

Abstract
Spinocerebellar ataxia 1 (SCA1) is a dominantly inherited neurodegenerative disease associated with progressive ataxia resulting from the loss of cerebellar Purkinje cells (PCs) and neurons in the brainstem. In PCs of SCA1 transgenic (Tg) mice, the disease causing ataxin-1 protein mediates the formation of S100B containing cytoplasmic vacuoles and further self-aggregates to form intranuclear inclusions. The exact function of the ataxin-1 protein is not fully understood. However, the aggregation and neurotoxicity of the mutant ataxin-1 protein is dependent on the phosphorylation at serine 776 (S776). Although protein kinase A (PKA) has been implicated as the S776 kinase, the mechanism of PKA/ataxin-1 regulation in SCA1 is still not clear. We propose that a dopamine D2 receptor (D2R)/S100B pathway may be involved in modulating PKA activity in PCs. Using a D2R/S100B HEK stable cell line transiently transfected with GFP-ataxin-1[82Q], we demonstrate that stimulation of the D2R/S100B pathway caused a reduction in mutant ataxin-1 S776 phosphorylation and ataxin-1 aggregation. Activation of PKA by forskolin resulted in an enhanced S776 phosphorylation and increased ataxin-1 nuclear aggregation, which was suppressed by treatment with D2R agonist bromocriptine and PKA inhibitor H89. Furthermore, treating SCA1 Tg PC slice cultures with forskolin induced neurodegenerative morphological abnormalities in PC dendrites consistent with those observed in vivo. Taken together our data support a mechanism where PKA dependent mutant ataxin-1 phosphorylation and aggregation can be regulated by D2R/S100B signaling.
Introduction
SCA1 is one of the nine-inherited neurodegenerative diseases that belong to a group of trinucleotide repeat disorders of the nervous system. SCA1 is caused by a mutation in the ataxin-1 protein and is associated with progressive ataxia resulting from the loss of cerebellar PCs and neurons in the brainstem (Zoghbi et al. 2000; Koeppen 2005; Matilla et al. 2007). The exact function of the ataxin-1 protein has yet to be deciphered (Orr et al. 1993; Banfi et al. 1994; Matilla et al. 2007). Overexpression of the human mutant ataxin-1 gene in PCs of SCA1 Tg mice results in a progressiveataxia and PC pathology very similar towhat are seen in SCA1 patients (Burright et al. 1995). A prominent feature of PC degeneration in SCA1 Tg mice is the development of cytoplasmic vacuoles and intranuclear inclusions. The vacuolar development occurs much earlier, preceding the appearance of nuclear aggregates and the onset of behavioral abnormalities (Skinner et al. 2001; Vig et al. 2006; Vig et al. 2009). The immunohistochemical analysis revealed that PC vacuoles of both SCA1 mice and human patients localize S100B protein, which otherwise is exclusively expressed in Bergmann glia (Vig et al. 2006; Vig et al. 2009). However, a definite role of glial proteins in SCA1 pathology has yet to be described.
S100B belongs to a family of Ca2+-modulated proteins of the EF-hand type (Donato 1991, 1999; Zimmer et al. 2005). S100B is abundantly expressed in the nervous system as a cytokine with both neurotrophic and neurotoxic effects (Reeves et al. 1994; Huttunen et al. 2000; Rothermundt et al. 2003; Zimmer et al. 2005). Glial expressed S100B released from astrocytes has been shown to promote neuronal survival and development (Winningham-Major et al. 1989; Barger et al. 1995; Whitaker-Azmitia et al. 1997). It has been recently reported that S100B interacts with D2Rs and enhances dopamine signaling in HEK 293 cells co-expressing D2R and S100B proteins (Liu et al. 2008). S100B binds to the amino terminus of the third cytoplasmic loop of the D2R (Liu et al. 2008). Further, the D2R has a S100B-binding motif similar to TRTK12, a S100B inhibitory peptide (Ivanenkov et al. 1995; Bianchi et al. 1996; Liu et al. 2008). D2Rs are highly localized to cerebellar PCs and interestingly, D2Rs are down-regulated in mice lacking ataxin-1 as well as in SCA1 Tg mice (Goold et al. 2007). Dopamine is widely distributed in the central nervous system and is involved in the control of movement, cognition, endocrine responses and reward. Furthermore, dopaminergic abnormalities lead to many psychiatric and neurological disorders including SCA1 (Kish et al. 1997; Goold et al. 2007). D2Rs, via G-protein-coupling, inhibit adenylate cyclase and cAMP accumulation (Watts et al. 1996; Neve et al. 2004; Beaulieu et al. 2005; Liu et al. 2008).
cAMP-dependent protein kinase, PKA, is a serine/threonine protein kinase, which regulates many cellular processes including metabolism, cell cycle and cellular response to extracellular stimuli. The PKA holoenzyme consists of both regulatory and catalytic subunits. Adenylate cyclase production of cAMP increases PKA activity and the phosphorylation of PKA substrates. Therefore, factors resulting in an increase or decrease in cAMP levels are modulators of PKA activity. Here, we report that the stimulation of PKA increases both normal and mutant ataxin-1 phosphorylation and intranuclear inclusion formation in a cell culture model. Since ataxin-1 is phosphorylated by PKA at S776 (Jorgensen et al. 2009), the inhibition of adenylate cyclase and/or accumulation of cAMP may play a critical role in the SCA1 pathogenesis. We believe that by targeting D2R/S100B signaling, cAMP levels can be reduced, which could be therapeutically beneficial to SCA1 patients.
Materials and Methods
Materials
Bromocriptine (BRC), forskolin (FSK), H89, culture media and β tubulin antibody were purchased from Sigma-Aldrich (St. Louis, MO, USA). Fetal bovine serum (FBS) for cell culture was purchased from HyClone (Logan, UT, USA). Fugene 6 transfection reagent and GFP antibody were purchased from Roche (Indianapolis, IN, USA). S100B and phospho-ataxin-1 S776 antibodies were purchased from Abcam (Cambridge, MA, USA). D2R antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-calbindin D-28K (CaB) was obtained from Millipore (Temecula, CA, USA). βIII tubulin antibody was purchased from Promega (Madison, WI, USA). WT-ataxin-1 antibody was obtained from Antibodies Incorporated (Davis, CA, USA).
Cell Culture and Transfections
D2R/S100B HEK cells, previously described (Liu et al. 2008), were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS, penicillin-streptomycin, appropriate selection antibiotics (G418 sulfate and puromycin, Sigma), and grown in an incubator at 37°C in the presence of 5% CO2. WT HEK 293 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). GFP CMV expression vector was purchased from Clone Tech (Mountain View, CA, USA). GFP-ataxin-1[30Q] and [82Q], mammalian CMV expression vectors were kindly provided by Dr. Huda Zoghbi, Howard Hughes Medical Institute, Baylor College of Medicine (Cummings et al. 1998; Emamian et al. 2003; Chen et al. 2003; Vig et al. 2007). GFP-ataxin-1 A776 mutants were kindly provided by Dr. Harry Orr, Institute of Human Genetics, University of Minnesota (Emamian et al. 2003). The identities of the various GFP-ataxin-1 constructs were verified by western blotting as shown previously (Vig et al. 2007) and by immunofluorescence, where A776 mutants show no reaction with the phospho-ataxin-1 S776 antibodies. The GFP-ataxin-1 constructs were transformed into E. coli D5Hα cells (Invitrogen) and selected by kanamycin (Sigma) for plasmid propagation according to the manufacturer’s guide lines. D2R/S100B HEK cells were transfected using Fugene 6 transfection reagent (Roche) according to the manufacture’s guide lines to express the various GFP-ataxin-1 constructs.
