

Abstract
A series of new substituted 7-phenyl-3H-pyrrolo[3,2-f]quinolin-9-ones were synthesized and evaluated for their antiproliferative activity. The most active derivatives showed high selectivity against human leukemia cell lines and potently inhibited their growth, with GI50 values in the nanomolar range. The active compounds strongly blocked tubulin assembly and colchicine binding to tubulin. Their activities were equal to or greater than that of the reference compound combretastatin A-4. Flow cytometry studies showed that the two most active compounds arrested Jurkat cells in the G2/M cell-cycle phase in a concentration-dependent manner. This effect was associated with apoptosis, mitochondrial depolarization, generation of reactive oxygen species, activation of caspase-3, and cleavage of the enzyme poly(ADP-ribose) polymerase.
Keywords: anticancer agents, antiproliferative activity, apoptosis, pyrroloquinolinones, tubulin
Introduction
For several years we have studied the pyrroloquinoline nucleus as a promising scaffold for antiproliferative agents,[1–3] leading to the discovery of the 3H-pyrrolo[3,2-f]quinoline (PyQ) scaffold (Figure 1) as the most promising framework for the design of potent anticancer compounds. In particular, we found several active agents with N-pyrrole alkyl substituents that are derivatives of 7-phenyl-3H-pyrrolo[3,2-f]quinolin-9-ones (7-PPyQs).[3] We showed that 7-PPyQs exhibit strong cytotoxic activity against a wide panel of human tumor cell lines, including multi-drug resistance (MDR)-positive cancer cells. Moreover, in vivo, the 7-PPyQs exhibit significant inhibition of tumor growth (83 %) in a syngenic hepatocellular carcinoma model in Balb/c mice.[2]
Figure 1.
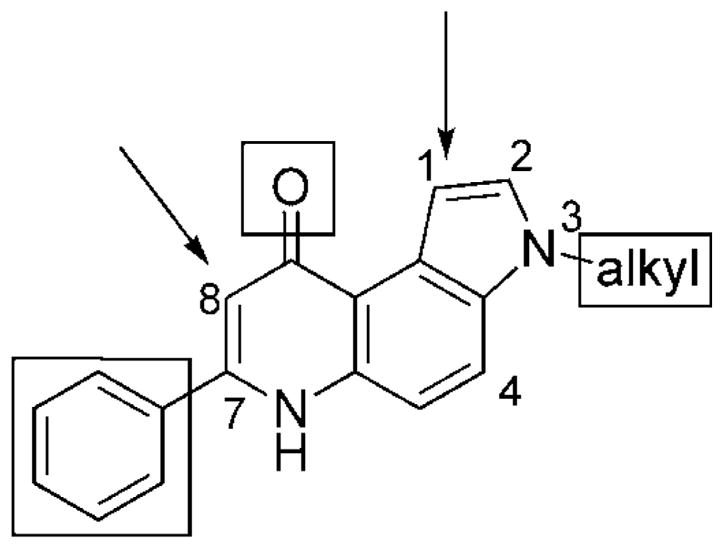
3H-Pyrrolo[3,2-f]quinolin-9(6H)-ones; SARs are described in the text.
Structure–activity relationship (SAR) studies of various pyrroloquinoline derivatives allowed us to establish some structural features that are crucial for antitumor activity. First, the [3,2-f] angular geometry is essential for significant activity, as is the carbonyl group at the 9-position and the side phenyl ring. Moreover, optimal activity requires the presence of an N-alkyl group on the pyrrole ring. Although active compounds were shown to have substituents ranging from ethyl to n-pentyl groups as well as a cyclopropylmethyl group, ionizable groups such as dimethylaminoethyl at position N3 or oxygen-containing substituents at positions C2 and C4 caused severe loss of activity. Because previously synthesized compounds[3] had caused cells to arrest in the G2/M phase of the cell cycle, we suspected that these compounds interact with tubulin, possibly at the colchicine site. Thus, the SAR observations suggested the presence of a deep hydrophobic pocket near the colchicine site.[3] Substituents on the phenyl ring at C7, either electron-withdrawing or -donating groups, had only minor effects on activity.
In an effort to produce additional highly active analogues, we synthesized and evaluated the effects of substitutions at other positions of the 3H-pyrrolo[3,2-f]quinolin-9-one nucleus. Herein we describe 3H-PyQ derivatives with small substituents at the C1 position and with the side phenyl ring shifted from C7 to C8. In addition to synthesizing the simple 8-phenyl-PyQ 33, substituents were also introduced onto template 33 to examine the effects on the bioactivity of the resulting compounds. We also focused on increasing water solubility relative to previous analogues by inserting suitable substituents at the 1-, 3-, and 7-positions.[3] Such compounds should be valuable for in vivo studies on pharmacokinetics, metabolism, and toxicity.
The newly prepared agents were assayed for their in vitro cytotoxicity against a panel of human solid tumors and leukemic cell lines. The most active compounds were confirmed as inhibitors of tubulin assembly through colchicine site binding, and were found to have major effects on cell-cycle distribution and induction of apoptosis.
Results and Discussion
Chemistry
The new PPyQs were synthesized as shown in Schemes 1–3, generally beginning with an appropriate 5-nitroindole derivative.[1–3] Scheme 1 shows the synthesis of 3,7-disubstituted PPyQs 11–13 by starting from commercial 5-nitroindole, which was first N-alkylated on the pyrrole ring to furnish derivatives 2–4.[3] The 5-nitroindole was alkylated by ethyl acrylate, trime-thoxybenzyl bromide or bromoethane in the presence of sodium hydride in N,N-dimethylformamide (DMF). Intermediates 2–4 were catalytically reduced (Pd/C 10 %, H2, ethanol) to amines 5–7.[3] These, in turn, were condensed with ethyl benzoylacetate or ethyl 3-oxo-3-(pyridin-3-yl)propanoate (1). We prepared the latter by modifying a published procedure[4] that furnished the β-ketoester 1 in good yield. This compound is present in solution as an equilibrium mixture of the diketo 1 a and enolic 1 b forms, as shown by the 1H NMR spectrum ([D6]DMSO). This showed two singlets at δ 6.08 (CH) and 12.52 (OH), consistent with an enolic form of the compound. The acrylate derivatives 8–10 were subjected to thermal cyclization (diphenyl ether, 250 °C) to yield compounds 11–13.
Scheme 1.

Synthesis of pyrroloquinolinone derivatives 11–13. Reagents and conditions: a) tBuOK, benzene, 80 °C, 6 h; b) NaH, ethyl acrilate or trimethoxybenzyl chloride, DMF, 70 °C, 4 h and 24–48 h, respectively; c) H2, Pd/C (10 %), EtOH, room temperature, 3 h; d) ethyl benzoylacetate or 1 a, EtOH, Drierite, reflux, 24 h; e) Ph2O, reflux, 30 min.
Scheme 3.

Synthesis of 8-phenylpyrroloquinolinone derivatives 33–37. Reagents and conditions: a) NaH, ethyl formate, room temperature, 24 h; b) EtOH, room temperature, 10–15 h; c) Ph2O, reflux, 15 min.
Scheme 2 illustrates the synthesis of 1,3-disubstitued 7-PPyQs 17, 21, and 22. Starting from the known 5-nitroindole 4,[3] carbaldehyde 14 was prepared by holding at reflux in DMF and phosphoryl chloride. Compound 14 was catalytically reduced (Pd/C 10 %) to 1-methyl-5-aminoindole 15, which then underwent condensation to yield 16. The final cyclization reaction, as in Scheme 1, yielded 1-methyl-3-ethyl-7-PPyQ 17. Carbaldehyde 14 was also transformed into the corresponding 1-carbonitrile derivative 18 in a one-pot reaction with hydroxyl-amine hydrochloride, pyridine, selenium dioxide, and magnesium sulfate,[5] followed by catalytic reduction to yield amine 19. Compound 19 was condensed with ethyl benzoylacetate to yield compound 20, which was thermally cyclized to 1-cyano-3-ethyl-7-PPyQ (21). The latter was transformed into 1-carboxy-amide-7-PPyQ 22 by treatment with a 1:1:1 mixture of acetic acid/sulfuric acid/water.
Scheme 2.

Synthesis of 7-phenylpyrroloquinolinone derivatives 17, 21, and 22. Reagents and conditions: a) POCl3, DMF, reflux, 3–5 h; b) and f) H2, Pd/C (10 %), EtOAc, 40 °C, 10–24 h; c) and g) ethyl benzoylacetate, EtOH, Drierite, reflux, 24 h; d) and h) Ph2O, reflux, 30 min; e) NH2OH·HCl, pyridine, 80 °C, 2 h, SeO2, Na2SO4, DMF, 80 °C, 2.5 h; i) CH3COOH/H2SO4, H2O, reflux, 16 h.
The synthetic route to 8-PPyQs 33–37 is similar to that previously reported for the synthesis of 3-phenylquinolinones (Scheme 3).[6] Commercial ethyl phenylacetates (R2 = H, m-methoxy, or p-bromo) were formylated with ethyl formate in the presence of sodium hydride[7] to afford intermediates 23[6]–25. These were condensed with 5-aminoindoles 7,[2] 26,[3] and 27[3] in ethanol at room temperature to yield enamine compounds 28–32. The crude products consisted of a mixture of the Z and E isomers, as observed by thin-layer chromatography and 1H NMR analysis. These compounds were not separated but underwent thermal cyclization (diphenyl ether, 250 °C) to 8-PPyQs 33–37.
Biological evaluation
In vitro antiproliferative activities
These new compounds were evaluated for their in vitro anti-proliferative activity against a panel of human tumor cell lines, including both solid and leukemic lines. The previously described[3] 3-ethyl-7-PPyQ (38) was used as a reference compound (Figure 2). After continuous drug exposure for 72 h, the compound concentration required for 50 % growth inhibition (GI50) was determined by the MTT colorimetric assay (Table 1). Of the compounds listed, the reference compound 38 showed nanomolar and sub-nanomolar GI50 values in all cell lines tested, whereas its regioisomer 3-ethyl-8-PPyQ 34 was inactive (GI50 > 50 μM), as were the other 8-PPyQ derivatives 33 and 35–37. Clearly, either removal of the side phenyl group from position 7 or shifting it to the 8-position of the tricyclic nucleus is deleterious for activity. Three of the new 7-PPyQ compounds, 11–13, are active in all cell lines, while an additional three (17, 21, and 22) have moderate activity only in the leukemic lines. In fact, compound 38 showed GI50 values 10–100-fold lower for leukemia cells (GI50: 0.5–2 nM) than for the solid tumor cells (GI50: 0.01–0.06 μM). This difference is even more dramatic for the two most active new compounds, 11 and 12, for which the GI50 values toward the leukemia cells are 0.5–10 nM and toward the solid tumor cells, 0.4–1.4 μM. Both of these compounds have more hydrophilic N-alkyl side chains than other 7-PPyQs.
