

Abstract
Ischaemic postconditioning (IPOC) is an intervention in which brief, intermittent periods of reocclusion at the onset of reperfusion (i.e. stuttering reperfusion) protect myocardium from lethal reperfusion injury. The mechanism underlying the cardioprotective effects of IPOC is incompletely understood. However, it is perceived that IPOC begins with specific cell-surface receptors responsible for activating a number of signalling pathways, many of which appear to converge at the mitochondrial level. IPOC has been demonstrated both in animal models and in patients with acute myocardial infarction (AMI) in small proof-of-concept trials. This intervention offers the possibility of further limiting infarct size in patients undergoing primary percutaneous coronary intervention (PCI). Here, we provide a brief overview of the concept of IPOC and the mechanisms underlying this phenomenon. (Neth Heart J 2010;18:389–93.)
Keywords: Myocardial, Myocardial Ischemia, Myocardial Reperfusion Injury, Signal Transduction
In patients presenting with acute myocardial infarction (AMI), infarct size can be limited by early myocardial reperfusion via percutaneous coronary intervention (PCI), thereby preserving left ventricular systolic function and improving clinical outcome. However, restoring blood flow after a prolonged ischaemic episode may also paradoxically cause irreversible damage to the myocardium. This so-called reperfusion injury leads to the death of cardiomyocytes that are still viable at the end of ischaemia and may account for up to 50% of the final infarct size in animal models.1 Although the experimental data regarding reperfusion injury limiting strategies are promising, translation into clear clinical benefit has proven difficult.2 Nevertheless, cardioprotective strategies that limit reperfusion injury and hence further enhance the benefit of reperfusion are of major clinical interest and are needed to improve the efficacy of primary PCI.
Shift from pre- to postconditioning
In 1986, Murry and colleagues demonstrated that four episodes of five-minute occlusions of the coronary artery, interspersed with five-minute episodes of reperfusion applied immediately prior to prolonged ischaemia, reduced myocardial infarct size by 75% in a canine heart.3 They termed this phenomenon ischaemic preconditioning (IPC), in which the mechanism involves the activation of a complex set of signalling cascades.1 Unfortunately, IPC has to be applied before the onset of ischaemia which has restricted its clinical application to elective procedures, in particular cardiac surgery, in which the ischaemic episode can be anticipated. Importantly, however, a few years ago it was shown that a similar intervention applied at the time of reperfusion, which was termed ischaemic postconditioning (IPOC), could also limit ischaemia-reperfusion injury.4 Thus, interrupting myocardial reperfusion, starting 30 seconds after reperfusion, with three episodes of 30-sec coronary artery reocclusions reduced infarct size in canine hearts by 44%. This concept provided an intervention that could be applied at the time of myocardial reperfusion for patients presenting with an AMI and sparked the interest of clinical investigators.
Indeed, within two years of the study by Zhao et al.,4 the first clinical studies of IPOC in small groups of patients with AMI were published. In these studies, it was reported that interrupting reperfusion (with four cycles of 1-min low pressure inflations and deflations of the angioplasty balloon immediately after direct stenting of the infarct-related artery) improved myocardial reperfusion, reduced myocardial infarct size both acutely and at six months, and improved left ventricular ejection fraction up to 7% (percent points) at one year.5-7 Table 1 gives an overview of the clinical trials to date using IPOC in AMI. Although the results of these trials using surrogate endpoints (including ejection fraction, enzyme release, etc.) are encouraging, the effect of IPOC on clinical outcome remains to be determined.
Table 1.
Overview of clinical studies investigating ischaemic postconditioning in acute myocardial infarction.
