

Abstract
Stimulation of the serotonin 1A receptor (5-HT1A-R) causes activation of extracellular signal-regulated protein kinase (Erk) and protein kinase C alpha (PKCα) in both hippocampal HN2-5 cells and cultured hippocampal slices from postnatal day-15 (P15) mice. Our earlier studies demonstrated that PKCα is coimmunoprecipitated with Erk and the phosphorylation of PKCα in this Erk–PKCα complex is dependent on the Erk pathway. Furthermore, the T638 residue, which must be phosphorylated for the complete activation of PKCα, is within an authentic MAP kinase consensus domain (S/TP), and the PKCα protein also contains two docking sites for Erk such as KRGRIYL and KRGIIYRDLKL. Using Föster Resonance Energy Transfer (FRET) we have confirmed an association between Erk and PKCα. Employing PKCα and Erk mutants we next demonstrated that Erk causes direct phosphorylation and activation of PKCα. By mutating the phosphoinositide dependent kinase-1 (PDK-1)-promoted phosphorylation site (S497) and the kinase site (K368) in PKCα, we observed that both of these autophosphorylation-deficient mutants are phosphorylated at T638 in an Erk-dependent manner. To confirm that Erk indeed catalyzes phosphorylation of PKCα at T638, we used a mutant Erk construct in which a bulky amino acid residue in the ATP binding site (Q103) had been replaced with glycine, enabling this mutant to utilize a bulky analog of ATP, cyclopentyl ATP. An in vitro kinase assay using this mutant Erk protein, radiolabeled cyclopentyl ATP, and a synthetic oligopeptide containing the S/TP site of PKCα showed phosphorylation of the peptide by Erk1/2. These results confirm the novel possibility that PKCα is a direct substrate of Erk1/2 in neuronal cells and help link two important signaling molecules that regulate maturation and protection of hippocampal neurons as well as many other cell types.
Keywords: Hippocampus, PKCα, Erk, 5-HT1A receptor
Introduction
The serotonin 1A receptor (5-HT1A-R) is a heptahelical Gi- and Go-coupled receptor, which functions by inhibiting adenylyl cyclase, stimulating inwardly rectifying K+ channels, or modestly stimulating phospholipase C [1]. In the brain, this receptor is present in the raphé neurons as their target postsynaptic neurons in the front brain [1; 2]. The receptor at the presynaptic site (autoreceptor) mainly regulates serotonin release and at the postsynaptic site (heteroreceptor) regulates synaptic activities by stimulating inwardly rectifying K+ channels or directly inhibiting Ca2+ channels. Functionally, the 5-HT1A-R receptor is crucial in various aspects such as synaptic plasticity, neurogenesis, neuroprotection, and also in anxiety, depression, learning and memory [2]. In our earlier studies, we have shown that stimulation of the receptor with 8-OH-DPAT (a 5-HT1A agonist) causes activation of mitogen-activated protein kinase (MAPK) isozymes Erk1 and Erk2) and also Protein kinase C alpha (PKCα) in both in HN2-5 cells (a mouse hippocampal neuron-derived cell line [3]), and organotypic hippocampal slice cultures from postnatal day 15 (P15) mice [4]. Additonally, PKCαis also co-immunoprecipitated with Erk1/2 [4]. The phosphorylation of PKCα is dependent on Erk1/2 as MEK (Erk kinase) inhibitors PD98059 and U0126 inhibit PKCα activation but the general PKC inhibitor GFX does not abrogate activation of PKCα or ERk1/2. A similar Erk1/2 dependent activation of PKCαhas been reported in parathyroid induced opossum kidney cells [5], although in many studies, the PKCα activation is shown to be upstream of Erk1/2 [6; 7; 8]. The PKCα isoenzyme is synthesized as an inactive membrane bound precursor and undergoes phosphorylation at three sites T497 (Activation loop), T638 (Turn motif), and S657 (hydrophobic domain) for its activation. Phosphorylation of the activation loop is promoted by the enzyme phosphoinositide dependent kinase 1 (PDK-1), which is reported to be an essential event in the activation of PKC isoenzymes as well as other members of ABC family members of protein kinases [9; 10]. This activation loop phosphorylation stimulates autophosphorylation of both “turn motif” and “hydrophobic domain”, turning it into a catalytically competent and stable mature PKC [10]. Dephosphorylation of the “turn motif” abolishes PKC enzyme activity [11]. However the PKC molecule remains in an inactive state via association of the pseudo-substrate, which is a part of the PKC molecule itself, to the active site [12]. The final activation of PKC requires removal of the pseudo-substrate from its active site, which is carried out upon binding of DAG and calcium.