Figure 2.

Structures of compounds 38,[3] CA-4, and colchicine taken as references in the assays of antiproliferative activity, inhibition of tubulin polymerization and colchicine binding, respectively.
Table 1.
In vitro inhibitory effects on cell proliferation by compounds 11–13, 17, 20, 21, 33–37, and 38.
| GI50 [μm][a] | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compd | HT-29 | ARO | Hep G2 | IGROV | MCF-7 | HeLa | HL-60 | ML2 | K562 | RS 4;11 | Jurkat |
| 11 | 0.48 ± 0.08 | 0.55 ± 0.15 | 0.52 ± 0.10 | 0.62 ± 0.15 | 0.58 ± 0.18 | 0.45 ± 0.10 | 0.002 ± 0.0001 | 0.005 ± 0.002 | 0.19 ± 0.06 | 0.002 ± 0.0004 | 0.0005 ± 0.00001 |
| 12 | 0.45 ± 0.10 | 0.58 ± 0.15 | 0.95 ± 0.15 | 0.85 ± 0.20 | 1.40 ± 0.50 | 0.40 ± 0.05 | 0.002 ± 0.0001 | 0.01 ± 0.002 | 0.03 ± 0.007 | 0.001 ± 0.0001 | 0.003 ± 0.0001 |
| 13 | 0.85 ± 0.14 | 0.45 ± 0.07 | 0.50 ± 0.05 | 0.65 ± 0.15 | 0.85 ± 0.15 | 0.40 ± 0.05 | 0.03 ± 0.002 | 0.3 ± 0.04 | 0.24 ± 0.03 | 0.04 ± 0.007 | 0.05 ± 0.004 |
| 17 | >50 | >50 | 5.8 ± 1.1 | >50 | >50 | 4.5 ± 0.8 | 17.5 ± 2.3 | 14.4 ± 2.3 | 27.3 ± 0.34 | 6.5 ± 1.0 | 8.0 ± 1.2 |
| 21 | >50 | >50 | >50 | >50 | >50 | >50 | 0.21 ± 0.01 | 0.55 ± 0.03 | 0.46 ± 0.008 | 0.13 ± 0.02 | 0.3 ± 0.01 |
| 22 | >50 | >50 | >50 | >50 | >50 | >50 | 0.32 ± 0.03 | 0.09 ± 0.02 | 1.7 ± 0.5 | 0.03 ± 0.002 | 0.05 ± 0.006 |
| 33 | >50 | >50 | >50 | ND | ND | >50 | >50 | >50 | >50 | ND | ND |
| 34 | >50 | >50 | >50 | ND | ND | >50 | >50 | >50 | >50 | ND | ND |
| 35 | >50 | >50 | >50 | ND | ND | >50 | >50 | >50 | >50 | ND | ND |
| 36 | >50 | >50 | >50 | ND | ND | >50 | >50 | >50 | >50 | ND | ND |
| 37 | >50 | >50 | >50 | ND | ND | >50 | >50 | >50 | >50 | ND | ND |
| 38[3] | 0.03 ± 0.01 | 0.03 ± 0.01 | 0.05 ± 0.01 | 0.06 ± 0.01 | 0.04 ± 0.01 | 0.01 ± 0.008 | 0.0005 ± 0.00002 | 0.001 ± 0.0003 | 0.001 ± 0.0001 | 0.002 ± 0.0003 | 0.0005 ± 0.00002 |
GI50: compound concentration required to inhibit tumor cell proliferation by 50 %; data are expressed as the mean ± SE from the dose–response curves of at least three independent experiments; ND: not determined. See text for details of the cell lines examined; the first six cell lines listed are derived from human solid tumors, the last five are from human leukemias.
Of the three compounds bearing a substituent at the 1-position, 21 and 22, with cyano and carboxyamide groups, respectively, are completely inactive (GI50 > 50 μM) in the solid tumor cells, but show activity at sub-micromolar concentrations in the leukemia lines. Compound 17, with a methyl group, has only modest activity in the leukemia and HeLa cells. We conclude that a substituent at the 1-position does not abolish antiproliferative activity, as do substituents at position 2.[2] This finding indicates that suitable substituents at the 1-position of 7-PPyQs might result in compounds with high selectivity for leukemic diseases.
Finally, compound 13, with a pyridine ring in place of the side phenyl group and an ethyl group at the N3 position, like 38, shows slightly less selectivity toward the leukemic lines than compounds 38, 11, 12, 17, 21, and 22.
Inhibition of tubulin polymerization and colchicine binding
In a previous study,[3] we found that compound 38 and a number of other 7-PpyQs cause cells to arrest at the G2/M phase of the cell cycle. This finding suggests that the target of these agents is likely to be tubulin, as this is a universal observation in cells treated with anti-tubulin agents. To determine whether this is true, the most active compounds from the current series, 11–13 and 22, together with an active agent from the earlier studies (compound 38), were evaluated for their in vitro inhibition of tubulin polymerization and for their inhibitory effect on the binding of [3H]colchicine to tubulin (Table 2).[8,9] For comparison, combretastatin A-4 (CA-4) was examined in simultaneous experiments.
Table 2.
Inhibition of tubulin polymerization and colchicine binding by compounds 11–13, 22, 38, and CA-4.
| Compd | Tubulin Assembly[a] IC50 [μm] | Colchicine Binding[b] Inhibition [%] | ||
|---|---|---|---|---|
| 1 μm | 5 μm | 50 μm | ||
| 11 | 0.75 ± 0.04 | 44 ± 6 | 75 ± 4 | ND |
| 12 | 1.4 ± 0.2 | ND | 30 ± 4 | 56 ± 2 |
| 13 | 2.4 ± 0.4 | ND | 23 ± 6 | 65 ± 0.3 |
| 22 | >40 | ND | ND | ND |
| 38 | 0.57 ± 0.02 | 34 ± 2 | 73 ± 0.7 | ND |
| CA-4 | 1.2 ± 0.01 | 84 ± 3 | 98 ± 0.3 | ND |
Inhibition of tubulin polymerization; tubulin was present at 10 μM; error values are ± SD.
Inhibition of [3H]colchicine binding; tubulin and colchicine were present at 1 and 5 μM, respectively, and the tested compounds were present at 1, 5, or 50 μM, as indicated; ND: not determined.
In the assembly assay, compound 11 was found to be the most active (IC50 = 0.75 μM), and it is nearly twice as potent as CA-4 (IC50 = 1.2 μM). Compound 38 is also more inhibitory than CA-4, whereas 12 and 13 are less inhibitory than CA-4. Compound 22 showed only minimal inhibitory activity, with less than 50 % inhibition of tubulin assembly at the highest concentration (40 μM) examined. These effects on tubulin assembly are in good agreement with the relative antiproliferative activities of the compounds.
In the colchicine binding studies, compounds 11 and 38 were roughly equal in activity, but are less potent than CA-4 despite their greater potency as inhibitors of assembly. Such differences are commonly observed, and CA-4 is a particularly potent inhibitor of colchicine binding.[9] Compounds 12 and 13 are much less active as inhibitors of colchicine binding than are CA-4, 11, and 38, with convincing inhibition only observed when the inhibitor concentration was increased to 50 μM, 10-fold higher than the concentration of [3H]colchicine in the reaction mixtures.
Cell-cycle analysis
The effects of various concentrations of compounds 11, 12, 13, and 22 on cell-cycle progression were examined with Jurkat leukemic T-lymphocytes (Figure 3). Untreated Jurkat cells showed a classical pattern of proliferation, with large numbers of cells in the G1 (47.2 %) and S (43.4 %) phases, and fewer cells in the G2/M (9.4 %) phase. As in our earlier study,[3] the two most active compounds, 11 and 12, showed a clear G2/M arrest pattern in a concentration-dependent manner, with a concomitant decrease of cells in the other phases of the cell cycle after 24 h treatment. In particular, as shown in Figure 3, the G2/M cell population increased from 9.4 % in the control to 77 % with compound 11 at 0.06 μM. The G1 cells decreased from 47 % in the control to 3 %, while the S-phase cells decreased from 43 to 20 %. Similar behavior was observed for compound 12, but a similar arrest (65 %) in G2/M phase required 0.25 μM compound 12. The other two compounds, 13 and 22, only induced G2/M arrest at yet higher concentrations, in good agreement with their lower potency in the tubulin polymerization assay and their lower antiproliferative activity. Further experiments showed that the accumulation of mitotic cells occurs in a time-dependent manner associated with the appearance of a hypodiploid peak (sub-G1) indicative of apoptosis (data not shown). This observation prompted us to investigate the possible activation of apoptotic mechanisms in the presence of the most active derivatives.
Figure 3.

Effects of compounds A) 11, B) 12, C) 13, and D) 22 on cell cycle distribution and G2/M arrest in Jurkat cells after 24 h incubation (■: G1, ●: G2/M, ▲: S). Cells were treated with various concentrations of test compounds (0.015–1.0 mM). The cells were then fixed and stained with propidium iodide (PI) to analyze DNA content by flow cytometry. Data are expressed as the mean ±SEM for two independent experiments.
Loss of plasma membrane asymmetry during apoptosis
To better characterize drug-induced apoptosis, we performed a bi-parametric cytofluorimetric analysis using propidium iodide (PI) and annexin V–FITC, which stain DNA and phosphatidylserine (PS) residues, respectively.[10, 11] Annexin V is a Ca2+-dependent phospholipid binding protein with high affinity for PS. Annexin V staining precedes the loss of membrane integrity that accompanies the final stages of cell death resulting from either apoptotic or necrotic processes. Because the externalization of PS occurs in the earlier stages of apoptosis, annexin V staining identifies apoptosis at an earlier stage than the appearance of sub-G1 cells. Such cells represent a later stage of cell death involving nuclear changes such as DNA fragmentation.
After drug treatment at various concentrations and time intervals, Jurkat cells were labeled with the two dyes, and the resulting red (PI) and green (FITC) fluorescence was monitored by flow cytometry. Compounds 11 and 12 provoked a remarkable induction of apoptotic cells after 24 h treatment, whereas the less active compounds 13 and 22 showed lower efficacy (Figure 4). The percentage of annexin-V-positive cells increased further at 48 h. These findings prompted us to further investigate the apoptotic machinery after treatment with compounds 11 and 12.
Figure 4.