Immunofluorescence, Western Blotting and Phosphorylation Studies
D2R/S100B HEK cells were placed on 2 well Lab-Tek chamber slides (Fisher, Houston, TX, USA) or on 6 well plates (Fisher) and transfected with either GFP vector alone, GFP-ataxin-1[30Q], [82Q] or GFP-ataxin-1[85Q] A776; cells were subjected to treatments with varying concentrations of bromocriptine (50–100nM), forskolin (30–100μM), and H89 (10–50μM) followed by immunocytochemistry as previously described (Hearst et al. 2009). Fixed slides were probed with the primary GFP and phospho-ataxin-1 S776 antibodies and fluorescent secondary antibodies Alexa-488 and Alexa-546 (Invitrogen, Carlsbad, CA). Cellswere observed on an Olympus BX60 epifluorescence microscope. Scoring of GFP-ataxin-1 transfected cells was performed according to the method described by Parfitt and co-workers (2009), cells were scored as soluble diffuse localization within the nucleus, as small punctuate foci or as larger inclusions (Parfitt et al. 2009). For western blotting, lysates were generated by resuspending cells in RIPA buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% sodium deoxycholate and 1% NP-40, Sigma), then sonicated as previously described (Hearst et al. 2009). Lysates were subjected to SDS-PAGE (4–20% acrylamide gels; Bio-Rad Laboratories, Hercules, CA, USA) as described earlier (Vig et al. 2009). Equal amounts of proteins were loaded in each well. Proteins were transferred to polyvinylidene difluoride (PVDF) membrane (Bio-Rad), blocked for 1hr with blocking solution (Western Breeze, Invitrogen) and incubated overnight with appropriate concentration of GFP or phospho-ataxin-1 S776 antibodies. Immunoreactive proteins were visualized by incubation (for 1 hr) in alkaline phosphatase-labeled secondary antibody followed by reaction with the luminescent substrates (Western Breeze, Invitrogen). Western blots were analyzed using Image J software, a public domain Java image processing program provided by the Research Services Branch, National Institute of Mental Health, Bethesda, Maryland, USA.
SCA1 Transgenic Mice
The SCA1 Tg mice were generated by Drs. Harry Orr and Huda Zoghbi (Burright et al. 1995). We maintain colonies of SCA1 Tg mice in our animal facility. The heterozygous PS-82 BO5 line of mice were identified using a transgene specific polymerase chain reaction assay and were backcrossed to the parental FVB/N strain (N=10) to establish congenic line with homogeneous background strain (Vig et al. 2009). The B05 line expresses 30 copies of the transgene PS-82 to produce the mutant human ataxin-1 protein in PCs (Burright et al. 1995). These mice develop progressive loss of PCs and cerebellar function, where heterozygous mice become visibly ataxic at 12 weeks of age and homozygotes show ataxia at 6 weeks of age. Wild-type (WT, same background) mice were obtained from Jackson Labs, Bar Harbor, ME, USA. All animal protocols were approved by Institutional Animal Care and Use Committee (IACUC) at the University of Mississippi Medical Center (UMMC, Jackson, MS, USA).
Purkinje Cell Cultures
Cerebellar PC cultures were prepared from SCA1 Tg mice and WT mice. The whole cerebella were removed from 7 day old mouse pups. Meninges were carefully removed under the dissection microscope. Isolated cerebellar tissues were rinsed and placed in petri dishes containing cold Gey’s balanced salt solution (Gey’s solution, Sigma) containing 5% glucose. Tissues were cut into 250 μm slices using a McIlwain tissue chopper and resuspended in cold Gey’s solution containing 5% glucose. Two to three slices were grown on Millicell membrane inserts (Fisher) using 6-well culture plates containing 1 ml plating media (vol/vol: 5% 10X Basal Medium with Earle’s Salt, 2.5% 10X HBSS, 25% Horse Serum (Invitrogen), 1% 100X Pen-Strep-Glutamine, 4.5% 10% D-Glucose, 0.5% 7.5% Sodium bicarbonate and 61.5% sterile water, Sigma). Plates were incubated overnight at 35.5°C with 5% CO2 and 100% humidity. The next day, cells were flushed and treated with various concentrations of forskolin in plating media. Cultures were fixed for immunostaining using calbindin-D28k antibody or lysed in RIPA buffer for western blotting as described above.
Tissue Processing
SCA1 Tg mice at 5 weeks of age were anesthetized and perfused with 4% buffered paraformaldehyde according to the method described by Neuroscience Associates, Knoxville, TN, USA. The brains were removed and immersed in the perfusion fixative then transferred to phosphate-buffered and processed for vibratome sectioning or paraffin embedding as previously described (Vig et al. 2009). Sections were cut from the midline sagittal plane of the cerebella. Sections were immunostained with D2R and S100B antibodies using the immunohistochemical protocol as previously described (Vig et al. 2009). Western blot analysis of the 4 and 6 week old SCA1 Tg mice and WT mice cerebellar samples were performed according to the protocol as previously described (Vig et al. 2009). The western blot membranes were probed overnight with the appropriate concentration of D2R and βIII Tubulin antibodies followed by alkaline phosphatase-labeled secondary antibody and luminescent substrate. The blots were visualized on hyperfilm-ECL (Amersham Biosciences, Buckinghamshire, UK).
Results
Stimulating D2R/S100B Signaling and Inhibiting PKA Activity Reduces Ataxin-1 S776 Phosphorylation and Aggregation
Dopamine D2 receptor signaling coupled to G-inhibitory-proteins is a well-established mechanism of regulating adenylate cyclase activity. Activation of adenylate cyclase generates second messenger cAMP, which increases PKA activity and phosphorylation of PKA substrates. Previously, Liu and co-workers reported that stimulation of D2R/S100B signaling by D2R agonists reduces cAMP levels in a D2R/S100B HEK stable cell line (Liu et al. 2008). Since phosphorylation is a common mechanism known to regulate the assembly of self-associating nuclear proteins (Emamian et al. 2003; Grimmler et al. 2005; Bansal et al. 2009; Hearst et al. 2009), we explored the impact of D2R signaling on mutant ataxin-1 phosphorylation and self-aggregation by transfecting GFP-ataxin-1[82Q] into a D2R/S100B HEK stable cell line (Fig. 1 and 2A). D2/S100B HEK cells expressing GFP-ataxin-1[82Q], under various treatment conditions, were scored as soluble ataxin-1, small ataxin-1 foci or large ataxin-1 inclusions, as described previously by Parfitt and co-workers (Parfitt et al. 2009)(Fig. 3A). FSK is a well established cAMP stimulator that activates adenylate cyclase increasing PKA activity and phosphorylation of PKA substrates (Seamon et al. 1981; Battaglia et al. 1986; Chijiwa et al. 1990; Fujihara et al. 1993; Muroi et al. 1993; Wroblewska et al. 1993; Insel et al. 2003). Immunofluorescence of D2/S100B HEK cells expressing GFP-ataxin-1[82Q] showed a significant increase in nuclear inclusion formation after treatment with FSK (Fig. 2A and 3B). FSK induced large GFP-ataxin-1 [82Q] inclusions containing phosphorylated S776 ataxin-1 (Fig. 2A). Whereas, control cells expressing GFP-ataxin-1[82Q] showed diffuse soluble nuclear staining (Fig. 2A). To investigate the impact of D2R/S100B signaling on inclusion formation, we used BRC, a selective D2 agonist, previously shown to reduce cAMP levels in HEK cells expressing D2Rs (Watts et al. 1996). To explore the role of PKA in ataxin-1 inclusion formation, we used H89, a potent and selective PKA inhibitor (Chijiwa et al. 1990; Fujihara et al. 1993; Muroi et al. 1993; Lortet et al. 1999; Mao et al. 2007). We found that FSK induced inclusion formation was significantly reduced by 2hr pretreatment with BRC or H89 (Fig. 2A and 3B). EC50 for BRC, FSK and H89 were 50nM, 30μM and 5μM respectively. D2/S100B HEK cells expressing GFP-ataxin-1[82Q] treated with H89 showed reduced phospho-ataxin-1 S776 staining compared to control. In contrast, GFP-ataxin-1[85Q] A776 transfected cells showed no phospho-ataxin-1 S776 immunostaining and failed to form inclusions (Fig. 2A). As a control, we expressed GFP-ataxin-1[82Q] and [30Q] in WT HEK cells, not expressing D2R or S100B, and treated these cells under the previously described conditions (Fig. 2B and C). We found that FSK also stimulated both GFP-ataxin-1[82Q] and [30Q] inclusion formation in WT HEK cells, whereas BRC was not effective in reducing inclusions (Fig. 3C and D). Further, H89 significantly reduced FSK stimulated inclusion formation in both GFP-ataxin-1[82Q] and [30Q] expressing cells (Fig. 3C and D). To explore the importance of ataxin-1 S776 phosphorylation site in inclusion formation, we expressed GFP-ataxin-1[85Q] A776 in WT HEK cells and treated with FSK. FSK had no effect on the inclusion formation in cells expressing A776 mutant ataxin-1 protein (data not shown). Further, for semi-quantification D2/S100B HEK cells expressing GFP-ataxin-1[82Q] treated with FSK, BRC, BRC + FSK, H89 or H89 + FSK were subjected to western blot analysis (Fig. 4A and B). FSK stimulation of PKA activity resulted in a large increase in ataxin-1 S776 phosphorylation compared to control (Fig. 4C). Furthermore, activation of D2R by BRC or inhibition of PKA by H89 greatly reduced FSK stimulated ataxin-1 S776 phosphorylation (Fig. 4C). To ensure that the various drugs were not influencing the CMV promoter activity of the various GFP-ataxin-1 constructs, we expressed GFP-ataxin-1[30Q], [82Q] and the GFP vector alone in WT HEK cells followed by the 2hr drug treatments. Western blot analysis displayed no remarkable change in the soluble GFP-ataxin-1[30Q] (Fig. 4D), GFP-ataxin-1[82Q] (Fig. 4E) or GFP (Fig. 4F) protein levels with respect to β tubulin. Overall, the cell culture data suggest that both mutant and nonpathogenic ataxin-1 S776 phosphorylation and aggregation is PKA dependent and mutant ataxin-1 phosphorylation and aggregation can be modulated through D2R/S100B signaling.