The MAPK enzymes require a very specific phosphorylation sequence where a serine or threonine is followed by proline (S/TP) [13]. In addition, a proline at position -2 is favorable but not absolutely required [14]. The Turn Motif of PKCαat T638 is within the consensus Erk phosphorylation sequence PVXTP. A very similar Erk phosphorylation site is reported in peroxiredoxin-6 where PVATP is the phosphorylation motif [15]. Additionally the MAPK enzymes require docking sites in the substrates for high affinity interactions [14]. The PKCαmolecule also contains two Erk1/2 docking domains such as KRGRIYL and KRGIIYRDLKL. So, there is a possibility that the “turn motif” phosphorylation may be catalyzed by Erk1/2. We have used Föster Resonance Energy Transfer (FRET) to demonstrate the interaction of PKCα with Erk1/2. Furthermore, we have demonstrated that autophosphorylation-deficient mutant forms of PKCα are phosphorylated at T638 in an Erk1/2-dependent manner upon stimulation of the 5-HT1A-R in the HN2-5 cells. Finally, in order to validate Erk1/2 mediated phosphorylation of PKCαat T638, we have used a mutant Erk1/2 construct [16] in which a bulky amino acid residue in the ATP binding site (glutamine 103) has been replaced with glycine, enabling this mutant to utilize a bulky analog of ATP (cyclopentyl ATP). A kinase reaction assay using this mutant Erk1/2 protein in its activated state, radiolabeled cyclopentyl ATP, and a synthetic peptide from the PKCαprotein containing the S/TP site in a reaction buffer have been used to demonstrate mutant Erk1/2 mediated phosphorylation of this peptide.
Materials and Methods
Cell culture, transfection, and immunoprecipitation
HN2-5 cells were grown in DMEM media with 10% FBS, 1%(v/v) penicillin-streptomycin solution in a 5%-CO2, humidified incubator. Transfection of different vector constructs was carried out in six-well plates (3 μg DNA/well) using Turbofect Transfection Reagent (MBI, Fermentas). Overnight-transfected cells were differentiated by treatment with 5 μM retinoic acid in 1% FBS containing medium. After 24 h, cells were serum starved for 1 h then treated with different drugs with or without 1 μM 8-OH-DPAT (D) for 1 h (for the stimulation of PKC). The cells were washed once with ice cold PBS and were lysed in a buffer (0.5 ml) containing 20 mM HEPES pH 7.4, 2mm EGTA, 2 mM MgCl2, 200 mM Na3VO4, 1% Triton X-100, 2 mM PMSF and protease inhibitor cocktail (Roche, Mannheim, Germany). All manipulations were carried out on ice. The cell lysates were incubated overnight at 4 °C with a Myc antibody (Cell Signaling) at 1:200 dilution. Next, a 40 μL aliquot of a slurry of previously washed A/G agarose beads was added to each lysate sample and the mixture was again incubated with rocking overnight at 4 °C. Following this, the Protein A/G agarose-immunoprecipitated complex was centrifuged, the pellet washed eight times with the same lysis buffer (1 ml at a time). Finally each pellet was reconstituted in 30-μL 2X Laemmli treatment buffer, heated in boiling water for 10 min, centrifuged, and the supernatant resolved using SDS PAGE. Resolved proteins were then transferred to a nitrocellulose membrane, which was probed successively with