Flow cytometric analysis of apoptotic cells after treatment of Jurkat cells with compounds 11 (■), 12 (●), 13 (▲), or 22 (▼). After treatment for A) 24 or B) 48 h, cells were harvested and labeled with annexin V–FITC and PI and then analyzed by flow cytometry. The data are expressed as the mean percentage of annexin-V-positive cells ±SEM for three independent experiments.
Apoptosis is mediated by mitochondrial depolarization. Mitochondria play an essential role in the propagation of apoptosis.[12, 13] It is well established that, at an early stage, apoptotic stimuli alter the mitochondrial transmembrane potential (ΔΨmt). Δ Ψmt was monitored by the fluorescence of the dye JC-1.[14] In the presence of normal cells (high ΔΨmt), JC-1 forms fluorescent red (λ 590 nm) aggregates locally and spontaneously that are associated with a large shift in the emission; upon mitochondrial membrane depolarization (low ΔΨmt), JC-1 forms monomers that emit at λ 530 nm. Treated Jurkat cells in the presence of derivatives 11 and 12 (0.5 and 0.1 μM) exhibited a significant increase in the percentage of cells with low ΔΨmt in a concentration- and time-dependent manner relative to the control cells, indicating depolarization of the mitochondrial membrane potential (Figure 5A). The disruption of ΔΨmt is associated with the appearance of annexin V positivity in the treated cells when they are in early apoptotic stage. In fact, the dissipation of ΔΨmt is characteristic of apoptosis and has been observed with both microtubule destabilizing and stabilizing agents in various cell types.[15]
Figure 5.

Assessment of mitochondrial dysfunction after treatment with compounds 11 or 12. A) Induction of loss of mitochondrial membrane potential in Jurkat cells after 24 and 48 h incubation with 11 and 12 at 0.1 and 0.5 mM. Cells were stained with the fluorescent probe JC-1 and analyzed by flow cytometry. B) and C) Mitochondrial production of ROS in Jurkat cells after 24 and 48 h incubation with 11 and 12. Cells were stained with B) HE or C) H2-DCFDA and analyzed by flow cytometry. The data are expressed as a percentage of the fluorescence intensity relative to untreated control and represent the mean ± SEM of three independent experiments.
Mitochondrial generation of ROS
Mitochondrial membrane depolarization is associated with mitochondrial production of reactive oxygen species (ROS).[16] Therefore, we investigated whether ROS production increased after treatment with compounds 11 and 12. We used the fluorescence indicator hydroethidine (HE), the fluorescence of which appears if ROS are generated.[17, 18] HE is oxidized by the superoxide anion into the ethidium ion, which emits red fluorescence. Superoxide is produced by mitochondria due to a shift from the normal four-electron reduction of O2 to a one-electron reduction when cytochrome c is released from mitochondria. ROS generation was also measured with the dye 2,7-dichlorodihydrofluorescein diacetate (H2-DCFDA), which is oxidized to the fluorescent compound dichlorofluorescein (DCF) by a variety of peroxides including hydrogen peroxide.[18] As shown in Figure 5B,C, compounds 11 and 12, as expected, induced the production of ROS relative to control cells, in agreement with the dissipation of Δ Ψmt.
Caspase-3 activation, PARP cleavage, and Bcl-2 down-regulation
Caspases are the central executioners of apoptosis mediated by various inducers.[19, 20] Caspases are synthesized as proenzymes, which are activated by cleavage. Caspases-2, -8, -9, and -10 are termed apical and are usually the first to be stimulated in the apoptotic process. These, in turn, activate effector caspases, such as caspase-3 in particular.[21] Exposure of Jurkat cells to compounds 11 or 12 was found to activate caspase-3, as shown in Figure 6, in a time- and concentration-dependent manner.
Figure 6.

Caspase-3 activity induced by compounds 11 and 12. Jurkat cells were incubated in the presence of A) 11 or B) 12 at 0.1 and 0.5 mM. After treatment for 48 h, cells were harvested and stained with an anti-human active caspase-3 fragment monoclonal antibody conjugated with FITC. Data obtained by flow cytometry analysis are expressed as the percentage of caspase-3 active fragment positive cells. Data are expressed as the mean ± SEM of three independent experiments.
Poly(ADP-ribose) polymerase (PARP) is a 116 kDa nuclear protein that appears to be involved in apoptosis.[22] This protein is one of the main cleavage targets of caspase-3 both in vitro and in vivo.[22] As illustrated in Figure 7A, immunoblot analysis shows that the typical 89 kDa fragment of PARP increased in treated cells with both compounds in a time-dependent manner. Bcl-2 is a protein extensively investigated as a modulating agent of apoptosis, and it plays a major role in inhibiting apoptosis. Bcl-2 regulates the mitochondrial membrane potential and avoids the subsequent release of cytochrome c to prevent caspase activation.[23, 24] Therefore, we examined whether the induced apoptosis by 11 and 12 is associated with changes in Bcl-2 expression. As depicted in Figure 7B, flow cytometric analysis shows that both compounds decrease Bcl-2 expression. Altogether, these data indicate that the induction of apoptosis by these new derivatives is mediated through caspase-3 activation, PARP cleavage, and Bcl-2 down-regulation.
Figure 7.

Western blot analysis of PARP and flow cytometric analysis of Bcl-2 expression. A) Jurkat cells were treated with compounds 11 and 12 at the indicated concentrations for 24 and 48 h. The cells were then subjected to immunoblot analysis as described in the Experimental Section. B) Cells were treated as above, and after permeabilization were labeled with a Bcl-2 FITC-conjugated antibody. Shown are representative histograms after 24 h treatment with compound 11 at the indicated concentrations. C) Bcl-2 expression in Jurkat cells after treatment with compounds 11 and 12 at the indicated concentrations for 24 and 48 h. Data are expressed as the mean ± SEM of two independent experiments.
Conclusions
Several 7- and 8-phenylpyrroloquinolinone derivatives were prepared and assayed for their antiproliferative activity on a panel of solid tumor and leukemic cell lines. Depending on the position and nature of substitutions, new SARs on PPyQs were demonstrated that add to those already known. Among these, the most surprising was the inactivity of the regioisomer 8-PPyQs 33–37 toward all cell lines. This indicates that the interaction of the 2-phenylquinolin-4-one moiety in the colchicine site of tubulin either requires the C7 phenyl group or is abolished with a bulky C8 substituent.
Another important SAR is the finding that small hydrophilic groups such as cyano and carboxyamide at position 1 of the 3-ethyl-7-PPyQ 38 (compounds 21 and 22, respectively) retain good antiproliferative activity, but only toward leukemic cells. This should permit the future design of compounds specifically directed against leukemias. Finally, introduction of a less hydrophobic side chain at the N3 or C7 positions of the PyQ nucleus provided compounds 11–13, which maintain high activity and an antiproliferative profile similar to the reference compound 38, all of which have greater cytotoxicity against leukemia cells than toward solid tumor cells.
In particular, compound 11 exhibits potent inhibition of tubulin polymerization and is one of the most potent inhibitors of colchicine binding (IC50 = 0.76 μM for assembly, 44 % inhibition of [3H]colchicine binding when equimolar with tubulin and one fifth the concentration of colchicine in the reaction mixture). Flow cytometry demonstrated that 11 has cellular effects typical for microtubule-interacting agents, causing accumulation of cells in the G2/M phase of the cell cycle and increased numbers of apoptotic sub-G1 cells. Moreover, compound 11 is a potent inducer of multiple additional markers of apoptosis in the Jurkat cell line. Apoptosis induced by multiple antimitotic agents has been associated with alteration in a variety of cellular signaling pathways, and compound 11 had similar effects. This agent induced Bcl-2 down-regulation after 24 h treatment. Because Bcl-2 prevents the initiation of the cellular apoptotic program by stabilizing mitochondrial permeability, its loss has multiple consequences. These include loss of Δ Ψmt, which, in turn, results in an uncoupling of the respiratory chain and mitochondrial efflux of caspase-9 and the apoptosis-inducing factor (AIF). Thus, the proteolytic activation of caspase-3 occurred with subsequent PARP cleavage. Therefore, compound 11, with its apparent selectivity toward leukemic cell lines, merits further investigation as a potential chemotherapeutic agent.
Experimental Section
Chemistry
Melting points were determined on a Gallenkamp MFB 595 010 M/B capillary melting point apparatus and are not corrected. IR spectra were recorded on a PerkinElmer 1760 FTIR spectrometer using KBr pressed disks; all values are expressed in cm−1. UV/Vis spectra were recorded on a PerkinElmer Lambda UV/Vis spectrometer. 1H NMR spectra were recorded on Bruker spectrometers (300 MHz) with the indicated solvents; chemical shifts (δ) are reported in ppm downfield from (CH3)4Si as internal reference. Coupling constants (J) are given in Hz. In the case of multiplets, δ values were measured by starting from the approximate center. Integrals were satisfactorily in line with those expected on the basis of compound structure. Elemental analyses were performed in the Microanalytical Laboratory, Department of Pharmaceutical Sciences, University of Padova (Italy), using a PerkinElmer elemental analyzer model 240B; results fell in the range of calculated values ±0.4 %. The analytical data are presented in detail for each final compound. Mass spectra were obtained with a Mat 112 Varian Mat Bremen (70 eV) mass spectrometer and an Applied Biosystems Mariner System 5220 LC–MS (nozzle potential: 250.00). Column flash chromatography was performed on Merck silica gel (250–400 mesh ASTM); reactions were monitored by analytical thin-layer chromatography (TLC) using Merck silica gel 60 F254 glass plates. Solutions were concentrated in a rotary evaporator under reduced pressure. Starting materials were purchased from Aldrich Chimica and Acros, and solvents from Carlo Erba, Fluka, and Lab-Scan. DMSO was obtained anhydrous by distillation under vacuum and stored on molecular sieves.
Ethyl 3-oxo-3-(pyridin-3-yl)propanoate (1).[4]
Reagent 1 was obtained by reacting commercial ethyl nicotinate (2.71 mL, 19.8 mmol) with EtOAc (1.5 equiv) in the presence of tBuOK (2 equiv) in excess benzene at 80 °C for 6 h. At the end of the reaction, solvent was removed by evaporation, a mixture of ice and H2O was added, and the mixture was extracted with Et2O after neutralization with CH3COOH. The extract was washed with H2O, dried on anhydrous Na2SO4 and evaporated under vacuum. A light-yellow oil product was obtained that was purified by distillation. Yield 45 %; Rf = 0.52 (EtOAc/n-hexane 8:2); bp: 121–123 °C (0.4 mm); 1a: 1H NMR ([D6]DMSO): δ = 1.17 (t, 3 H, J = 7.1 Hz, CH3), 4.12 (q, 2 H, J = 7.1 Hz, CH2), 4.27 (s, 2 H, CH2), 7.58 (ddd, 1 H, J = 0.8, 4.8, and 8.0 Hz, 5-H), 8.29 (dt, 1H, J = 2.3 Hz, 8.01, 4-H), 8.76 (dd, 1 H, J = 4.7 and 6.5 Hz, 6-H), 9.12 (dd, 1H, J = 0.6 and 2.3 Hz, 2-H); 1b: 6.08 (s, 1H, CH), 12.52 (s, 1H, OH).