Figure 1.

The D2R/S100B HEK stable cell line showing co-expression of S100B and D2R proteins. Cells were immunostainfed as described in the methods section. Shown are: DAPI nuclear stain in blue, S100B in green, and D2R in red. Scale bar: 25μm.
Figure 2.



(A) D2R/S100B HEK cells were transfected with GFP-ataxin-1[82Q] S776 or GFP-ataxin-1[85Q] A776. Cells expressing GFP-ataxin-1[82Q] S776 were serum starved overnight in 2% FBS, then pretreated for 2hrs with 2% FBS as the control, BRC, or H89, then treated for additional 2hrs with FSK, BRC+FSK, BRC, or control in 2% FBS. Cells expressing GFP-ataxin-1 [85Q] A776 were treated for 4hr with 2% FBS. Cells show DAPI staining in blue, GFP in green and phospho-ataxin-1 S776 in red. H89 treated cells showed less phospho-ataxin-1 S776 fluorescence. GFP-ataxin-1[85Q] A776 showed no phospho-ataxin-1 S776 fluorescence and failed to from inclusions. WT HEK cells were transfected with GFP-ataxin-1[82Q] S776 (B) and GFP-ataxin-1[30Q] S776 (C) and treated under the former conditions. BRC treatment had no impact on FSK induced inclusion formation in both GFP-ataxin-1[82Q] and [30Q] expressing cells. Scale bars: 10μm.
Figure 3.
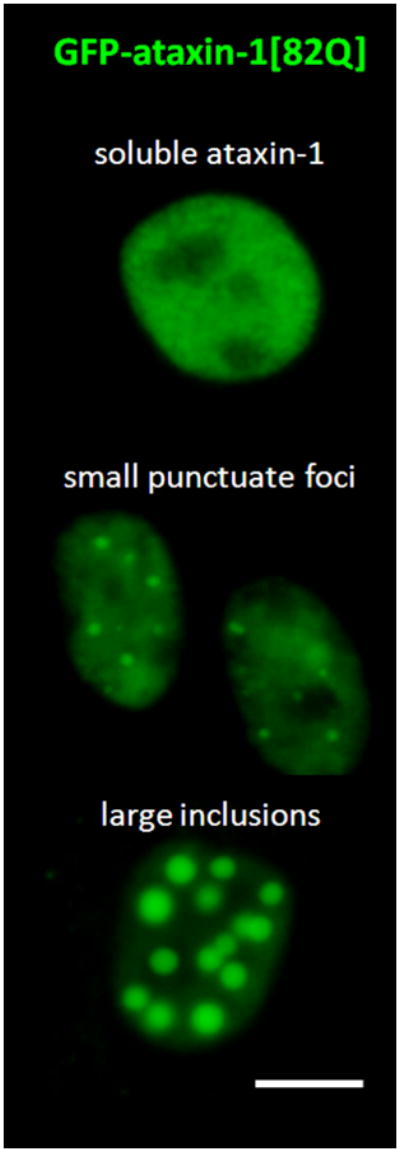
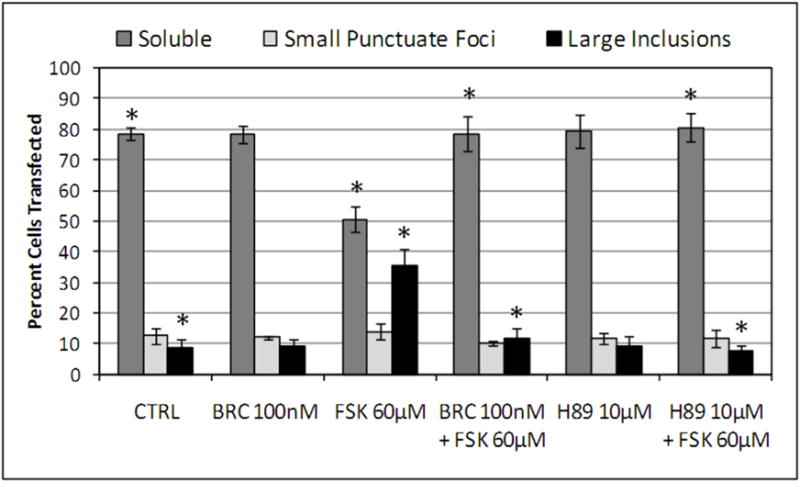
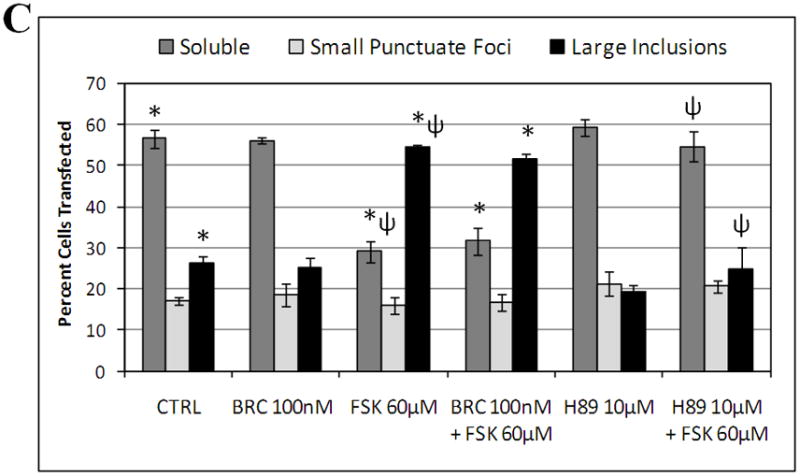
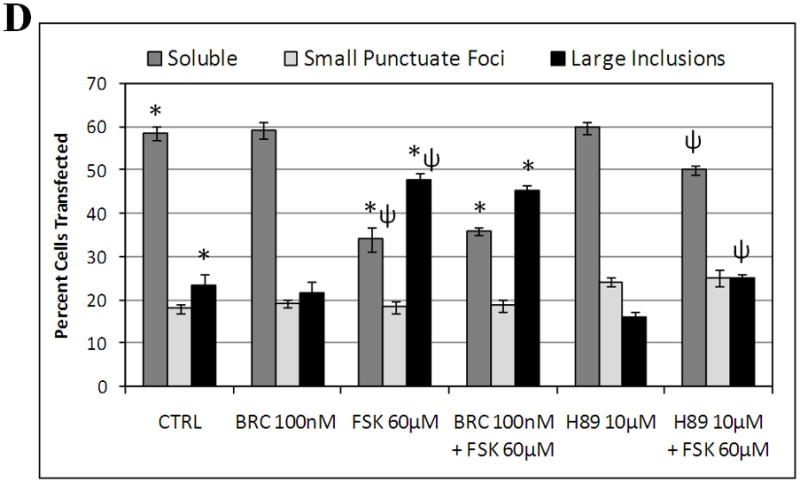
(A and B) D2R/S100B HEK cells transfected with GFP-ataxin-1[82Q] were scored as soluble ataxin-1, small ataxin-1 foci, or large ataxin-1 inclusions, as previously described by Parfitt and co-workers in 2009, under the previously described treatments (Fig. 2A). Data is represented as Mean ± SE. Statistics were calculated using Student’s t-test. Ctrl vs BRC, H89, BRC+FSK or H89+FSK, P>0.05. *FSK vs CRTL, BRC+FSK or H89+FSK, P<0.005. Scale bar: 10μm. (C) WT HEK cells transfected with GFP-ataxin-1[82Q] were scored as soluble ataxin-1, small ataxin-1 foci, or large ataxin-1 inclusions, under the various described treatments. FSK vs BRC+FSK, P>0.1. *Ctrl vs FSK or BRC+FSK, P<0.01. ψFSK vs H89+FSK, P<0.01. (D) WT HEK cells transfected with GFP-ataxin-1[30Q] were scored as soluble ataxin-1, small ataxin-1 foci, or large ataxin-1 inclusions, under the various described treatments. FSK vs BRC+FSK, P>0.1. *Ctrl vs FSK or BRC+FSK, P<0.01. ψFSK vs H89+FSK, P<0.01.