different antibodies. The membranes were incubated in blocking buffer [5% bovine serum albumin in TBS-T (20 mmol/L Tris–HCl, pH 7.4, 0.8% NaCl, 0.1% Tween 20)], and then probed with different primary antibodies, followed by treatment with HRP-linked respective secondary antibodies. Each nitrocellulose membrane was first probed with a rabbit polyclonal PPKCα/ß II (T638/641) antibody (1:1000). This was followed by stripping of the blot in 0.2 M glycine (pH 2.5), blocking and reprobing with the same Myc antibody (1:1000) as used for immunoprecipitation. Detection was carried out with the Supersignal luminol kit and bands were densitometrically quantified using a Fluorchem FC2 imaging system (Alpha Innotech, San Leandro, CA). The intensities of the luminescent bands were digitized using SPOT DENSO software, and the P-PKCα band intensities were normalized to the corresponding Myc band intensities. All the experiments were repeated 3 times and statistical analysis was performed using student's t-test and ANOVA.
FRET Analysis
HN2-5 cells, grown on poly-L-lysine-coated coverslips to 50-70% confluence, were cotransfected with Erk-CFP and PKCα-YFP vectors (1 μg each). The control cells were similarly cotransfected with the empty vectors, pECFP and pEYFP (1 μg each). After 24 h, cells were treated for 1 h with carrier or 8-OH-DPAT, fixed in 4% paraformaldehyde (30 min at room temperature), washed with PBS, and mounted on glass slides. Images were acquired using a Leica Confocal Scanning System (Exton, PA) TCS SP2. ECFP (Donor) and EYFP (Acceptor) fluorescence were excited with 458 and 514 nm lasers, respectively. We used acceptor photo bleaching method in which a small region of interest expressing both the flourophores of which the EYFP fluorescence channel was photobleached using a 514-nm laser to 30% of the original intensity and an increase in the intensity of the donor Erk-CFP was recorded. ECFP and EYFP images were taken both before and after acceptor photobleaching [17]. FRET efficiency (E) was calculated using Leica software using the following equation: for all Dpost > Dpre, where Dpre and Dpost are the ECFP emission spectra before and after regional photobleaching.
Cloning and site-directed mutagenesis
The PKCα coding sequence, without the stop codon, was cloned into a pGEM-T vector (Promega, USA) and subcloned into a pcDNA6A vector in frame with Myc and His tags (Invitrogen). The cloned DNA was used as template for PCR based site-directed mutagenesis using primers: Muta-497-Fwd-5′TCACGACCAGGGCCTTCTGTG3′ and Muta-497-Rvs-5′ CACAGAAGGCCCTGGTCGTGA3′. The mutated bases (italicized) yield T497→A mutant PKCα (ACC→ GCC). Overlapping PCR products were generated using primer pairs (EcoR1-Fwdα and mutα-497-rvs) and (muta-497-Fwdα and XHO-Rvsα). The two PCR products were gel purified and mixed together. 10 ng of the mixed DNA was used as template in a PCR reaction containing the primers ECoR1-Fwdα and XHO-Rvsα. Sequencing was performed by Genewiz laboratory (NJ, USA). A Flag-tagged mutant Erk-pCDNA3 vector used in this study has been described previously [16]. The full length mouse Erk and PKC cDNAs were cloned into pECFP and pEYFP respectively by standard procedures.