Ethyl 3-(5-nitro-1H-indol-1-yl)propanoate (2)
An amount corresponding to 35 mmol NaH (60% dispersion in mineral oil) was placed in a 100 mL round-bottomed flask and washed with toluene (3 × 10 mL). A solution of commercial 5-nitroindole (12 mmol) in dry toluene (60 mL) was dropped into the flask, and the initial yellow color changed to red, with the formation of H2 gas. After 30 min at room temperature, a solution of ethyl acrylate (12 mmol) in dry toluene (10 mL) was added, and the reaction mixture was held at reflux for 4 h. Next, the reaction mixture was cooled to 0 °C, a few drops of absolute EtOH were added, the volatiles were removed, and the residue was partitioned between H2O (15 mL) and CHCl3 (60 mL). The organic extract was evaporated under vacuum. Yellow–orange solid, yield 76 %; mp: 65–66 °C; Rf = 0.71 (eluent benzene/n-hexane/EtOAc 1:1:1); 1H NMR ([D6]DMSO): δ = 1.08 (t, 3 H, J = 7.1 Hz, CH3), 2.88 (t, 2H, J = 6.7 Hz, CH2), 3.99 (q, 2 H, J = 7.1 Hz, CH2), 4.56 (t, 2H, J = 6.7 Hz, CH2), 6.80 (d, 1 H, J2,3 = 3.2 Hz, 2-H), 7.81 (m, 2H, 7-H, 3-H), 8.02 (dd, 1H, J4,6 = 2.2 Hz, J6,7 = 9.1 Hz, 6-H), 8.58 (d, 1H, J4,6 = 2.2 Hz, 4-H); HRMS (ESI) calcd for C13H15N2O4 [M+H]+ m/z 263.0987, found 263.1012.
1-(3,4,5-Trimethoxybenzyl)-5-nitro-1H-indole (3)
An amount corresponding to 37 mmol NaH (60 % dispersion in mineral oil) was placed in a three-necked 100 mL round-bottomed flask and washed with toluene (3 × 10 mL). A solution of commercial 5-nitro-indole (12.3 mmol) in 20–30 mL dry DMF was dropped into the flask, and the initial yellow color changed to red, with the formation of H2 gas. After 30 min at room temperature, a solution of trimethoxybenzyl chloride (25 mmol) in dry DMF was added, and the reaction mixture was left stirring for 20–48 h with monitoring of the ongoing reaction by TLC analysis (eluent: toluene/n-hexane/EtOAc 1:1:0.3). At the end, the solvent was evaporated, and the residue was extracted with EtOAc. The organic phase, washed with H2O and dried over anhydrous Na2SO4, was dried under vacuum. Yield 98 %; Rf = 0.50 (benzene/n-hexane/EtOAc 1:1:1); mp: 158–160 °C; 1H NMR ([D6]DMSO): δ = 3.59 (s, 3H, 4′-OCH3), 3.69 (s, 6 H, 3′- and 5′-OCH3), 5.41 (s, 2 H, CH2), 6.65 (s, 2 H, 2′- and 6′-H), 6.80 (d, 1 H, J2,3 = 3.2 Hz, 2-H), 7.81 (m, 2 H, 7-H and 3-H), 8.02 (dd, 1H, J4,6 = 2.2 Hz, J6,7 = 9.1, 6-H), 8.58 (d, 1H, J4,6 = 2.2 Hz, 4-H); HRMS (ESI) calcd for C18H19N2O5 [M+H]+ m/z 343.1249, found 343.1364.
General procedure for the synthesis of 5-aminoindole derivatives 5–7[3]
A solution of a nitroindole 2–4[3] (5.87 mmol) in 400 mL of EtOH was dropped into a suspension of 10 % Pd/C (125 mg) saturated with H2 in 200 mL EtOH. The mixture was stirred at room temperature with hydrogen at atmospheric pressure for 3 h, and the reaction was monitored by TLC analysis (EtOAc/n-hexane 1:3). The catalyst was then removed by filtration, and the filtrate was evaporated under reduced pressure to dryness to give the corresponding aminoindole. The nearly pure residue (90% yield) was used in the next reaction without purification.
Ethyl 3-(5-amino-1H-indol-1-yl)propanoate (5)
Brown dense liquid; yield 98 %; Rf = 0.48 (benzene/n-hexane/EtOAc 1:1:1); 1H NMR ([D6]DMSO): δ = 1.12 (t, 3H, J = 7.1 Hz, CH3), 2.75 (t, 2H, J = 6.7 Hz, CH2), 4.01 (q, 2H, J = 7.1 Hz, CH2), 4.63 (t, 2 H, J = 6.7 Hz, CH2), 5.18 (bs, 2H, NH2), 6.15 (dd, 1 H, J3,2 = 3.1 Hz, J3,4 = 0.8 Hz, 3-H), 6.52 (dd, 1 H, J6,7 = 8.6 Hz, J6,4 = 2.1 Hz, 6-H), 6.67 (dd, 1 H, J4,6 = 2.1 Hz, J4,3 = 0.6 Hz, 4-H), 7.16 (d, 1H, J7,6 = 8.6 Hz, 7-H), 7.19 (d, 1 H, J2,3 = 3.0 Hz, 2-H).
1-(3,4,5-Trimethoxybenzyl)-5-amino-1H-indole (6)
Reddish waxy solid; yield 90 %; Rf = 0.58 (EtOAc/n-hexane 7:3); 1H NMR ([D6]DMSO): δ = 3.59 (s, 3 H, 4′-OCH3), 3.69 (s, 6H, 3′- and 5′-OCH3), 4.40 (bs, 2 H, NH2), 5.18 (s, 2 H, CH2), 6.53 (s, 2 H, 2′- and 6′-H), 6.64 (m, 3- and 7-H), 6.78 (d, 1 H, J4,6 = 2.1 Hz, 4-H), 7.22 (dd, 1H, J6,7 = 8.6 Hz, 6-H), 7.32 (d, 1 H, J2,3 = 3.2 Hz, 2-H).
General procedure for the synthesis of ethyl 3-(1-substituted-in-doleamino)-3-phenylacrylates (8–10)
In a 50 mL round-bottomed flask, 3–4 mmol 3-substituted aminoindole 5–7[2] in 10–20 mL absolute EtOH were condensed with an equimolar quantity of commercial ethyl benzoylcetate for 8 and 9 or ethyl 3-oxo-3-(pyridin-3-yl)-propanoate for 10 with 1 mL glacial CH3COOH and 100 mg Drierite. The mixture was held at reflux for ~24 h, the reaction being monitored by TLC analysis (CH2Cl2/EtOAc 8:2). As the reaction did not come to completion, after 24 h the mixture was cooled and filtered to remove the Drierite. The resulting solution was evaporated to dryness under vacuum, and the residue was purified by flash chromatography.
Ethyl 3-[1-(3-ethoxy-3-oxopropyl)-1H-indol-5-ylamino]-3-phenyl-acrylate (8)
Brown dense liquid; yield 68 %; Rf = 0.83 (CH2Cl2/EtOAc 8:2); 1H NMR ([D6]DMSO): δ = 1.11 (t, 3H, J = 7.1 Hz, CH3), 1.16 (t, 3 H, J = 7.1 Hz, CH3), 2.80 (t, 2 H, J = 6.7 Hz, CH2), 4.01 (q, 2 H, J = 7.1 Hz, CH2), 4.13 (q, 2H, J = 7.1 Hz, CH2), 4.38 (t, 2 H, J = 6.7 Hz, CH2), 4.83 (s, 1H, CH), 6.20 (d, 1H, J3,2 = 3.1 Hz, 3-H), 6.60 (dd, 1 H, J6,7 = 8.8 Hz, J6,4 = 2.1 Hz, 6-H), 6.97 (d, 1 H, J4,6 = 2.1 Hz, 4-H), 7.30 (m, 7 H, ar), 10.25 (s, 1 H, NH); HRMS (ESI) calcd for C24H27N2O4 [M+H]+ m/z 407.1926, found 407.1997.
Ethyl 3-phenyl-3-[1-(3,4,5-trimethoxybenzyl)-1H-indol-5-ylamino]acrylate (9)
Dark-green liquid; yield 70 %; Rf = 0.96 (CH2Cl2/EtOAc 8:2); 1H NMR ([D6]DMSO): δ = 1.23 (t, 3 H, J = 7.1 Hz, CH3), 3.59 (s, 3 H, 4′-OCH3), 3.69 (s, 6 H, 3′- and 5′-OCH3), 4.12 (q, 2 H, J = 7.1 Hz, CH2), 4.83 (s,1 H, CH), 5.18 (s, 2 H, CH2), 6.20 (d, 1H, J3,2 = 3.1 Hz, 3-H), 6.60 (dd, 1 H, J6,7 = 8.8 Hz, J6,4 = 2.1 Hz, 6-H), 6.53 (s, 2 H, 2′- and 6′-H), 6.97 (d, 1 H, J4,6 = 2.1 Hz, 4-H), 7.30 (m, 7 H, ar), 10.20 (s, 1H, NH); HRMS (ESI) calcd for C29H31N2O5 [M+H]+ m/z 487.2188, found 487.2410.
Ethyl 3-(1-ethyl-1H-indol-5-ylamino)-3-(pyridin-3-yl)acrylate (10)
Brown dense liquid; yield 40 %; Rf = 0.75 (EtOAc/n-hexane 8:2); 1H NMR ([D6]DMSO): δ = 1.23 (t, 3H, J = 7.3 Hz, CH3), 1.29 (t, 3 H, CH3), 4.11 (m, 4H, J = 7.1 Hz, 2 × CH2), 4.90 (s, 1H, CH), 6.22 (dd, 1 H, J3,2 = 3.1 and J3,4 = 0,6 Hz, 3-H), 6.62 (dd, 1H, J6,7 = 8.8 and J6,4 = 2.01 Hz, 6-H), 6.97 (d, 1 H, J4,6 = 2.0 Hz, 4-H), 7.31 (m, 4H, ar), 7.40 (d, 1 H, J3,2 = 3.1 Hz, 2-H), 7.46 (d, 1 H, J6,7 = 8.8 Hz, 7-H), 10.35 (s, 1 H, NH); HRMS (ESI) calcd for C20H22N3O2 [M+H]+ m/z 336.1667, found 336.1749.