Figure 4.
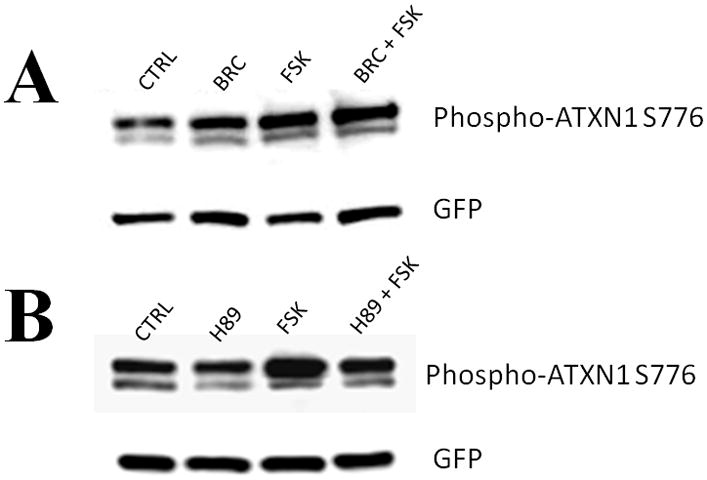
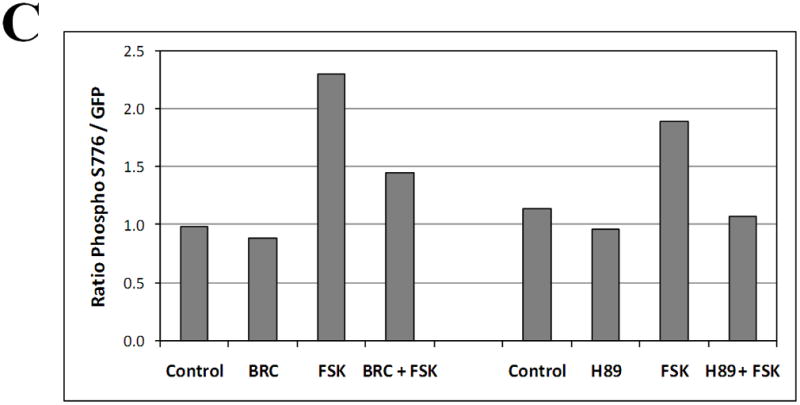
Western blot of lysates from D2/S100B HEK cells expressing GFP-ataxin-1[82Q] treated as previously described (Fig. 3B) and detected with GFP and phospho-ataxin-1 S776 antibodies. (A) Lanes: Control, 100nM BRC, 60μM FSK, BRC + FSK. (B) Lanes: Control, 10μM H89, 60μM FSK, H89 + FSK. (C) Optical densities of the protein bands were taken using Image J software. FSK increased ataxin-1 S776 phosphorylation compared to control, while BRC and H89 impeded FSK stimulated S776 phosphorylation of ataxin-1. Western blots from the lysates of WT HEK cells expressing GFP-ataxin-1[30Q] (D), [82Q] (E) and GFP alone (F) were used to monitor the effect of drug treatments on the CMV promoter activity. Blots were probed with GFP and β Tubulin antibodies.
Forskolin Induces Morphological Changes in SCA1 Purkinje Cell Cultures
To further evaluate the role of PKA in the SCA1 pathology, we cultured cerebellar slices from 7 day old SCA1 Tg and WT mouse pups. PCs were identified based on CaB localization and failure to express glial fibrillary acidic protein (Vig et al. 2009). The slice cultures were treated with FSK followed by CaB immunostaining. FSK induced neurodegenerative morphological changes in SCA1 PCs as demonstrated by a decrease in dendritic arborizations consistent with those seen in vivo (Fig. 5A and B; Vig et al. 2009). We saw no dendritic atrophy in WT PCs treated with FSK (Fig. 5C and D). Furthermore, to confirm our immunofluorescent data, we measured changes in PC dendritic length in both SCA1 and WT slice cultures treated with FSK compared to the controls (Fig. 5E). In SCA1 PCs, FSK significantly decreased dendritic length, while FSK had no effect on dendritic length of WT PCs. The FSK sensitivity seen in the SCA1 PC cultures further signifies that the up-regulation of PKA activity may be contributing to SCA1 PC degeneration as seen in vivo.
Figure 5.