Mutant Erk (Erk*) mediated in-vitro kinase assay
Mutant Erk mediated in vitro kinase assay was followed, as previously described [16]. In the Flag-tagged mutant Erk construct, the normal amino acid residue (glutamine 103) is changed to a glycine residue (glycine 103), which causes the ATP binding site to be bulky and therefore, allows the utilization of an analog of ATP (cyclopentyl ATP) that cannot be used by wild-type Erk2 or other cellular kinases. The empty vector and the mutant FLAG-Erk vector were transiently transfected into 70-80% confluent HN2-5 cells using EXGen500 (MBI Fermentas, USA) transfection reagent. After 24 h, the cells were differentiated using 5 μM retinoic acid in 1% serum for 18 h, serum starved for 1 h, and then treated with 1 μM 8-OH-DPAT for 30 min (for the stimulation of Erk1/2). The cells were lysed in a hypotonic lysis buffer (20 mM HEPES pH 7.4, 2 mM EGTA, 2 mM MgCl2, 200 mM Na3VO4, 2 mM PMSF and protease inhibitor cocktail). The expressed mutant FLAG-Erk protein was immunoprecipitated using anti-FLAG-M2- agarose beads (Sigma). Both the immunoprecipitated products from the empty vector and mutant Erk vector transfected cells were used for a kinase reaction assay by mixing 32P-labeled cyclopentyl ATP, a synthetic, 17-amino acid oligopeptide from the putative Erk phosphorylation site of PKCα protein (FFTRGQPVLTPPDQLVIA), in a reaction buffer and incubating for 20 min at 30°C. The reaction mixture was heat inactivated and spotted onto a high performance thin layer chromatography (HPTLC) plate. The labeled and unlabeled oligopeptides were separated by developing the plate in n-butanol: acetic acid: water: 4:2:2. The resolved, radiolabeled peptide was visualized by autoradiography.
Results and Discussion
Physical interactions between Erk and PKCα
We employed the FRET microscopy method to monitor the in-cell physical association of Erk and PKCα. Insignificant FRET efficiency (<0.4%) was observed in the empty vector-transfected cells. Representative confocal images for FRET are shown in Figure 1. From five independent experiments, we obtained a mean FRET efficiency of 7.19±0.75%. This strongly indicated that Erk and PKCα protein molecules are held in a close physical association. It should be noted, that activated Erk distributes in both cytosol as well as nucleus (Fig. 1, upper panel, cyan fluorescence) whereas PKCα always remains in the cytoplasm (Fig. 1, lower panel, yellow fluorescence). Non-specific association accounted for <0.4% FRET efficiency.
Figure 1. Significant physical association occurs between Erk and PKCα molecules in the HN2-5 cells.
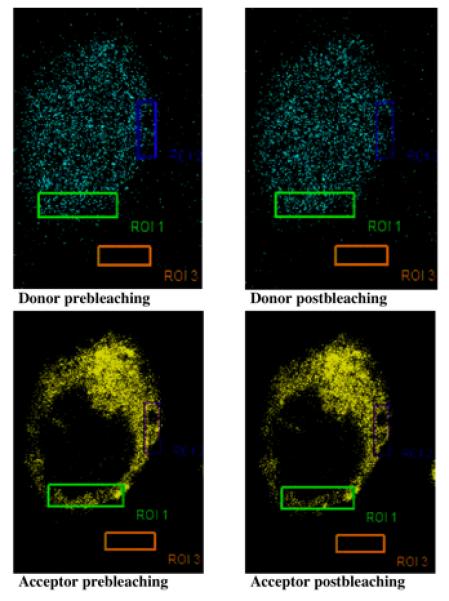
Erk-CFP and PKC-YFP vector were cotransfected into HN2-5 cells grown on coverslips. After 24 h, the cells were fixed, mounted on slides, and analyzed by laser confocal microscopy using a Leica Confocal Microscope equipped with a FRET software package. Two small regions of interest were photobleached using YFP excitation (514 nm). The images were acquired before and after photobleaching. The florescence of the donor showed an increase in intensity in the post-photobleaching image, whereas the fluorescence of the acceptor underwent a decrease. Mean FRET efficiency determined from six discrete samples used in three discrete experiments was 7.19±0.75%.