General procedure for the synthesis of 3-substituted 7-phenyl-and 7-pyridin-3H-pyrrolo[3,2-f]quinolin-9-(6H)ones 11–13
In a two-necked round-bottomed flask, 50 mL Ph2O was heated at boiling point; 1–2 mmol of acrylates 8–10 were then added portion-wise, and the mixture was held at reflux for 30 min. After cooling to 60 °C, the separated precipitate was collected by filtration and washed several times with Et2O. In all cases, the collected products were purified by flash chromatography (EtOAc/MeOH 8:2) or re-crystallized from a suitable solvent.
Ethyl 3-(9-oxo-7-phenyl-6H-pyrrolo[3,2-f]quinolin-3(9H)-yl)propanoate (11)
Yellow solid; yield 45 %; mp: 202–204 °C; Rf = 0.60 (EtOAc/MeOH 9:1); mp: 249–250 °C (dec.); 1H NMR (CD3OD): δ = 1.17 (t, 3 H, CH3), 2.91 (d, 2 H, CH2), 4.09 (q, 2 H, CH2), 4.64 (d, 2 H, CH2), 6.69 (s, 1 H, 8-H), 7.46 (d, 1H, J1,2 = 3.1 Hz, 1-H), 7.59 (m, 4 H, ar), 7.67 (d, 1 H, J1,2 = 3.1 Hz, 2-H) 7.82 (m, 2 H, 2′- and 6′-H), 7.93 (d, 1 H, J4,5 = 8.9 Hz, 4-H); 13C NMR (CD3OD): δ = 14.8, 36.7, 43.5, 62.3, 105.9, 109.8, 113.6, 118.3, 126.1, 128.1, 130.4, 131.8, 136.4, 173.3, 180.2; HRMS (ESI) calcd for C22H20N2O3 [M+H]+ m/z 361.1547, found 361.1532; anal. calcd for C22H20N2O3: C 73.32, H 5.59, N 7.77, found: C 73.56, H 5.42, N 7.43.
7-Phenyl-3-(3,4,5-trimethoxybenzyl)-3H-pyrrolo[3,2-f]quinolin-9(6H)-one (12)
Light-brown solid; yield 40 %; mp: 249–250 °C; Rf = 0.55 (eluent EtOAc/MeOH 9:1); 1H NMR (CD3OD): δ = 3.91 (s, 9 H, 3 × OCH3), 5.70 (s, 2 H, CH2), 6.60 (s, 2 H, 2′- and 6′-H), 6.90 (s, 1 H, 8-H), 7.77 (m, 5 H, ar), 7.92 (d, 1H, J1,2 = 3.1 Hz, 2-H), 8.03 (m, 3 H, 4-H, 2″- and 6″-H); 13C NMR (CD3OD): δ = 51.2, 56.5, 61.1, 105.1, 128.6, 130.7, 135.9, 154.8, 180.3; HRMS (ESI) calcd for C27H24N2O4 [M+H]+ m/z 441.1809, found 441.1668; anal. calcd for C27H24N2O4: C 73.62, H 5.49, N 6.36, found: C 73.81, H 5.39, N 6.15.
3-Ethyl-7-(pyridin-3-yl)-3H-pyrrolo[3,2-f]quinolin-9(6H)-one (13)
Beige solid; yield 35 %; mp: 278–280 °C (dec.) (MeOH); Rf = 0.46 (eluent EtOAc/MeOH 8:2); 1H NMR (CD3OD): δ = 1.38 (t, 3 H, CH3), 4.25 (q, 2 H, CH2), 6.56 (bs, 1 H, 8-H), 7.35 (d, 1H, J1,2 = 2.9 Hz, 1-H), 7.44 (d, 1 H, J4,5 = 9.0 Hz, 5-H), 7.53 (m, 2H, 2- and 5′-H), 7.79 (d, 1 H, J4,5 = 9.0 Hz, 4-H), 8.14 (m, 1 H, 6′-H), 8.60 (dd, 1H, J = 1.5 and 4.9 Hz, 4′-H), 8.88 (d, 1H, J = 2.3 Hz, 2′-H); 13C NMR (CD3OD): δ = 16.5, 42.2, 105.7, 117.5, 125.5, 129.5, 133.3, 148.7, 151.7, 182.9; HRMS (ESI) calcd for C18H15N3O [M+H]+ m/z 290.1288, found 290.1385; anal. calcd for C18H15N3O: C 74.72, H 5.23, N 14.52, found: C 74.95, H 5.36; 14.11
1-Ethyl-5-nitro-1H-indole-3-carbaldehyde (14)
A solution of 1-ethyl-5-nitro-1H-indole 4[3] (1.5634 g, 8.22 mmol) in DMF was dropped into a mixture of 2 mL POCl3 and 3 mL DMF cooled at 10 °C. The temperature was raised to 35–40 °C with stirring, and the reaction continued until the starting material was no longer detectable by TLC. The volatile products were removed in vacuo, and the residue was diluted with a mixture of H2O and ice, followed by prior basification with 6 % NaOH. A heavy precipitate formed, which was removed by filtration and washed with a mixture of H2O and EtOAc. The organic phase of the filtrate was separated and evaporated to give a yellow residue (1.0052 g). The precipitate was resuspended in EtOAc, and the mixture was stirred overnight. It was then filtered, and the EtOAc solution was dried under vacuum to give another 0.512 g of product. Light-yellow solid; yield 87 %; mp: 178–180°C; Rf = 0.46 (EtOAc/n-hexane 7:3); 1H NMR ([D6]DMSO): δ = 1.44 (t, 3H, J = 7.1 Hz, CH3), 4.40 (q, 2 H, J = 7.1 Hz, CH2), 7.90 (d, 1 H, J6,7 = 9.1 Hz, 7-H), 8.20 (dd, 1 H, J4,6 = 2.2 and J6,7 = 9.1 Hz, 6-H), 8.65 (s 1 H, 2-H), 8.95 (d, 1H, J4,6 = 2.1 Hz, 4-H), 10.00 (s, 1 H, HCO); HRMS (ESI) calcd for C11H11N2O3 [M+H]+ m/z 219.0725, found 219.0757.
1-Ethyl-3-methyl-1H-indole-5-amine (15)
As with the aminoindole compounds 5–7, the nitroindole carbaldehyde 14 was catalytically reduced, at 40–50 °C for 10–14 h, to give a dense, red liquid product. Yield 77 %; Rf = 0.74 (EtOAc/n-hexane 8:2); 1H NMR ([D6]DMSO): δ = 1.05 (t, 3 H, CH3), 2.11 (s, 3 H, CH3), 3.99 (q, 2 H, CH2), 4.35 (bs, 2 H, NH2), 6.49 (dd, 1 H, J6,7 = 8.2 and J6,4 = 2.3 Hz, 6-H), 6.60 (d, 1H, J6,4 = 2.3, 4-H), 6.91 (s, 1H, 2-H), 7.07 (bd, 1 H, J6,7 = 8.2 Hz, 7-H).
Ethyl 3-(1-ethyl-3-methyl-1H-indol-5-ylamino)-3-phenylacrylate (16)
As with the acrylate compounds 8–10, aminoindole 15 was condensed with ethyl benzoylacetate to give a brown liquid product. Yield 56 %; Rf = 0.91 (CH2Cl2/EtOAc 8:2); 1H NMR ([D6]DMSO): δ = 1.23 (t, 3 H, J = 7.3 Hz, CH3), 1.29 (t, 3 H, CH3), 2.03 (s, 3H, CH3), 4.11 (m, 4 H, J = 7.1 Hz, 2 × CH2), 4.83 (s, 1 H, CH), 6.54 (dd, 1 H, J6,7 = 8.7 and J6,4 = 1.8 Hz, 6-H), 6.87 (d, 1 H, J4,6 = 1.9 Hz, 4-H), 7.16 (d, 1 H, J7,6 = 8.8 Hz, 7-H), 7.31 (m, 6H, ar), 10.25 (s, 1 H, NH); HRMS (ESI) calcd for C22H25N2O2 [M+H] + m/z 349.1871, found 349.2023.
3-Ethyl-1-methyl-7-phenyl-3H-pyrrolo[3,2-f]quinolin-9(6H)-one (17)
As with the pyrroloquinolinone derivatives 11–13, the acrylate 16 was cyclized to give a light-brown solid product. Yield 30 %; Rf = 0.77 (EtOAc/MeOH 8:2); mp: 163–165 °C; 1H NMR (CD3OD): δ = 1.58 (t, 3 H, J = 7.3 Hz, CH3), 2.92 (s, 3H, CH3), 4.31 (q, 2H, J = 7.3 Hz, CH2), 6.73 (s, 1H, 2-H), 7.33 (bs, 1 H, 8-H), 7.56 (d, 1H, J = 8.9 Hz, 5-H), 7.64 (m, 3 H, 3′-, 4′- and 5′-H), 7.85 (m, 3 H, 4-, 2′- and 6′-H); 13C NMR (CD3OD): δ = 12.3, 16.2, 42.3, 105.6, 125.6, 129.5, 148.8, 180.0; HRMS (ESI) calcd for C20H18N2O [M+H]+ m/z 303.1492, found 303.1722; anal. calcd for C20H18N2O: C 79.44, H 6.00, N 9.26, found: C 79.61, H 6.23, N 9.01.
1-Ethyl-5-nitro-1H-indole-3-carbonitrile (18)
In a round-bottomed flask, 5-nitroindolo-3-carbaldehyde 14 (1.539 g, 7.05 mmol) was dissolved in 5 mL anhydrous DMF. To this was added 0.514 g (7.4 mmol) NH2OH·HCl and 0.57 mL (7.4 mmol) anhydrous pyridine. The reaction mixture was stirred at 80 °C under N2 for ~ 2 h, until the starting material disappeared and another spot with higher Rf formed (TLC, EtOAc/n-hexane 7:3). At this point, 0.821 g of SeO2 and 0.400 g of MgSO4 were added to the hot reaction mixture. Stirring continued at 80 °C for another 2.5 h. The reaction mixture was cooled and filtered, and the filtered solution was evaporated to dryness under vacuum. The residue was resuspended in cold H2O, and the suspension was re-filtered. The collected yellow solid was washed several times with H2O and finally dried. Yield 60 %; Rf = 0.53 (EtOAc/n-hexane 7:3); mp: 158–161°C; 1H NMR ([D6]DMSO): δ = 1.41 (t, 3H, J = 7.1 Hz, CH3), 4.38 (q, 2 H, J = 7.1 Hz, CH2), 7.96 (d, 1 H, J6,7 = 9.1 Hz, 7-H), 8.20 (dd, 1 H, J4,6 = 2.2 and J6,7 = 9.1 Hz, 6-H), 8.51 (d, 1H, J4,6 = 2.1 and J6,7 = 9.1 Hz, 4-H), 8.64 (s, 1 H, 2-H); HRMS (ESI) calcd for C11H10N3O2 [M+H]+ m/z 216.0728, found 216.0783.