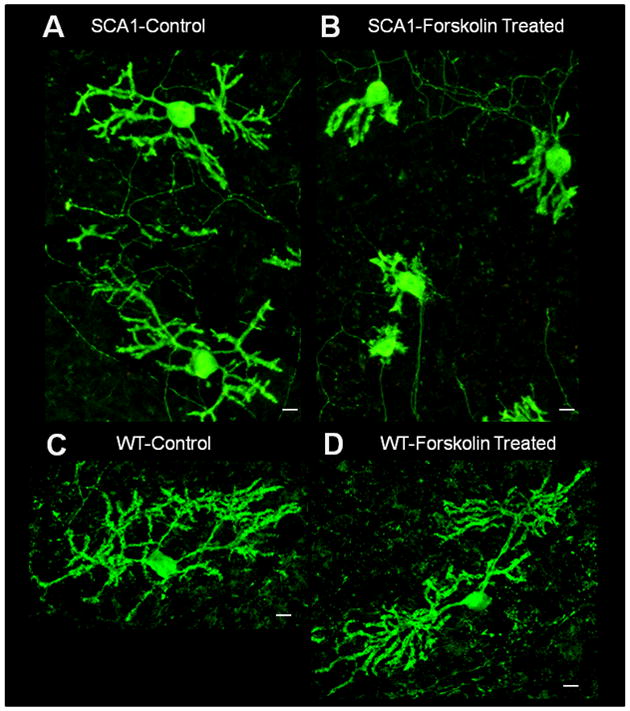
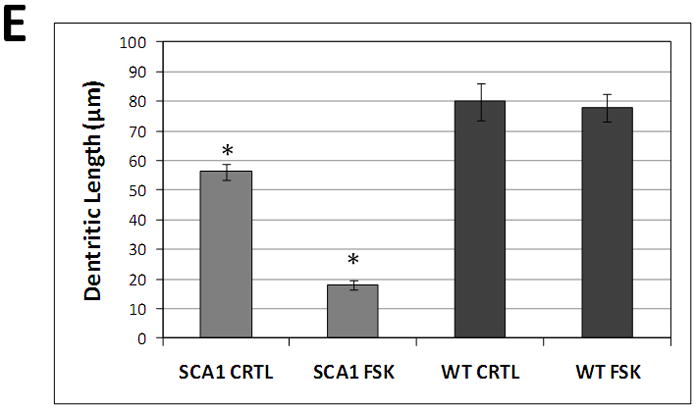
Calbindin-D28k immunofluorescence of organotypic cerebellar slice cultures prepared from 7 day old SCA1 Tg mice (A and B) and WT mice (C and D). 250 μm thick slices were grown on Millicell membrane inserts using 6-well culture plates in 25% horse serum. One day old in vitro slices were incubated with or without 10μM FSK for 16hrs. These slices were grown for additional 5 days in the growth media followed by fixation and immunocytochemistry. The digitized image show abnormally developed dendritic processes in 10μM FSK treated SCA1 slices (B) as compared with SCA1 control slices (A). We saw no changes in dendritic processes in 10μM FSK treated WT slices (D) as compared to WT control slices (C). Scale bar: 10μm. (E) Changes in PC dendritic length of SCA1 and WT slice cultures treated with FSK, from above digitized images, were semi-quantified using Image J software. Measurements were taken along the dendrite, from the PC soma to distal dendritic end, (n=20). Data is represented as Mean ± SE. Statistics were calculated using Student’s t-test. *SCA1 slices cultures treated with FSK displayed a significant decrease in dendritic length compared to SCA1 Controls, P<0.001. WT slices cultures showed no significant changes in dendritic length with FSK treatment compared to Controls, P>0.05.
D2R/S100B Vacuoles in SCA1 Purkinje Cells
Cytoplasmic S100B vacuolar development in PCs is the earliest morphologic change seen in SCA1 Tg mice, which precedes ataxin-1 inclusion formation (Skinner et al. 2001; Vig et al. 2006; Vig et al. 2009). Recently, we found that S100B containing PC vacuoles also localized autophagic marker protein, microtubule-associated protein light chain 3 (LC3) (Vig et al. 2009). In addition, LC3-I and LC3-II, ratios were significantly altered in SCA1 mice, indicating an upregulation of autophagic processes (Vig et al. 2009). Autophagic clearance of toxic proteins has also been demonstrated in Huntington’s and Alzheimer’s diseases (Iwata et al. 2005; Lee 2009; Rami 2009). Mutant ataxin-1 degradation may follow a similar process, as cytoplasmic clearance of mutant ataxin-1 has been shown to involve interaction with Atg proteins (Iwata et al. 2005). Interestingly, we found that the cerebellar sections prepared from 5 week old SCA1 Tg mice displayed co-localization of D2R to S100B positive autophagic PC vacuoles (Fig. 6A). In Figure 6A, triangles point to S100B positive Bergmann glia and arrows point to S100B and D2R positive PC vacuoles. D2R co-localization to S100B vacuoles indicates that autophagic processes could be associated with the loss of D2Rs in SCA1 PCs. In addition, Goold and co-workers reported a decrease in D2R protein levels at 5 weeks in SCA1 Tg mice (Goold et al. 2007). We observed almost complete loss of D2R protein levels by 6 weeks of age (Fig. 6C and D), yet D2R protein expression remained similar to WT animals up to 4 weeks of age (Fig. 6B), suggesting that this age could be ideal for therapeutic intervention.
Figure 6.
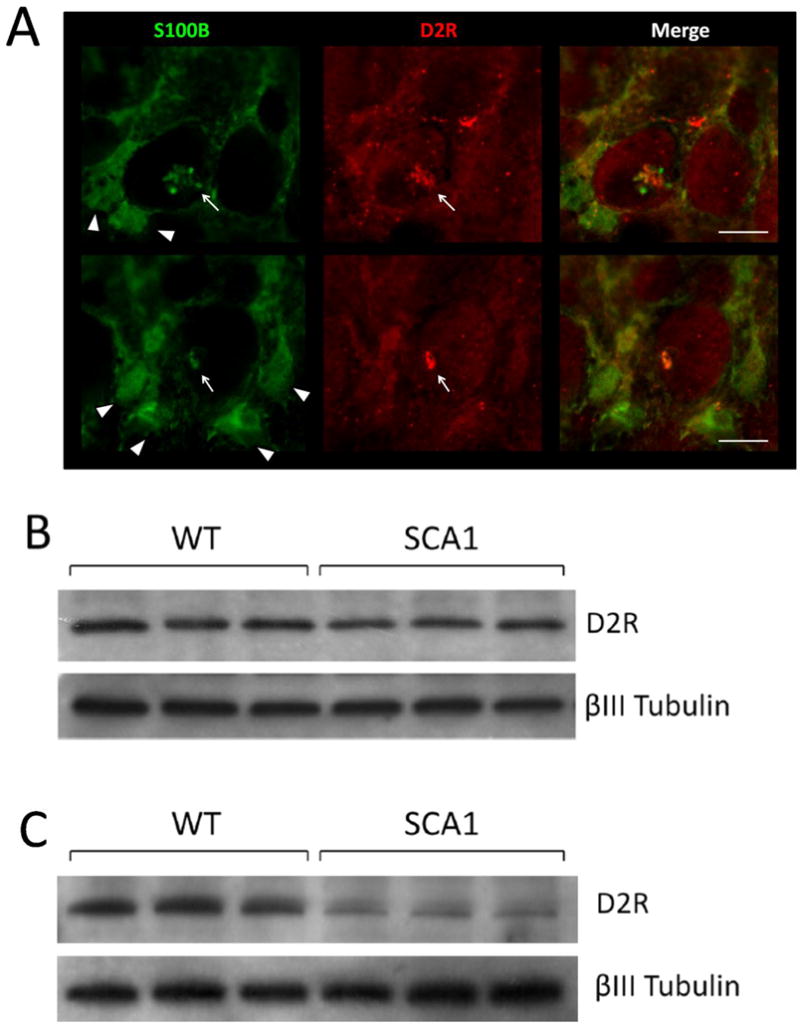

(A) 5 week old SCA1 Tg mice were perfused as previously described (Vig et al. 2009) and 50 μm vibratome sections were immunostained for D2R and S100B. S100B immunostaining is shown in green and D2R in red. Triangles point to S100B positive Bergmann Glia and arrows point to S100B and D2R positive PC vacuoles. Scale bar: 10μm. (B) 4 week old and (C) 6 week old SCA1transgenic mice and WT mice cerebellar extracts were subjected to western blotting loading 25μg per well followed by detection with D2R and βIII Tubulin antibodies. (D) 6 week old SCA1 Tg mice and WT mice cerebellar sections were immunostained with D2R and βIII Tubulin antibodies followed by HRP secondary staining. D2R protein levels are visibility reduced in SCA1 PCs compared to WT, although the number of βIII Tubulin positive PCs remains constant. Arrows point to SCA1 PC vacuoles. Scale bar: 10μm.