| Study | Year | IPOC/control(n) | Ischaemia time(hours) | IPOC protocol* | Results | |
|---|---|---|---|---|---|---|
| Enzymatic infarct size | Functional parameters | |||||
| Laskey et al. | 2005 | 10/7 | 5.7 | 2 x 90 s | No difference on peak CK | Improved ST-segment resolutionImproved CFVR |
| Staat et al.6 | 2005 | 14/16 | 5.3 | 4 x 60 s | 36% Reduction 72 h CK | Improved ST-segment resolutionImproved MBG |
| Ma et al.20 | 2006 | 47/47 | 6.6 | 3 x 30 s | Decreased peak CK and CK-MB (p=NS) | Improved WMSI and endothelial functionDecreased MDA |
| Yang et al.21 | 2007 | 23/18 | 5.2 | 3 x 30 s | 27% Reduction 72 h CK | 27% reduction IS SPECT 1 weekImproved LVEF 44-54% 1 week (p=NS) |
| Thibault et al.7 | 2008 | 17/21 | 4.7 | 4 x 60 s | 40% Reduction CK, 47% Reduction TnI | 39% IS Reduction SPECT 6 monthsImproved LVEF 49-56% 1 yearImproved WMSI |
| Laskey et al.22 | 2008 | 12/12 | 3.8 | 2 x 90 s | Decreased peak CK | Improved ST-segment resolutionImproved CFVR |
| Zhao et al.23 | 2009 | 24 and 25/26 | 5.8/4.8 | 3 x 30 s/3 x 60 s | Reduced TnI 1 week (60s>30s>routine) | Improved LVEF 1 week (60s>30s>routine)Decreased apoptosis |
| Lønborg et al.24 | 2010 | 59/59 | 4.0 | 4 x 30 s | Decreased peak TnT (p=NS) | 19% Reduction IS MRI 3 monthsImproved NYHA class 3 months |
* IPOC protocol: number of balloon inflations x inflation duration. CK=creatine kinase, CK-MB=creatine kinase isoenyzme MB, CFVR=coronary flow velocity reserve, IS=infarct size, LVEF=left ventricular ejection fraction, MBG=myocardial blush grade, MDA=malondialdehyde, MRI=magnetic resonance imaging, NYHA = New York Heart Association, SPECT=single photon emission computed tomography, TnI=troponin I, TnT=troponin T, WMSI=wall motion score index.
Mechanisms underlying ischaemic postconditioning
When the concept of IPOC was first described, the mechanism of protection was initially attributed to reduction of oxidative stress, decreasing intracellular calcium overload, improving endothelial function and reducing inflammation.4 Since then, mechanisms underlying the cardioprotective effects of IPOC have been the subject of intense investigation, including inhibition of mitochondrial permeability transition pore (mPTP) opening during the first minutes of reperfusion.1 This crucial inhibitory event is mediated by activation of survival signalling pathways (reperfusion injury salvage kinases, RISKs), which in turn are triggered by the activation of specific cell-surface receptors. A detailed overview is shown in figure 1 and is briefly described below.
Figure 1.
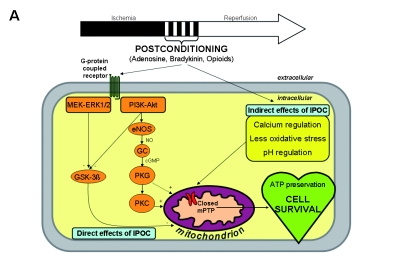
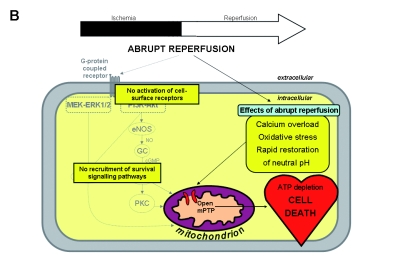
A) Overview of the potential mechanisms linking ischaemic postconditioning (IPOC) to the mitochondrial permeability transition pore (mPTP). These can be divided into direct and indirect effects of IPOC. The direct effects start with a number of autocoids including adenosine, bradykinin and opioids accumulating in the postconditioned myocardium and acting on their cell-surface receptors. The activation of cell-surface receptors recruits signalling cascades including the Akt and extracellular signal-regulated kinase (ERK1/2) components of the reperfusion injury salvage kinase (RISK) pathway. Activated Akt phosphorylates multiple substrates, including endothelial nitric oxide synthase (eNOS) and glycogen synthase kinase 3β (GSK-3β). Phosphorylation of eNOS activates the protein kinase G (PKG) through elevated guanosine 3’,5’-cyclic monophosphate (cGMP), thereby recruiting protein kinase C (PKC) and possibly inhibiting the mPTP. The ERK1/2 pathway may be crucial in inactivation of GSK-3β (through phosphorylation) and thereby preventing mPTP opening, an effect that may also be activated by Akt. The indirect effects of IPOC include calcium regulation, attenuation of oxidative stress and delaying pH correction at reperfusion. Ultimately, mPTP opening during early reperfusion is inhibited by these various effects, leading to ATP preservation and cell survival. B) Abrupt restoration of the blood flow to the ischaemic myocardium can cause irreversible damage through effects such as calcium overload, oxidative stress and a rapid restoration of neutral pH, among others, which are factors known to induce mPTP opening at the time of reperfusion. Ultimately, mPTP opening during early reperfusion is induced by these various effects, resulting in ATP depletion and cell death. GC=guanylyl cyclase, MAPK = mitogen-activated protein kinase, NO=nitric oxide, PI3K=phosphoinositide 3 kinase.