5-Amino-1-ethyl-1H-indole-3-carbonitrile (19)
As with the aminoindole compounds 5–7 and 15, the nitroindole carbonitrile 18 was catalytically reduced at 40–50 °C for 10–24 h to give a yellow semi-solid product. Yield 98 %; Rf = 0.66 (EtOAc/n-hexane 8:2); 1H NMR ([D6]DMSO): δ = 1.36 (t, 3H, J = 7.2 Hz, CH3), 4.15 (q, 2 H, J = 7.2 Hz, CH2), 4.96 (bs, 2 H, NH2), 6.65 (dd, 1 H, J4,6 = 1.9 and J6,7 = 8.7 Hz, 6-H), 6.72 (d, 1 H, J6,4 = 1.9 Hz, 4-H), 7.35 (d, 1 H, J6,7 = 8.7 Hz, 7-H), 8.02 (s, 1 H, HC-2); HRMS (ESI) calcd for C11H12N3 [M+H]+ m/z 186.0987, found 186.1047.
Ethyl 3-(3-cyano-1-ethyl-1H-indol-5-ylamino)-3-phenylacrylate (20)
As with the acrylate compounds 8–10 and 16, aminoindole 19 was condensed with benzoylacetate to give a dense yellow liquid. Yield 52 %; Rf = 0.84 (EtOAc/n-hexane 8:2); 1H NMR ([D6]DMSO): δ = 1.23 (t, 3 H, J = 7.3 Hz, CH3), 1.29 (t, 3 H, CH3), 4.11 (m, 4 H, J = 7.1 Hz, 2 × CH2), 4.92 (s, 1H, CH), 6.67 (dd, 1H, J6,7 = 8.7 and J6,4 = 2.0 Hz, 6-H), 6.95 (d, 1 H, J4,6 = 1.9 Hz, 4-H), 7.35 (d, 1 H, J7,6 = 8.7 Hz, 7-H), 7.31 (m, 6 H, ar), 10.25 (s, 1 H, NH).
3-Ethyl-9-oxo-7-phenyl-6,9-dihydro-3H-pyrrolo[3,2-f]quinoline-1-carbonitrile (21)
As with the pyrroloquinolinone derivatives 8–10 and 17, the acrylate 20 was cyclized to give a brown solid. Yield 24 %; mp: 209 °C; Rf = 0.33 (EtOAc/MeOH 9:1, light-blue fluorescent spot at λ365 nm); 1H NMR (CD3OD): δ = 1.63 (t, 3 H, J = 7.3 Hz, CH3), 4.55 (q, 2H, J = 7.3 Hz, CH2), 6.76 (s, 1H, 2-H), 7.70 (m, 3H, 3′-, 4′- and 5′-H), 7.84 (d, 1 H, J4,5 = 8.9 Hz, 4-H), 7.93 (m, 2 H, 2′- and 6′-H), 8.11 (d, 1H, J4,5 = 9.1 Hz, 5-H), 8.31 (s, 1 H, 2-H); 13C NMR (CD3OD): δ = 15.9 (CH3), 43.4 (CH2), 89.9, 118.8 (CN), 128.8 (C-2′, C-3′, C-5′, C-6′); HRMS (ESI) calcd for C20H15N3O2 [M+H]+ m/z 314.1288, found 314.1301; anal. calcd for C20H15N3O: C 76.66, H 4.82, N 13.41, found: 76.94, H 4.53, N 13.11.
3-Ethyl-9-oxo-7-phenyl-6,9-dihydro-3H-pyrrolo[3,2-f]quinoline-1-caboxamide (22)
CH3COOH, H2SO4, and H2O (2 mL each) were added in sequence to a 100 mL flask containing 0.188 g cyano compound 21 (0.60 mmol). The mixture was heated at 70 °C for 16 h. At the end (followed by TLC, EtOAc/MeOH 8:2), the reaction was stopped by adding a mixture of H2O and ice and left at 4 °C for 4 h. The formed precipitate was collected, washed with H2O, and dried to yield a brown solid. Yield 37 %; mp: 223–225 °C; Rf = 0.56 (EtOAc/MeOH 9:1, light-blue fluorescent spot at λ365 nm); 1H NMR (CD3OD): δ = 1.53 (t, 3 H, J = 7.3 Hz, CH3), 4.43 (q, 2 H, J = 7.3 Hz, CH2), 6.91 (s, 1H, 2-H), 7.61 (d, 1H, J4,5 = 9.1 Hz, 5-H), 7.82 (m, 3H, 3′-, 4′- and 5′-H), 8.11 (d, 1H, 6-H), 8.22 (s, 1H, 8-H); HRMS (ESI) calcd for C20H17N3O2 [M+H]+ m/z 332.1541, found 332.1444; anal. calcd for C20H17N3O2: C 72.49, H 5.17, N 12.68, found: C 72.61, H 4.92, N 12.29.
General procedure for the synthesis of ethyl 3-oxo-2-phenylpropanoate derivatives (23[7]–25)
NaH (60% in oil, 4 equiv in mmol) was added portionwise to a mixture of commercial ethyl phenylacetate, m-methoxyphenylacetate or p-bromophenylacetate (3.1–6.1 mmol) and ethyl formate (61.4–121.3 mmol). This mixture was stirred at room temperature under N2 for 24 h (TLC, CHCl3/MeOH 9.9:0.1), then poured into a mixture of ice and H2O, acidified with HCl, and extracted with EtOAc. The extract was dried over anhydrous Na2SO4 and concentrated under reduced pressure to yield colored oils.
Ethyl 3-oxo-2-(m-methoxyphenyl)propanoate (24)
Orange oil; yield 87 %; Rf = 0.84 (EtOAc/n-hexane 8:2); 1H NMR ([D6]DMSO): δ = 1.18 (t, 3H, J = 7.1 Hz, CH3), 3.72 (s, 1H, OCH3), 4.09 (q, 2H, J = 7.1 Hz, CH2), 6.78 (dd, 1H, J = 2.7 and 8.1, 4-H), 6.85 (m, 2H, 2- and 6-H), 7.21 (dd, 1H J = 8.1, 5-H),7.84 (s, 1H, CH), 10.95 (bs, 1 H, CHO); HRMS (ESI) calcd for C12H15O4 [M+H]+ m/z 223.0926, found 223.0939.
Ethyl 3-oxo-2-(p-bromophenyl)propanoate (25)
Yellow oil; yield 67 %; Rf = 0.86 (EtOAc/n-hexane 8:2); 1H NMR ([D6]DMSO): δ = 1.18 (t, 3 H, J = 7.1 Hz, CH3), 4.10 (q, 2 H, J = 7.1 Hz, CH2), 7.26 (d, 1 H, J = 8.4 Hz, 5- and 6-H), 7.49 (d, 1H, J = 8.6 Hz, 2- and 3-H), 7.87 (s, 1 H, CH), 11.194 (bs, 1 H, CHO); HRMS (ESI) calcd for C11H11O3Br [M+H]+ m/z 272.9949, found 270.9987 and 272.9994.
General procedure for the synthesis of 3-(1H-indol-5-ylamino-2-phenylpropanoate derivatives (28–32)
A solution of aminoindole (7,[2] 26,[3] or 27[3]) (1.9–2.2 mmol) in 20 mL absolute EtOH was added to aldehyde compounds (23,[7] 24, or 25) in equimolar ratio dissolved in a minimal quantity of EtOH. Drierite (100 mg) was added to the mixture, and the mixture was stirred at room temperature for 10–15 h. At the end (TLC, CHCl3/MeOH 9:1) the reaction mixture was filtered, and the filtrate was concentrated under vacuum to give semi-solid products.
Ethyl 3-(1H-indol-5-ylimino)-2-phenylpropanoate (28)
Dark oil; yield 57 %; Rf = 0.46 and 0.83 (EtOAc/n-hexane 8:2); 1H NMR([D6]DMSO): δ = 1.28 (t, 3H, J = 7.2 Hz, CH3), 4.14 (q, 2 H, J = 7.2 Hz, CH2), 7.44–7.18 (m, 10 H, ar), 7.64 (d, 1H, J = 13.0 Hz, =CH), 8.05 (d, 1H, J = 13.9 Hz, CH), 8.43 (d, 1H, J = 13.6 Hz, CH), 10.34 (d, 1 H, J = 13.0 Hz, =CH), 11.04 (bs, 1 H, NH); HRMS (ESI) calcd for C19H19N2O2 [M+H]+ m/z 307.1402, found 307.1444.
Ethyl 3-(1-ethyl-1H-indol-5-ylimino)-2-phenylpropanoate (29)
Black oil; yield 74 %; Rf = 0.30 and 0.85 (EtOAc/n-hexane 8:2); 1H NMR ([D6]DMSO): δ = 1.20 (m, 6H, J = 7.2 Hz, 2 × CH3), 4.14 (m, 6 H, J = 7.2 Hz, 2 0 CH2), 6.34 (t, 2 H, J = 2.5 Hz, 1- and 2-H), 7.27–7.45 (m, 8 H, ar), 7.65 (d, 1 H, J = 12.9 Hz, =CH), 8.05 (d, 1H, J = 13.5 Hz, NH), 8.45 (d, 1 H, J = 13.5 Hz, NH), 10.35 (d, 1 H, J = 12.9 Hz, =CH); HRMS (ESI) calcd for C21H23N2O2 [M+H]+ m/z 335.1715, found 335.1707.
Ethyl 3-(1-cyclopropylmethyl-1H-indol-5-ylimino)-2-phenylpropanoate (30)
Black liquid; yield 95 %; Rf = 0.58 and 0.92 (EtOAc/n-hexane 8:2); 1H NMR ([D6]DMSO): δ = 0.33 (m, 4H, 2 × CH2), 0.47 (m, 4 H, 2 × CH2), 1.30–1.90 (m, 12 H, 2 × CH), 4.15–4.41 (m, 6 H, 2 × CH2), 7.14–7.47 (m, 10 H, ar), 8.06 (d, 1H, J = 13.0 Hz, =CH), 8.40 (d, 1 H, J = 13.7 Hz, NH), 8.60 (d, 1H, J = 13.7 Hz, NH), 10.33 (d, 1H, J = 13.0 Hz, =CH).