Discussion
Our data supports the argument that phosphorylation of mutant ataxin-1 S776 is critical for ataxin-1 mediated toxicity (Emamian et al. 2003; Chen et al. 2003; Jorgensen et al. 2009). In cell culture models, ataxin-1 S776 phosphorylation is coupled to inclusion formation, whereas mutant A776 ataxin-1 protein fails to form inclusions (Emamian et al. 2003; Chen et al. 2003; Jorgensen et al. 2009). Further evidence comes from the fact that SCA1 A776 Tg mice show lesser pathology (Chen et al. 2003). We demonstrate the first mutant ataxin-1 S776 phosphorylation inhibitory mechanism, where mutant ataxin-1 S776 phosphorylation and aggregation can be reduced by D2R/S100B signaling or by PKA inhibition (Fig. 3, 4 and 8). The fact that the PKA phosphorylation site motif at S776 is highly conserved implies that ataxin-1 is a PKA substrate and that ataxin-1’s phosphorylation status may directly be dependent on PKA activity (Glass et al. 1986; Orr et al. 1993; Banfi et al. 1996; Gossen et al. 1996; Chen et al. 2003) (Fig. 7). Interestingly, in our cell culture model, even 30Q ataxin-1 formed FSK induced nuclear inclusions. This has been reported earlier in cultured HEK cells (Tsai et al. 2004) and is probably due to the high levels of the epsilon isoform of 14-3-3, which favors inclusions formation in cell culture, verses the predominant zeta isoform of 14-3-3 found in PCs (Umahara and Uchihara 2010). We postulate that PKA inhibitory pathways, such as D2R/S100B signaling, may impact S776 phosphorylation of both normal and mutant ataxin-1 and may be targeted to reduce ataxin-1 neurotoxicity in SCA1 (Fig. 8).
Figure 8.

D2R/S100B Signaling Pathway: D2R agonist or BRC activate G-protein-coupled inhibitory receptor, D2R, causing Gi proteins to bind and inhibit adenylate cyclase cAMP production, lowering PKA activity and decreasing S776 phosphorylation of PKA substrate ataxin-1, reducing inclusion bodies and neurodegeneration. FSK or other adenylate cyclase activator increases cAMP production and PKA activity, increasing S776 phosphorylation of PKA substrate ataxin-1, producing inclusion bodies and increasing neurodegeneration. H89 inhibits PKA S776 phosphorylation of ataxin-1. BRC could be potentially therapeutic by reducing mutant ataxin-1 S776 phosphorylation and reducing SCA1 neurodegeneration.
Figure 7.

Ataxin-1 homologs from different species have a highly conserved PKA phosphorylation site motif at S776 according to PKA motif established by Glass and coworkers in 1986 (Glass et al. 1986; Orr et al. 1993; Banfi et al. 1996; Gossen et al. 1996; Chen et al. 2003).
The importance of D2Rs in motor function and coordination has been demonstrated in D2R knockout mice, which have impaired locomotion and coordinated movements (Baik et al. 1995; Aoyama et al. 2000; Wang et al. 2000). Recent reports suggest that ataxin-1 may be an essential component of D2R gene expression in PCs of the cerebellum. Furthermore, ataxin-1 has been depicted as a transcriptional co-regulator, which alters the expression of many genes including D2R in both SCA1 Tg mice and Atxn1-null mice (Goold et al. 2007). SCA1 Tg mice start losing D2Rs in PCs at around 4–6 wks postnatally, which corresponds to the onset of behavioral abnormalities (Vig et al. 2006; Goold et al. 2007; Fig. 6). The loss of D2Rs may be a major contributing factor causing an upregulation of adenylate cyclase-cAMP-PKA pathways increasing neurotoxic mutant ataxin-1 S776 phosphorylation. We recently showed that S100B is internalized in cultured PCs (Vig et al. 2009). Since S100B complexes with D2R to inhibit adenylate cyclase and control cAMP levels (Liu et al. 2008), we speculate that under physiologic conditions, S100B inside PCs interacts with D2R, enhancing the regulation of PKA activity.
We have recently demonstrated that the cytoplasmic vacuoles in SCA1 PCs contain S100B protein and are formed only when mutant ataxin-1 is expressed in both SCA1 Tg mice as well as in SCA1 patients (Vig et al. 2009). In addition, the process of vacuolar formation may be associated with autophagy (Vig et al. 2009). Considering that our data demonstrates D2Rs co-localized with S100B in PC vacuoles (Fig. 6A), the continuous expression of mutant ataxin-1 to form vacuoles may be depleting functional D2Rs from the PC soma, dysregulating cAMP production and increasing PKA activity. Further evidence implicating D2Rs in the SCA1 pathogenesis comes from the beneficial effects of lithium therapy to SCA1 Tg mice (Watase et al. 2007). The stimulatory effects of lithium on the transcription of the D2R gene was shown in rats fed chow containing 0.2% LiCO3, which resulted in an increased D2R mRNA expression level and an increased transcription rate of the D2R gene in the rat brain striatum (Kameda et al. 2001). It is tempting to speculate that the beneficial effects of lithium in SCA1 Tg mice could be as a result of increased D2R mRNA expression in PCs, leading to down-regulation of PKA activity.
Dopamine receptors localize not only to PC, but also to many different neurons in other regions of the brain. Dopamine distortions have been reported in SCA1, where dopamine (DA) levels were greatly reduced (−76%) in the putamen of seven patients with SCA1 (Kish et al. 1997). Kish and coworkers (Kish et al. 1997) further concluded that the degeneration in nigrostriatal DA neurons begins at the nerve ending in SCA1. Parkinson’s disease (PD) like symptoms have been observed in SCA patients and recent report s by Socal and co-workers (Socal et al. 2009) indicated a phenotypic association between PD and CAG expansions among SCA2 and SCA3 patients. Interestingly, several SCA patients with Parkinsonian symptoms have responded well to levodopa and dopamine agonists (Geschwind et al. 1997; Schöls et al. 2004; Manto 2005). D2R agonist BRC has been used to treat PD and may be potentially therapeutic for SCA1 patients (Calne et al. 1974; van Hilten et al. 2000). Therefore, we postulate that down-regulation of the D2R pathway may enhance ataxin-1 toxicity in PCs and/or in other susceptible regions of the brain in SCA1. Taken together our data suggests that D2R agonists could be used to modulate cAMP levels and reduce PKA induced neurotoxicity in SCA1.
Acknowledgments
This work was partly supported by grants from the National Institutes of Health and National Ataxia Foundation. We would like to thank Dr. Kim Neve, Department of Behavioral Neuroscience, Oregon Health & Science University and Portland Veterans Affairs Medical Center for providing the D2R/S100B HEK cell lines and for his advice in preparing this manuscript. In addition, we would like to thank Dr. Michael Hebert, Department of Biochemistry, the University of Mississippi Medical Center, for his help with cell culture and ataxin-1 plasmid propagation.
