, Albitar M. Mechanisms of constitutive activation of Janus kinase 2-V617F revealed at the atomic level through molecular dynamics simulations. Cancer. 2009;115:1692-700 [DOI] [PubMed] [Google Scholar]
- 35. Lu X, Huang LJ, Lodish HF. Dimerization by a cytokine receptor is necessary for constitutive activation of JAK2V617F. J Biol Chem. 2008;283:5258-66 [DOI] [PubMed] [Google Scholar]
- 36. Zhao L, Ma Y, Seemann J, Huang LJ. A regulating role of JAK2 FERM domain in hyperactivation of JAK2(V617F). Biochem J. 2010;426:91-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356:459-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen G, Zeng W, Miyazato A, Billings E, Maciejewski JP, Kajigaya S, et al. Distinctive gene expression profiles of CD34 cells from patients with myelodysplastic syndrome characterized by specific chromosomal abnormalities. Blood. 2004;104:4210-8 [DOI] [PubMed] [Google Scholar]
- 39. Sloand EM, Yong AS, Ramkissoon S, Solomou E, Bruno TC, Kim S, et al. Granulocyte colony-stimulating factor preferentially stimulates proliferation of monosomy 7 cells bearing the isoform IV receptor. Proc Natl Acad Sci U S A. 2006;103:14483-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Haase D, Germing U, Schanz J, Pfeilstöcker M, Nösslinger T, Hildebrandt B, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood. 2007;110:4385-95 [DOI] [PubMed] [Google Scholar]
- 41. Rosenfeld S, Follmann D, Nunez O, Young NS. Antithymocyte globulin and cyclosporine for severe aplastic anemia: association between hematologic response and long-term outcome. JAMA. 2003;289:1130-1135 [DOI] [PubMed] [Google Scholar]
- 42. Maciejewski JP, Risitano A, Sloand EM, Nunez O, Young NS. Distinct clinical outcomes for cytogenetic abnormalities evolving from aplastic anemia. Blood. 2002;99:3129-3135 [DOI] [PubMed] [Google Scholar]
- 43. Haase D. Cytogenetic features in myelodysplastic syndromes. Ann Hematol. 2008;87:515-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Touw IP, Dong F. Severe congenital neutropenia terminating in acute myeloid leukemia: disease progression associated with mutations in the granulocyte-colony stimulating factor receptor gene. Leuk Res. 1996;20:629-631 [DOI] [PubMed] [Google Scholar]
- 45. Hexner EO, Serdikoff C, Jan M, Swider CR, Robinson C, Yang S, et al. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood. 2008;111:5663-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lasho TL, Tefferi A, Hood JD, Verstovsek S, Gilliland DG, Pardanani A. TG101348, a JAK2-selective antagonist, inhibits primary hematopoietic cells derived from myeloproliferative disorder patients with JAK2V617F, MPLW515K or JAK2 exon 12 mutations as well as mutation negative patients. Leukemia. 2008;22:1790-2 [DOI] [PubMed] [Google Scholar]
- 47. Mesa RA, Tefferi A. Emerging drugs for the therapy of primary and post essential thrombocythemia, post polycythemia vera myelofibrosis. Expert Opin Emerg Drugs. 2009;14(3):471-9 [DOI] [PubMed] [Google Scholar]
- 48. Pardanani A, Lasho T, Smith G, Burns CJ, Fantino E, Tefferi A. CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia. 2009;23:1441-5 [DOI] [PubMed] [Google Scholar]
- 49. Mathur A, Mo JR, Kraus M, O’Hare E, Sinclair P, Young J, et al. An inhibitor of Janus kinase 2 prevents polycythemia in mice. Biochem Pharmacol. 2009;78:382-9 [DOI] [PubMed] [Google Scholar]
- 50. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561-6 [DOI] [PubMed] [Google Scholar]
- 51. Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399-401 [DOI] [PubMed] [Google Scholar]
- 52. Blum G, Gazit A, Levitzki A. Substrate competitive inhibitors of IGF-1 receptor kinase. Biochemistry. 2000;39:15705-12 [DOI] [PubMed] [Google Scholar]
- 53. Reddy MV, Pallela VR, Cosenza SC, Mallireddigari MR, Patti R, Bonagura M, et al. Design, synthesis and evaluation of (E)-alpha-benzylthio chalcones as novel inhibitors of BCR-ABL kinase. Bioorg Med Chem. 2010;18:2317-26 [DOI] [PubMed] [Google Scholar]
- 54. Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, Lapidot Z, et al. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature. 1996;379:645-8 [DOI] [PubMed] [Google Scholar]
- 55. Pedranzini L, Dechow T, Berishaj M, Comenzo R, Zhou P, Azare J, et al. Pyridone 6, a pan-Janus-activated kinase inhibitor, induces growth inhibition of multiple myeloma cells. Cancer Res. 2006;66:9714-21 [DOI] [PubMed] [Google Scholar]