Ethyl 3-(1-ethyl-1H-indol-5-ylimino)-2-(3-methoxyphenyl)propanoate (31)
Black liquid; yield 75 %; mp: 249–250 °C; Rf = 0.64 and 0.85 (EtOAc/n-hexane 9:1); 1H NMR ([D6]DMSO): δ = 1.33 (m, 6 H, 2 × CH3), 4.14 (m, 4 H, 2 × CH2), 6.82–7.45 (m, 9 H, ar), 7.66 (d, 1 H, J = 13.2 Hz, =CH), 8.04 (d, 1 H, J = 13.6 H z, CH), 8.45 (d, 1 H, J = 13.9 Hz, CH), 10.36 (d, 1 H, J = 13.2 Hz, =CH); HRMS (ESI) calcd for C22H25N2O3 [M+H]+ m/z 365.1820, found 365.1796.
Ethyl 2-(4-Bromophenyl)-3-(1-ethyl-1H-indol-5-ylimino)propanoate (32)
Black oil; yield 73.93 %; Rf = 0.52 and 0.92 (EtOAc/n-hexane 8:2); 1H NMR ([D6]DMSO): δ = 1.30 (m, 6H, 2 × CH3), 4.17 (m, 4 H, 2 × CH2), 7.57–7.07 (m, 9 H, ar), 7.69 (d, 1H, J = 13.2 Hz, =CH), 8.05 (d, 1H, J = 13.6 Hz, NH), 8.65 (d, 1H, J = 13.6 Hz, NH), 10.38 (d, 1H, J = 13.2 Hz, =CH); HRMS (ESI) calcd for C21H22N2O2Br [M+H]+ m/z 414.0766, found 413.0823 and 415.0799.
General procedure for the synthesis of 8-phenyl-pyrrolo[3,2-f]quinolinones (33–37)
In a two-necked round-bottomed flask, Ph2O (30 mL) was heated at boiling point; 1–2 mmol of propanoates 28–32 were then added in portions, and the mixture was held at reflux for 15 min. After cooling to 30°C, the light precipitate slowly separated and was then collected by filtration and washed several times with Et2O. In all cases, the collected products were purified by recrystallization from a suitable solvent.
8-Phenyl-3H-pyrrolo[3,2-f]quinolin-9(6H)-one (33)
Light-brown solid; yield 87.98 %; mp: 178–190°C (MeOH); Rf = 0.28 (EtOAc/n-hexane 8:1, violet fluorescent spot at λ365 nm); 1H NMR ([D6]DMSO): δ = 7.25 (dd, 1 H, J = 7.4 Hz, 4′-H), 7.29 (dd, 1 H, J = 2.7 Hz, 1-H), 7.39 (d, 2 H, J = 7.4 Hz, 2′- and 6′-H), 7.45 (dd, 1 H, J = 2.7 Hz, 2-H), 7.61 (dd, 2 H, J = 8.4 Hz, 4-H and 5-H), 7.76 (d, 2 H, J = 7.4 Hz, 3′-H and 5′-H), 8.06 (d, 1H, J = 6.1 Hz, 7-H), 11.50 (bs, 1 H, NH), 11.98 (d, 1H, J = 5.7 Hz, NH); 13C NMR ([D6]DMSO): δ = 104.8, 111.9, 117.5, 119.2, 120.5, 123.5, 125.6, 126.3, 128.0, 128.9, 131.8, 135.5, 135.6, 137.3, 175 (CO); HRMS (ESI) calcd for C17H13N2O [M+H]+ m/z 261.0975, found 261.1272; anal. calcd for C17H12N2O: C 78.44, H 4.65, N 10.76, found: C 78.31, H 4.52, N 10.56.
3-Ethyl-8-phenyl-3H-pyrrolo[3,2-f]quinolin-9(6H)-one (34)
Light-brown solid; yield (93%); mp: 248–250 °C (EtOH); Rf = 0.45 (EtOAc/n-hexane 8:1, violet fluorescent spot at λ365 nm); 1H NMR ([D6]DMSO): δ = 1.40 (t, 3H, J = 7.3 Hz, CH3), 4.32 (q, 2 H, J = 7.1 Hz, CH2), 7.27 (dd, 1H, J = 7.4 Hz, 4′-H), 7.36 (dd, 1H, J = 8.5 Hz, 5-H), 7.40 (dd, 2 H, J = 7.6 Hz, 2′-H and 6′-H), 7.51 (d, 1 H, J = 2.7 Hz, 1-H), 7.60 (d, 1H, J = 2.7 Hz, 2-H), 7.78 (d, 2 H, J = 7.8 Hz, 3′- and 5′-H), 7.88 (d, 1 H, J = 8.5 and 0.6 Hz, 4-H), 8.07 (s, 1 H, 7-H), 12.02 (bs, 1 H, NH); 13C NMR ([D6]DMSO): δ = 16.3 (CH3), 40.9 (CH2), 104.2, 112.1, 115.7, 119.3, 120.6, 124.1, 126.4, 128.1, 128.3, 128.8, 131.1, 135.58, 135.7, 137.2, 175.8 (CO); HRMS (ESI) calcd for C19H17N2O [M+H]+ m/z 289.1288, found 289.1266; anal. calcd for C19H16N2O: C 79.14, H 5.59, N 9.72, found: C 78.94, H 5.34, N 9.60.
3-Methylcyclopropyl-8-phenyl-3H-pyrrolo[3,2-f]quinolin-9(6H)-one (35)
Grey solid; Yield 60 %; mp: 197–198°C (70 % EtOH); Rf = 0.57 (EtOAc/n-hexane 8:2, violet fluorescent spot at λ365 nm); 1H NMR ([D6]DMSO): δ = 0.38 (m, 2 H, CH2), 0.51 (m, 2 H, CH2), 1.24 (m, 2 H, CH), 3.98 (d, 2H, J = 6.87 Hz, CH2), 7.27 (dd, 1 H, J = 7.3 Hz, 4′-H), 7.38 (m, 3 H, 2′-, 6′- and 5-H), 7.55 (dd, 1H, J = 2.9 Hz, 2-H), 7.62 (dd, 1 H, J = 3.6 Hz, 1-H), 7.78 (d, 2H, J = 1.3 Hz, 5′ and 3′-H), 7.91 (d, 1 H, J = 8.8 Hz, 4-H), 8.07 (d, 1 H, J = 6.3 Hz, 7-H), 12.0 (d, 1 H, J = 6.10 Hz, NH); 13C NMR ([D6]DMSO): δ = 4.1 (2 × CH2), 12.2 (CH), 50.2 (CH2), 100.4, 104.1, 110.1, 114.8, 115.9, 118.9, 120.6, 124.0, 126.4, 128.1, 128.5, 128.9, 128.9, 130.4, 135.5, 137.3, 175.9 (CO); HRMS (ESI) calcd for C21H20N2O [M+H]+ m/z 315.1445, found 315.1422; anal. calcd for C21H19N2O: C 80.23, H 5.77, N 8.91, found: C 79.91, H 5.46, N 8.81.
3-Ethyl-8-(3-methoxyphenyl)-3H-pyrrolo[3,2-f]quinolin-9(6H)-one (36)
Light-yellow solid; yield 60 %; mp: 262–264°C (MeOH); Rf = 0.65 (EtOAc/n-hexane 9:1, violet fluorescent spot at λ365 nm); 1H NMR ([D6]DMSO): δ = 1.40 (t, 3H, J = 7.2 Hz, CH3), 3.8 (s, 3 H, OCH3), 4.32 (q, 2 H, J = 7.2 Hz, CH2), 6.85 (m, 1 H, J = 1.5 Hz, 4′-H), 7.28 (d, 1 H, J = 8.3 Hz, 5-H), 7.33 (d, 1 H, J = 1.9 Hz, 6′-H), 7.36 (d, 1 H, J = 1.9 Hz, 5′-H), 7.43 (m, 1 H, J = 1.5 Hz, 2′-H), 7.45 (d, 1H, J = 2.9 Hz, 1-H), 7.61 (d, 1 H, J = 2.9 Hz, 2-H), 7.88 (dd, 1 H, J = 8.3 and 0.8 Hz, 4-H), 8.09 (s, 1H, 7-H), 12.00 (bs, 1H, NH); 13C NMR ([D6]DMSO): δ = 16.26 (CH3), 40.9 (CH2), 55.4 (OCH3), 104.1, 111.9, 111.9, 114.6, 115.8, 120.3, 121.2, 124.0, 129.0, 129.1, 131.7, 135.4, 135.8, 138.6, 159.2, 175.8 (CO); HRMS (ESI) calcd for C20H19N2O2 [M+H]+ m/z 333.1511, found 333.1516; anal. calcd for C20H18N2O2: C 75.45, H 5.70, N 8.80, found: C 75.18, H 5.35, N 8.71.
8-(4-Bromophenyl)-3-ethyl-3H-pyrrolo[3,2-f]quinolin-9(6H)-one (37)
Grey solid; yield 50 %; mp: 216–218 °C (MeOH/EtOH 80:20); Rf = 0.65 (EtOAc/n-hexane 9:1, violet fluorescent spot at λ365 nm); 1H NMR ([D6]DMSO): δ = 1.40 (t, 3 H, J = 7.2 Hz, CH3), 4.32 (q, 2 H, J = 7.2 Hz, CH2), 7.34 (d, 1 H, J = 3.2 Hz, 1-H), 7.36 (d, 1 H, J = 3.1 Hz, 2-H), 7.56 (dd, 2 H, J = 8.6 and J = 2.0 Hz, 2′- and 6′-H), 7.60 (d, 1 H, J5,4 = 8.4 Hz, 5-H), 7.81 (dd, 2H, J = 8.6 and 1.97 Hz, 3′and 5′-H), 7.89 (dd, 1 H, J5,4 = 8.4 and J4,1 = 0.6 Hz, 4-H), 8.14 (s, 1 H, 7-H), 12.07 (bs, 1 H, NH); 13C NMR ([D6]DMSO): δ = 16.3 (CH3), 40.9 (CH2), 100.3, 110.3, 114.9, 118.9, 119.2, 119.3, 123.8, 128.5, 130.4, 130.8, 130.9, 132.3, 136.4, 153.8, 175.7 (CO); HRMS (ESI) calcd for C19H16BrN2O [M+H]+ m/z 383.0538, found 332.0870; anal. calcd for C19H15BrN2O: C 62.14, H 4.12, N 7.63, found: C 62.01, H 3.89, N 7.51.