References
- Aoyama S, Kase H, Borrelli E. Rescue of locomotor impairment in dopamine D2 receptor-deficient mice by an adenosine A2A receptor antagonist. J Neurosci. 2000;15:5848–5852. doi: 10.1523/JNEUROSCI.20-15-05848.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baik JH, Picetti R, Saiardi A, Thiriet G, Dierich A, Depaulis A, Le Meur M, Borrelli E. Parkinsonian-like locomotor impairment in mice lacking dopamine D2 receptors. Nature. 1995;377:424–428. doi: 10.1038/377424a0. [DOI] [PubMed] [Google Scholar]
- Banfi S, Servadio A, Chung MY, Kwiatkowski JrTJ, McCall AE, Duvick LA, Shen Y, Roth EJ, Orr HT, Zoghbi HY. Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat Genet. 1994;7:513–520. doi: 10.1038/ng0894-513. [DOI] [PubMed] [Google Scholar]
- Banfi S, Servadio A, Chung M, Capozzoli F, Duvick LA, Elde R, Zoghbi HY, Orr HT. Cloning and developmental expression analysis of the murine homolog of the spinocerebellar ataxia type 1 gene (Sca1) Hum Mol Genet. 1996;5:33–40. doi: 10.1093/hmg/5.1.33. [DOI] [PubMed] [Google Scholar]
- Bansal PK, Mishra A, High AA, Abdulle R, Kitagawa K. Sgt1 dimerization is negatively regulated by protein kinase CK2-mediated phosphorylation at Ser361. J Biol Chem. 2009;284:18692–18698. doi: 10.1074/jbc.M109.012732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barger SW, Van Eldik LJ, Mattson MP. S100β protects hippocampal neurons from damage induced by glucose deprivation. Brain Res. 1995;677:167–170. doi: 10.1016/0006-8993(95)00160-r. [DOI] [PubMed] [Google Scholar]
- Battaglia G, Norman AB, Hess EJ, Creese I. Forskolin potentiates the stimulation of rat striatal adenylate cyclase mediated by D-1 dopamine receptors, guanine nucleotides, and sodium fluoride. J Neurochem. 1986;46:1180–1185. doi: 10.1111/j.1471-4159.1986.tb00635.x. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Bianchi R, Garbuglia M, Verzini M, Giambanco I, Ivanenkov VV, Dimlich RV, Jamieson GA, Jr, Donato R. S-100 (alpha and beta) binding peptide (TRTK-12) blocks S-100/GFAP interaction: identification of a putative S-100 target epitope within the head domain of GFAP. Biochim Biophys Acta. 1996;1313:258–267. doi: 10.1016/0167-4889(96)00098-5. [DOI] [PubMed] [Google Scholar]
- Burright EN, Clark HB, Servadio A, Matilla T, Feddersen RM, Yunis WS. SCA-1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell. 1995;82:937–948. doi: 10.1016/0092-8674(95)90273-2. [DOI] [PubMed] [Google Scholar]
- Calne DB, Teychenne PF, Leigh PN, Bamji AN, Greenacre JK. Treatment of parkinsonism with bromocriptine. Lancet. 1974;2:1355–1356. doi: 10.1016/s0140-6736(74)92219-3. [DOI] [PubMed] [Google Scholar]
- Chen HK, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, Aitken A, Skoulakis EM, Orr HT, Botas J, Zoghbi HY. Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell. 2003;113:457–468. doi: 10.1016/s0092-8674(03)00349-0. [DOI] [PubMed] [Google Scholar]
- Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- Donato R. Perspectives in S-100 protein biology. Cell Calcium. 1991;12:713–726. doi: 10.1016/0143-4160(91)90040-l. [DOI] [PubMed] [Google Scholar]
- Donato R. Functional roles of S100 proteins, calcium-binding proteins of the EF-hand type. Biochim Biophys Acta. 1999;1450:191–231. doi: 10.1016/s0167-4889(99)00058-0. [DOI] [PubMed] [Google Scholar]
- Emamian ES, Kaytor MD, Duvick LA, Zu T, Tousey SK, Zoghbi HY, Clark HB, Orr HT. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron. 2003;38:375–387. doi: 10.1016/s0896-6273(03)00258-7. [DOI] [PubMed] [Google Scholar]
- Fujihara M, Muroi M, Muroi Y, Ito N, Suzuki T. Mechanism of lipopolysaccharide-triggered junB activation in a mouse macrophage-like cell line (J774) J Biol Chem. 1993;268:14898–14905. [PubMed] [Google Scholar]
- Glass DB, El-Maghrabi MR, Pilkis SJ. Synthetic peptides corresponding to the site phosphorylated in 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase as substrates of cyclic nucleotide-dependent protein kinases. J Biol Chem. 1986;261:2987–2993. [PubMed] [Google Scholar]
- Goold R, Hubank M, Hunt A, Holton J, Menon RP, Revesz T, Pandolfo M, Matilla-Dueñas A. Down-regulation of the dopamine receptor D2 in mice lacking ataxin-1. Hum Mol Genet. 2007;17:2122–2134. doi: 10.1093/hmg/ddm162. [DOI] [PubMed] [Google Scholar]
- Gossen M, Schmitt I, Obst K, Wahle P, Epplen JT, Riess O. cDNA cloning and expression of rsca1, the rat counterpart of the human spinocerebellar ataxia type 1 gene. Hum Mol Genet. 1996;5:381–389. doi: 10.1093/hmg/5.3.381. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Perlman S, Figueroa CP, Treiman LJ, Pulst SM. The prevalence and wide clinical spectrum of the spinocerebellar ataxia type 2 trinucleotide repeat in patients with autosomal dominant cerebellar ataxia. Am J Hum Genet. 1997;60:842–850. [PMC free article] [PubMed] [Google Scholar]
- Grimmler M, Bauer L, Nousiainen M, Körner R, Meister G, Fischer U. Phosphorylation regulates the activity of the SMN complex during assembly of spliceosomal U snRNPs. EMBO Rep. 2005;6:70–76. doi: 10.1038/sj.embor.7400301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearst SM, Gilder AS, Negi SS, Davis MD, George EM, Whittom AA, Toyota CG, Husedzinovic A, Gruss OJ, Hebert MD. Cajal-body formation correlates with differential coilin phosphorylation in primary and transformed cell lines. J Cell Sci. 2009;122:1872–1881. doi: 10.1242/jcs.044040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttunen HJ, Kuja-Panula J, Sorci G, Agneletti AL, Donato R, Rauvala H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J Biol Chem. 2000;275:40096–40105. doi: 10.1074/jbc.M006993200. [DOI] [PubMed] [Google Scholar]
- Insel PA, Ostrom RS. Forskolin as a tool for examining adenylyl cyclase expression, regulation, and G protein signaling. Cell Mol Neurobiol. 2003;23:305–314. doi: 10.1023/A:1023684503883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanenkov VV, Jamieson GA, Jr, Gruenstein E, Dimlich RV. Characterization of S-100b binding epitopes. Identification of a novel target, the actin capping protein, CapZ. J Biol Chem. 1995;270:14651–14658. doi: 10.1074/jbc.270.24.14651. [DOI] [PubMed] [Google Scholar]
- Iwata A, Christianson JC, Bucci M, Ellerby LM, Nukina N, Forno LS, Kopito RR. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc Natl Acad Sci USA. 2005;102:13135–13140. doi: 10.1073/pnas.0505801102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen ND, Andresen JM, Lagalwar S, Armstrong B, Stevens S, Byam CE, Duvick LA, Lai S, Jafar-Nejad P, Zoghbi HY, Clark HB, Orr HT. Phosphorylation of ATXN1 at Ser776 in the cerebellum. J Neurochem. 2009;110:675–686. doi: 10.1111/j.1471-4159.2009.06164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameda K, Miura J, Suzuki K, Kusumi I, Tanaka T, Koyama T. Effects of lithium on dopamine D2 receptor expression in the rat brain striatum. J Neural Transm. 2001;3:321–334. doi: 10.1007/s007020170078. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Guttman M, Robitaille Y, El-Awar M, Chang LJ, Levey AI. Striatal dopamine nerve terminal markers but not nigral cellularity are reduced in spinocerebellar ataxia type 1. Neurology. 1997;48:1109–1111. doi: 10.1212/wnl.48.4.1109. [DOI] [PubMed] [Google Scholar]
- Koeppen AH. The pathogenesis of spinocerebellar ataxia. Cerebellum. 2005;4:62–73. doi: 10.1080/14734220510007950. [DOI] [PubMed] [Google Scholar]