- 56. Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346-7 [DOI] [PubMed] [Google Scholar]
- 57. Adrián FJ, Ding Q, Sim T, Velentza A, Sloan C, Liu Y, et al. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat Chem Biol. 2006;2:95-102 [DOI] [PubMed] [Google Scholar]
- 58. Samanta AK, Chakraborty SN, Wang Y, Kantarjian H, Sun X, Hood J, et al. Jak2 inhibition deactivates Lyn kinase through the SET-PP2A-SHP1 pathway, causing apoptosis in drug-resistant cells from chronic myelogenous leukemia patients. Oncogene. 2009;28:1669-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Delhommeau F, Pisani DF, James C, Casadevall N, Constantinescu S, Vainchenker W. Oncogenic mechanisms in myeloproliferative disorders. Cell Mol Life Sci. 2006;63:2939-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tefferi A, Gilliland DG. Oncogenes in myeloproliferative disorders. Cell Cycle. 2007;6:550-66 [DOI] [PubMed] [Google Scholar]
- 61. Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105:2640-53 [DOI] [PubMed] [Google Scholar]
- 62. Mian AA, Schüll M, Zhao Z, Oancea C, Hundertmark A, Beissert T, et al. The gatekeeper mutation T315I confers resistance against small molecules by increasing or restoring the ABL-kinase activity accompanied by aberrant transphosphorylation of endogenous BCR, even in loss-of-function mutants of BCR/ABL. Leukemia. 2009;23:1614-21 [DOI] [PubMed] [Google Scholar]
- 63. Xie S, Wang Y, Liu J, Sun T, Wilson MB, Smithgall TE, et al. Involvement of Jak2 tyrosine phosphorylation in Bcr-Abl transformation. Oncogene. 2001;20:6188-95 [DOI] [PubMed] [Google Scholar]
- 64. Xie S, Lin H, Sun T, Arlinghaus RB. Jak2 is involved in c-Myc induction by Bcr-Abl. Oncogene. 2002;21:7137-46 [DOI] [PubMed] [Google Scholar]
- 65. Samanta AK, Lin H, Sun T, Kantarjian H, Arlinghaus RB. Janus kinase 2: a critical target in chronic myelogenous leukemia. Cancer Res. 2006;66:6468-72 [DOI] [PubMed] [Google Scholar]
- 66. Ferrajoli A, Faderl S, Van Q, Koch P, Harris D, Liu Z, et al. WP1066 disrupts Janus kinase-2 and induces caspase-dependent apoptosis in acute myelogenous leukemia cells. Cancer Res. 2007;67:11291-9 [DOI] [PubMed] [Google Scholar]
- 67. Jallades L, Hayette S, Tigaud I, Johnston A, Coiffier B, Magaud J-P, et al. Emergence of therapy-unrelated CML on a background of BCRABL-negative JAK2 V617F-positive chronic idiopathic myelofibrosis. Leuk Res. 2008;32:1608-10 [DOI] [PubMed] [Google Scholar]
- 68. Krämer A, Reiter A, Kruth J, Erben P, Hochhaus A, Müller M, et al. JAK2-V617F mutation in a patient with Philadelphia-chromosomepositive chronic myeloid leukaemia. Lancet Oncol. 2007;8:658-60 [DOI] [PubMed] [Google Scholar]
- 69. Inami M, Inokuchi K, Okabe M, Kosaka F, Mitamura Y, Yamaguchi H, et al. Polycythemia associated with the JAK2V617F mutation emerged during treatment of chronic myelogenous leukemia. Leukemia. 2007;21:1103-4 [DOI] [PubMed] [Google Scholar]
- 70. Hussein K, Bock O, Seegers A, Flasshove M, Henneke F, Buesche G, et al. Myelofibrosis evolving during imatinib treatment of a chronic myeloproliferative disease with coexisting BCR-ABLtranslocation and JAK2V617F mutation. Blood. 2007;109:4106-7 [DOI] [PubMed] [Google Scholar]
- 71. Krämer A. JAK2-V617F and BCR-ABL—double jeopardy? Leuk Res. 2008;32:1489-90 [DOI] [PubMed] [Google Scholar]
- 72. Xie S, Wang Y, Liu J, Sun T, Wilson MB, Smithgall TE, et al. Involvement of Jak2 tyrosine phosphorylation in Bcr-Abl transformation. Oncogene. 2001;20:6188-95 [DOI] [PubMed] [Google Scholar]
- 73. Wilson-Rawls J, Liu J, Laneuville P, Arlinghaus RB. P210 Bcr-Abl interacts with the interleukin-3 beta c subunit and constitutively activates Jak2. Leukemia. 1997;Suppl 3:428-31 [PubMed] [Google Scholar]
- 74. Neviani P, Santhanam R, Trotta R, Notari M, Blaser BW, Liu S, et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell. 2005;8:355-68 [DOI] [PubMed] [Google Scholar]
- 75. Sloand EM, Kim S, Fuhrer M, Risitano AM, Nakamura R, Maciejewski JP, et al. Fas-mediated apoptosis is important in regulating cell replication and death in trisomy 8 hematopoietic cells but not in cells with other cytogenetic abnormalities. Blood. 2002;100:4427-32 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.