Biology
Growth inhibitory activity
Human T-leukemia (Jurkat), promyelocytic leukemia (HL-60), chronic myelogenous leukemia (K562), myeloid leukemia (ML-2), and acute lymphoblastic leukemia (RS 4;11) cells, the latter with a t(4;11) translocation, were grown in RPMI-1640 medium (Gibco Milan, Italy). Human breast adenocarcinoma (MCF-7), cervix carcinoma (HeLa), ovarian cancer (IGROV), anaplastic thyroid (ARO), hepatoma (HepG2), and colon adenocarcinoma (HT-29) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with penicillin G (115 U mL−1; Gibco), streptomycin (115 mg mL−1; Invitrogen, Milan, Italy) and 10 % fetal bovine serum (Invitrogen). Individual wells of a 96-well tissue culture microtiter plate were inoculated with 100 mL complete medium containing 8 × 103 cells. The plates were incubated at 37 °C in a humidified incubator containing 5% CO2 for 18 h. At this time, the initial medium was removed from each well, and drug solutions (100 μL each), dissolved in complete medium, at various concentrations, was added to each well and incubated at 37°C for 72 h. Cell viability was assayed by the [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (MTT) test as previously described.[25] The GI50 value is defined as the compound concentration required to inhibit cell proliferation by 50 %.
Effects on tubulin polymerization and on colchicine binding to tubulin
Bovine brain tubulin was purified as described previously. To evaluate the effect of the compounds on tubulin assembly in vitro, various concentrations were pre-incubated with 10 mM tubulin in glutamate buffer at 30 °C and then cooled to 0 °C. After addition of GTP, the mixtures were transferred to 0 °C cuvettes in a recording spectrophotometer and warmed to 30°C, and the assembly of tubulin was observed turbidimetrically. The IC50 value is defined as the compound concentration required to inhibit the extent of assembly by 50 % after incubation for 20 min. The capacity of the test compounds to inhibit colchicine binding to tubulin was measured as described,[8,9] except that the reaction mixtures contained 1 mM tubulin, 5 mM [3H]colchicine, and 1 mM test compound.
Flow cytometric analysis of cell-cycle distribution and apoptosis
For flow cytometric analysis of DNA content, 5 × 105 Jurkat cells in exponential growth were treated with various concentrations of the test compounds for 24 and 48 h. After an incubation period, the cells were collected, centrifuged, and fixed with ice-cold ethanol (70 %). The cells were then treated with lysis buffer containing RNase A and Triton X-100 (0.1 %), and then stained with PI. Samples were analyzed on a Cytomic FC500 flow cytometer (Beckman Coulter). DNA histograms were analyzed using MultiCycle® for Windows (Phoenix Flow Systems, CA, USA).
Annexin V assay
Surface exposure of PS by apoptotic cells was measured by flow cytometry with a Coulter Cytomics FC500 (Beckman Coulter, USA) by adding annexin V–FITC to cells according to the manufacturer’s instructions (annexin V Fluos, Roche Diagnostics). The cells were simultaneously stained with PI. Excitation was set at λ 488 nm, and the emission filters were set at λ 525 and 585 nm.
Assessment of mitochondrial changes
The mitochondrial membrane potential was measured with the lipophilic cation 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1, Molecular Probes, USA), as described.[25] Briefly, after various times of treatment, the cells were collected by centrifugation and resuspended in Hank’s Balanced Salt Solution (HBSS) containing JC-1 at a concentration of 1 mM. The cells were then incubated for 10 min at 37 °C, centrifuged, and resuspended in HBSS. The production of ROS was measured by flow cytometry using HE (Molecular Probes) and H2-DCFDA (Molecular Probes).
After various treatment times, the cells were collected by centrifugation and resuspended in HBSS containing the fluorescence probes HE and H2-DCFDA at concentrations of 2.5 and 0.1 mM, respectively. The cells were then incubated for 30 min at 37°C, centrifuged, and resuspended in HBSS. The fluorescence was recorded directly with the flow cytometer using an excitation wavelength of λ 488 nm and emission at λ 585 and 530 nm for HE and H2-DCFDA, respectively.
Caspase-3 assay
Caspase-3 activation in Jurkat cells was evaluated by flow cytometry using a human active caspase-3 fragment antibody conjugated with FITC (BD Pharmingen) as described.[26] Briefly, after various incubation times in the presence of test compounds, the cells were collected by centrifugation and resuspended in Cytofix™ (BD Pharmingen) buffer for 20 min, washed with Perm/Wash™ (BD Pharmingen) and then incubated for 30 min with the antibody. After this period the cells were washed and analyzed by flow cytometry. The results are expressed as a percentage of caspase-3 active fragment positive cells.
Flow cytometric analysis of Bcl-2 expression
Bcl-2 expression was evaluated in Jurkat cells by flow cytometry using a monoclonal antibody conjugated with FITC (DAKO). Briefly, after various incubation times in the presence of test compounds, the cells were collected by centrifugation and permeabilized using a Fix&Perm cell permeabilization kit (Caltag Laboratories, Burlingame, CA, USA). The cells were then labeled with Bcl-2 antibody, washed, and analyzed by flow cytometry.
Western blot analysis
Jurkat cells were incubated in the presence of test compounds and, at various time points, were collected, centrifuged, and washed twice with ice-cold PBS. The pellet was then resuspended in lysis buffer. After the cells were lysed on ice for 30 min, lysates were centrifuged at 15 000 g at 4°C for 10 min. The protein concentration in the supernatant was determined using BCA protein assay reagents (Pierce, Italy). Equal amounts of protein (20 μg) were resolved by SDS-PAGE (12 % acrylamide) and transferred to PVDF Hybond-p membrane (GE Healthcare). Membranes were blocked with I-block (Tropix) overnight, under rotation at 4 °C. Membranes were then incubated with a primary antibody against the cleaved fragment of PARP (rabbit, 1:1000, Cell Signaling), or β-actin (mouse, 1:10 000, Sigma) for 2 h at room temperature. Membranes were next incubated with peroxidase-labeled goat anti-rabbit IgG (1:100 000, Sigma) or peroxidase-labeled goat anti-mouse IgG (1:100 000, Sigma) for 60 min. All membranes were visualized using ECL Advance (GE Healthcare) and exposed to Hyper film MP (GE Healthcare). To ensure equal protein loading, each membrane was stripped and re-probed with anti-β-actin antibody.
Acknowledgments
This work was supported by grants from the Italian Ministry of Education, University and Research (MIUR).
References
- 1.Ferlin MG, Chiarelotto G, Gasparotto V, Barzon L, Palù G, Castagliuolo I. J Med Chem. 2005;48:3417–3427. doi: 10.1021/jm049387x. [DOI] [PubMed] [Google Scholar]
- 2.Gasparotto V, Castagliuolo I, Chiarellotto G, Pezzi V, Montanaro D, Brun P, Palù G, Viola G, Ferlin MG. J Med Chem. 2006;49:910–1915. doi: 10.1021/jm0510676. [DOI] [PubMed] [Google Scholar]
- 3.Gasparotto V, Castagliuolo I, Ferlin MG. J Med Chem. 2007;50:5509–5513. doi: 10.1021/jm070534b. [DOI] [PubMed] [Google Scholar]
- 4.Burrus HO, Powell G. J Am Chem Soc. 1945;67:1468–1472. [Google Scholar]
- 5.Owa T, Yoscino H, Okauchi K, Yoshimatsu K, Ozawa Y, Sugi NH, Nagasu T, Koyanagi N, Kitoh K. J Med Chem. 1999;42:3789–3799. doi: 10.1021/jm9902638. [DOI] [PubMed] [Google Scholar]
- 6.Huang LJ, Hsieh MC, Teng CM, Lee KH, Kuo SC. Bioorg Med Chem. 1998;6:1657–1662. doi: 10.1016/s0968-0896(98)00141-2. [DOI] [PubMed] [Google Scholar]
- 7.Beccalli EM, La Rosa C, Marchesini A. J Org Chem. 1984;49:4287–4290. [Google Scholar]
- 8.Hamel E. Cell Biochem Biophys. 2003;38:1–21. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 9.Verdier-Pinard P, Lai JY, Yoo HD, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Mol Pharmacol. 1998;53:62–76. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- 10.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 11.Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, Laface DM, Green DR. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Green DR, Kroemer G. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 13.Ly JD, Grubb DR, Lawen A. Apoptosis. 2003;8:115–128. doi: 10.1023/a:1022945107762. [DOI] [PubMed] [Google Scholar]
- 14.Salvioli S, Ardizzoni A, Franceschi C, Cossarizza A. FEBS Lett. 1997;411:77–82. doi: 10.1016/s0014-5793(97)00669-8. [DOI] [PubMed] [Google Scholar]
- 15.Mollinedo F, Gajate C. Apoptosis. 2003;8:413–450. doi: 10.1023/a:1025513106330. [DOI] [PubMed] [Google Scholar]
- 16.Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B, Kroemer G. J Exp Med. 1995;182:367–377. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staniek K. Biochem Pharmacol. 2005;69:719–723. doi: 10.1016/j.bcp.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 18.Rothe G, Valet G. J Leukocyte Biol. 1990;47:440–448. [PubMed] [Google Scholar]
- 19.Earnshaw WC, Martins LM, Kaufmann SH. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- 20.Denault JB, Salvesen GS. Chem Rev. 2002;102:4489–4499. doi: 10.1021/cr010183n. [DOI] [PubMed] [Google Scholar]
- 21.Porter G, Janicke RU. Cell Death Differ. 1996;3:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 22.Soldani C, Scovassi AI. Apoptosis. 2002;7:321–328. doi: 10.1023/a:1016119328968. [DOI] [PubMed] [Google Scholar]
- 23.Kluck RM, Bossy-Wetzel E, Green DR. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 24.Knudson CM, Korsmeyer SJ. Nat Genet. 1997;16:358–363. doi: 10.1038/ng0897-358. [DOI] [PubMed] [Google Scholar]
- 25.Viola G, Cecconet L, Leszl A, Brun P, Salvador A, Dall’Acqua F, Basso G, Diana P, Barraja P, Cirrincione G. Cancer Chemother Pharmacol. 2009;64:1235–1251. doi: 10.1007/s00280-009-0994-9. [DOI] [PubMed] [Google Scholar]
- 26.Romagnoli R, Baraldi PG, Carrion MD, Cruz-Lopez O, Cara CL, Balzarini J, Hamel E, Canella A, Fabbri E, Gambari R, Basso G, Viola G. Bioorg Med Chem Lett. 2009;19:2022–2028. doi: 10.1016/j.bmcl.2009.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]