Cell-surface receptor activation
A number of experimental studies have linked the activation of cell-surface receptors at the time of reperfusion with protection by IPOC. Similar to IPC, the IPOC stimulus also appears to result in the activation of specifically G protein-coupled receptors (GPCRs) by endogenous ligands, such as adenosine, opioids, and bradykinin, providing the initial trigger for recruitment of survival signalling pathways.8 The adenosine receptor was the first GPCR to be linked with IPOC. Thus, the reduction in myocardial infarct size elicited by IPOC was abolished in the presence of 8-p-(sulphophenyl) theophylline, a non-specific adenosine receptor blocker.9 A subsequent study showed that the IPOC stimulus delayed the washout of adenosine generated during ischaemia-reperfusion in mouse hearts, likely resulting in greater activation of adenosine receptors during reperfusion.10
Survival signalling pathways
A number of different signalling pathways have been reported to mediate the protective effects of IPOC, most notably the RISK pathway.11 The RISK pathway describes a group of survival protein kinases (ERK1/2 and PI3K/Akt), which when activated at the time of reperfusion confer powerful cardioprotection. Pharmacological agents, including growth factors, GPCR agonists, cytokines, natriuretic peptide, adipocytokines and statins, administered at the immediate onset of reperfusion activate either ERK1/2 or PI3K/Akt and accordingly reduce myocardial infarct size (pharmacological postconditioning) in animal models of AMI.11 Tolerance to one stimulus does not necessarily rule out the effectiveness of another pharmacological postconditioning agent, as described for IPC.12 In recent years, the activation of these kinases has been linked to the inhibition of mPTP opening.13 Although the exact mechanism through which mPTP inhibition is mediated remains incompletely understood, it might be related to (antiapoptotic) downstream signalling components of the RISK pathway, such as glycogen synthase kinase 3β (GSK-3β), a protein kinase linked to the regulation of glycogen metabolism and cellular survival.13
Mitochondria as the end effectors
In the past years, mPTP inhibition has emerged as a potential convergence point for many cardioprotective signalling pathways and a final common end-effector for IPC and IPOC. Additive effects of both interventions are probably not operative, since IPOC does not afford additional protection in preconditioned hearts.14 A variety of different mechanisms may provide the link between IPOC and mPTP inhibition. These include indirect beneficial effects of IPOC on factors which influence mPTP opening such as calcium regulation, oxidative stress, ATP levels and pH regulation and a direct mPTP inhibiting effect depending on signal transduction pathways and phosphorylation and activation of protein kinases or other signalling components.13 The mPTP is a non-selective pore localised in the inner mitochondrial membrane. Opening of this pore, in response to the abrupt reperfusion of ischaemic myocardium, allows transfer of small molecules into the matrix of the mitochondria leading to the collapse of the mitochondrial membrane potential and uncoupling of oxidative phosphorylation, resulting in ATP depletion, mitochondrial swelling and cell death.15,16 Argaud et al. were the first to link IPOC with the inhibition of mPTP opening, by demonstrating increased calcium retention in mitochondria isolated from postconditioned in vivo rabbit hearts compared with control hearts.17 Furthermore, mice lacking mitochondrial cyclophilin D, a presumed regulatory component of the mPTP, are resistant to cardioprotective effects of both IPC and IPOC,18 confirming the regulatory role of mPTP. More recently, it has been shown that administration of a single bolus of cyclosporin A, an inhibitor of mPTP opening, at the time of reperfusion was associated with a smaller infarct size in patients with AMI.19
Conclusion
Cardioprotective strategies that limit lethal reperfusion injury in patients with AMI are of major clinical interest in order to further enhance the beneficial effects of early reperfusion. In this regard, IPOC represents a novel treatment strategy for reducing lethal reperfusion injury and further limiting myocardial infarct size in those patients undergoing primary PCI. IPOC constitutes a form of modified myocardial reperfusion which affords cardioprotection by recruitment of pro-survival signalling pathways that appear to converge on the mitochondria. Although the beneficial effects of IPOC have been shown in small clinical studies, large multicentre trials are needed to determine whether these cardioprotective effects should be pursued in the standard treatment of AMI.