- Lee JA. Autophagy in neurodegeneration: two sides of the same coin. BMB Rep. 2009;42:324–330. doi: 10.5483/bmbrep.2009.42.6.324. [DOI] [PubMed] [Google Scholar]
- Liu Y, Buck DC, Neve KA. Novel interaction of the dopamine D2 receptor and the Ca2+ binding protein S100B: role in D2 receptor function. Mol Pharmacol. 2008;74:371–378. doi: 10.1124/mol.108.044925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lortet S, Samuel D, Had-Aissouni L, Masmejean F, Kerkerian-Le Goff L, Pisano P. Effects of PKA and PKC modulators on high affinity glutamate uptake in primary neuronal cell cultures from rat cerebral cortex. Neuropharmacology. 1999;38:395–402. doi: 10.1016/s0028-3908(98)00193-2. [DOI] [PubMed] [Google Scholar]
- Manto MU. The wide spectrum of spinocerebellar ataxias (SCAs) Cerebellum. 2005;4:2–6. doi: 10.1080/14734220510007914. [DOI] [PubMed] [Google Scholar]
- Mao LM, Tang Q, Wang JQ. Protein kinase C-regulated cAMP response element-binding protein phosphorylation in cultured rat striatal neurons. Brain Res Bull. 2007;72:302–308. doi: 10.1016/j.brainresbull.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matilla-Dueñas A, Goold R, Giunti P. Clinical, genetic, molecular, and pathophysiological insights into spinocerebellar ataxia type 1. Cerebellum. 2007;7:1–9. doi: 10.1007/s12311-008-0009-0. [DOI] [PubMed] [Google Scholar]
- Muroi M, Suzuki T. Role of protein kinase A in LPS-induced activation of NF-kappa B proteins of a mouse macrophage-like cell line, J774. Cell Signal. 1993;5:289–298. doi: 10.1016/0898-6568(93)90019-i. [DOI] [PubMed] [Google Scholar]
- Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- Orr HT, Chung MY, Banfi S, Kwiatkowski TJ, Jr, Servadio A, Beaudet AL, McCall AE, Duvick LA, Ranum LP, Zoghbi HY. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4:221–226. doi: 10.1038/ng0793-221. [DOI] [PubMed] [Google Scholar]
- Parfitt DA, Michael GJ, Vermeulen EG, Prodromou NV, Webb TR, Gallo JM, Cheetham ME, Nicoll WS, Blatch GL, Chapple JP. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum Mol Genet. 2009;18:1556–1565. doi: 10.1093/hmg/ddp067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rami A. Review: autophagy in neurodegeneration: firefighter and/or incendiarist? Neuropathol Appl Neurobiol. 2009;5:449–461. doi: 10.1111/j.1365-2990.2009.01034.x. [DOI] [PubMed] [Google Scholar]
- Reeves RH, Yao J, Crowley MR, Buck S, Zhang X, Yarowsky P, Gearhart JD, Hilt DC. Astrocytosis and axonal proliferation in the hippocampus of S100b transgenic mice. Proc Natl Acad Sci USA. 1994;91:5359–5363. doi: 10.1073/pnas.91.12.5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothermundt M, Peters M, Prehn JH, Arolt V. S100B in brain damage and neurodegeneration. Microsc Res Tech. 2003;60:614–632. doi: 10.1002/jemt.10303. [DOI] [PubMed] [Google Scholar]
- Schöls L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3:291–304. doi: 10.1016/S1474-4422(04)00737-9. [DOI] [PubMed] [Google Scholar]
- Seamon KB, Daly JW. Forskolin: a unique diterpene activator of cyclic AMP-generating systems. J Cyclic Nucleotide Res. 1981;7:201–224. [PubMed] [Google Scholar]
- Skinner PJ, Vierra-Green CA, Clark HB, Zoghbi HY, Orr HT. Altered Trafficking of membrane proteins in Purkinje cells of SCA1 transgenic mice. Am J Pathol. 2001;159:905–913. doi: 10.1016/S0002-9440(10)61766-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socal MP, Emmel VE, Rieder CR, Hilbig A, Saraiva-Pereira ML, Jardim LB. Intrafamilial variability of Parkinson phenotype in SCAs: novel cases due to SCA2 and SCA3 expansions. Parkinsonism Relat Disord. 2009;15:374–378. doi: 10.1016/j.parkreldis.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Tsai CC, Kao HY, Mitzutani A, Banayo E, Rajan H, McKeown M, Evans RM. Ataxin-1, a SCA1 neurodegenerative disorder protein, is functionally linked to the silencing mediator of retinoid and thyroid hormone receptors. Proc Natl Acad Sci U S A. 2004;101:4047–4052. doi: 10.1073/pnas.0400615101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umahara T, Uchihara T. 14-3-3 Proteins and Spinocerebellar Ataxia Type 1: from Molecular Interaction to Human Neuropathology. Cerebellum. 2010;9 doi: 10.1007/s12311–010–0158–9. [DOI] [PubMed] [Google Scholar]
- van Hilten JJ, Ramaker CC, Stowe R, Ives NJ. Bromocriptine versus levodopa in early Parkinson’s disease. Cochrane Database Syst Rev. 2000;3:CD002258. doi: 10.1002/14651858.CD002258.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig PJS, Lopez ME, Wei J, D’Souza DR, Subramony SH, Henegar J. Glial S100B positive vacuoles in Purkinje cells: earliest morphological abnormality in SCA1 transgenic mice. J Neurol Sci [Turk] 2006;23:166–174. doi: 10.1901/jaba.2006.23-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig PJ, Wei J, Shao Q, Hebert MD, Subramony SH, Sutton LT. Role of tissue transglutaminase type 2 in calbindin-D28k interaction with ataxin-1. Neurosci Lett. 2007;420:53–57. doi: 10.1016/j.neulet.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig PJ, Shao Q, Subramony SH, Lopez ME, Safaya E. Bergmann glial S100B activates myo-inositol monophosphatase 1 and Co-localizes to purkinje cell vacuoles in SCA1 transgenic mice. Cerebellum. 2009;8:231–244. doi: 10.1007/s12311-009-0125-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xu R, Sasaoka T, Tonegawa S, Kung MP, Sankoorikal EB. Dopamine D2 long receptor-deficient mice display alterations in striatum-dependent functions. J Neurosci. 2000;22:8305–8314. doi: 10.1523/JNEUROSCI.20-22-08305.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watase K, Gatchel JR, Sun Y, Emamian E, Atkinson R, Richman R, Mizusawa H, Orr HT, Shaw C, Zoghbi HY. Lithium therapy improves neurological function and hippocampal dendritic arborization in a spinocerebellar ataxia type 1 mouse model. PLoS Med. 2007;5:e182. doi: 10.1371/journal.pmed.0040182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts VJ, Neve KA. Sensitization of endogenous and recombinant adenylate cyclase by activation of D2 dopamine receptors. Mol Pharmacol. 1996;50:966–976. [PubMed] [Google Scholar]
- Whitaker-Azmitia PM, Vingate M, Borella A, Gerlai R, Roder J, Azmitia EC. Transgenic mice overexpressing the neurotrophic factor S-100β show neuronal cytoskeletal and behavioral signs of altered aging processes: implications for Alzheimer’s disease and Down’s syndrome. Brain Res. 1997;776:51–60. doi: 10.1016/s0006-8993(97)01002-0. [DOI] [PubMed] [Google Scholar]
- Winningham-Major F, Staecker JL, Barger SW, Coats S, Van Eldik LJ. Neurite extension and neuronal survival activities of recombinant S100β proteins that differ in the content and position of cysteine residues. J Cell Biol. 1989;109:3064–3071. doi: 10.1083/jcb.109.6.3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroblewska B, Wroblewski JT, Saab OH, Neale JH. N-acetylaspartylglutamate inhibits forskolin-stimulated cyclic AMP levels via a metabotropic glutamate receptor in cultured cerebellar granule cells. J Neurochem. 1993;61:943–948. doi: 10.1111/j.1471-4159.1993.tb03606.x. [DOI] [PubMed] [Google Scholar]
- Zimmer DB, Chaplin J, Baldwin A, Rast M. S100-mediated signal transduction in the nervous system and neurological diseases. Cell Mol Biol. 2005;51:201–214. [PubMed] [Google Scholar]
- Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]