References
- 1.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121-35. [DOI] [PubMed] [Google Scholar]
- 2.Dirksen MT, Laarman GJ, Simoons ML, Duncker DJ. Reperfusion injury in humans: a review of clinical trials on reperfusion injury inhibitory strategies. Cardiovasc Res. 2007;74:343-55. [DOI] [PubMed] [Google Scholar]
- 3.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124-36. [DOI] [PubMed] [Google Scholar]
- 4.Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579-88. [DOI] [PubMed] [Google Scholar]
- 5.Laskey WK. Brief repetitive balloon occlusions enhance reperfusion during percutaneous coronary intervention for acute myocardial infarction: a pilot study. Catheter Cardiovasc Interv. 2005;65:361-7. [DOI] [PubMed] [Google Scholar]
- 6.Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L'Huillier I, et al. Postconditioning the human heart. Circulation. 2005;112:2143-8. [DOI] [PubMed] [Google Scholar]
- 7.Thibault H, Piot C, Staat P, Bontemps L, Sportouch C, Rioufol G, et al. Long-term benefit of postconditioning. Circulation. 2008;117:1037-44. [DOI] [PubMed] [Google Scholar]
- 8.Vinten-Johansen J. Postconditioning: a mechanical maneuver that triggers biological and molecular cardioprotective responses to reperfusion. Heart Fail Rev. 2007;12:235-44. [DOI] [PubMed] [Google Scholar]
- 9.Yang XM, Philipp S, Downey JM, Cohen MV. Postconditioning's protection is not dependent on circulating blood factors or cells but involves adenosine receptors and requires PI3-kinase and guanylyl cyclase activation. Basic Res Cardiol. 2005;100:57-63. [DOI] [PubMed] [Google Scholar]
- 10.Kin H, Zatta AJ, Lofye MT, Amerson BS, Halkos ME, Kerendi F, et al. Postconditioning reduces infarct size via adenosine receptor activation by endogenous adenosine. Cardiovasc Res. 2005;67:124-33. [DOI] [PubMed] [Google Scholar]
- 11.Hausenloy DJ, Yellon DM. Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection. Heart Fail Rev. 2007;12:217-34. [DOI] [PubMed] [Google Scholar]
- 12.Liem DA, te Lintel Hekkert M, Manintveld OC, Boomsma F, Verdouw PD, Duncker DJ. Myocardium tolerant to an adenosine-dependent ischemic preconditioning stimulus can still be protected by stimuli that employ alternative signaling pathways. Am J Physiol Heart Circ Physiol. 2005;288:H1165-72. [DOI] [PubMed] [Google Scholar]
- 13.Hausenloy DJ, Ong SB, Yellon DM. The mitochondrial permeability transition pore as a target for preconditioning and postconditioning. Basic Res Cardiol. 2009;104:189-202. [DOI] [PubMed] [Google Scholar]
- 14.Manintveld OC, Hekkert ML, van der Ploeg NT, Verdouw PD, Duncker DJ. Interaction between pre- and postconditioning in the in vivo rat heart. Exp Biol Med (Maywood). 2009;234:1345-54. [DOI] [PubMed] [Google Scholar]
- 15.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658-62. [DOI] [PubMed] [Google Scholar]
- 16.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652-8. [DOI] [PubMed] [Google Scholar]
- 17.Argaud L, Gateau-Roesch O, Raisky O, Loufouat J, Robert D, Ovize M. Postconditioning inhibits mitochondrial permeability transition. Circulation. 2005;111:194-7. [DOI] [PubMed] [Google Scholar]
- 18.Lim SY, Davidson SM, Hausenloy DJ, Yellon DM. Preconditioning and postconditioning: the essential role of the mitochondrial permeability transition pore. Cardiovasc Res. 2007;75:530-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473-81. [DOI] [PubMed] [Google Scholar]
- 20.Ma X, Zhang X, Li C, Luo M. Effect of postconditioning on coronary blood flow velocity and endothelial function and LV recovery after myocardial infarction. J Interv Cardiol. 2006;19:367-75. [DOI] [PubMed] [Google Scholar]
- 21.Yang XC, Liu Y, Wang LF, Cui L, Wang T, Ge YG, et al. Reduction in myocardial infarct size by postconditioning in patients after percutaneous coronary intervention. J Invasive Cardiol. 2007;19:424-30. [PubMed] [Google Scholar]
- 22.Laskey WK, Yoon S, Calzada N, Ricciardi MJ. Concordant improvements in coronary flow reserve and ST-segment resolution during percutaneous coronary intervention for acute myocardial infarction: a benefit of postconditioning. Catheter Cardiovasc Interv. 2008;72:212-20. [DOI] [PubMed] [Google Scholar]
- 23.Zhao WS, Xu L, Wang LF, Zhang L, Zhang ZY, Liu Y, et al. A 60-s postconditioning protocol by percutaneous coronary intervention inhibits myocardial apoptosis in patients with acute myocardial infarction. Apoptosis. 2009;14:1204-11. [DOI] [PubMed] [Google Scholar]
- 24.Lonborg J, Kelbaek H, Vejlstrup N, Jorgensen E, Helqvist S, Saunamaki K, et al. Cardioprotective effects of ischemic postconditioning in patients treated with primary percutaneous coronary intervention, evaluated by magnetic resonance. Circ Cardiovasc Interv. 2010;3:34-41. [DOI] [PubMed] [Google Scholar]