PKCα is phosphorylated and activated when the HN2-5 cells were treated with the 5-HT1A agonist 8-OH-DPAT
Previously we have demonstrated that stimulation of 5-HT1A-R causes Erk1/2-dependent phosphorylation of PKCα in both mouse hippocampus (P15) and hippocampal derived HN2-5 cells [4]. Here we show that transiently expressed wild type Myc-tagged PKCα protein in HN2-5 cells also undergoes phosphorylation at T638 in an Erk1/2 dependent manner. To achieve this, we transfected HN2-5 cells with a wild type PKCα construct and after 18 h, the cells were differentiated with retinoic acid for 16 h, then serum starved for 1 h, followed by treatment for 1 h with carrier or 8-OH-DPAT (a 5-HT1A agonist) (1 μM) in the absence or presence of Way100635 (a 5-HT1A antagonist), U0126 (an MEK inhibitor), and GFX (a general PKC inhibitor) for 1 h. The protein products were immunoprecipitated with a Myc antibody and analyzed by Western blotting as described in methods. As shown in Figure 2, 8-OH-DPAT treatment caused an induction in Myc-tagged P-T638-PKCα, which was eliminated in the presence Way100635 or U0126, but not GFX. Thus, 5-HT1A-R-linked phosphorylation of PKCα was Erk-dependent, but not PKC-dependent in the hippocampal neuron-derived HN2-5 cells.
Figure 2. Transiently expressed wild type PKCαundergoes phosphorylation at T638 in an Erk dependent manner.
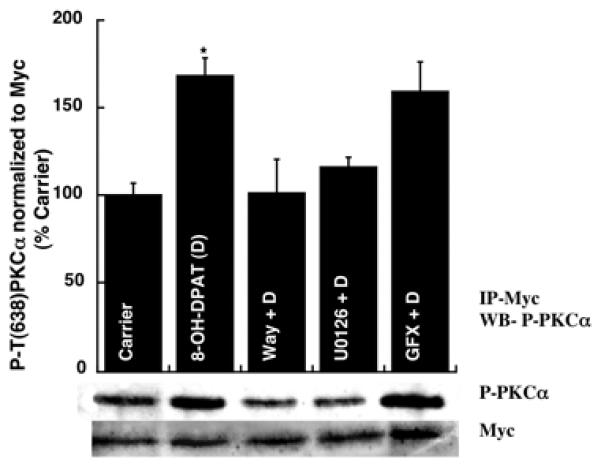
HN2-5 cells were transfected into wild type pcDNA6-PKCα construct, differentiated by retinoic acid, treated with drugs and lysed. Lysates were subjected to immunoprecipitation with a Myc tag antibody. Immunoprecipitates were analyzed by Western blotting using a P-PKCα/ß II (T638/641) antibody (upper panel) and then a Myc antibody (lower panel). *p < 0.05 with respect to all other sets except GFX + D. There was no significant difference between D and GFX + D.
Phosphorylation of PKCα at T638 is not PDK-1 dependent
It has been reported that the activation loop of PKCα is phosphorylated by a phosphoinositide dependent kinase 1 enzyme (PDK-1). The PDK-1 enzyme binds to the C-terminal hydrophobic loop of the newly synthesized PKCα molecule, which serves as the docking site [18], and then phosphorylates the T497 residue of PKCα. Once this T497 is phosphorylated, the PKCα is reported to undergo autophosphorylation of T638. We mutated the PDK-1 phosphorylation site of PKCα (T497 to A). This mutant, Myc-tagged PKCα cDNA construct was transfected into HN2-5 cells, the cells were differentiated and treated with carrier or 8-OH-DPAT (1 μM) in the absence or presence of Way100635 (4 μM), or U0126 (10 μM). The cells were lysed and immunoprecipitated using a Myc antibody. Western blotting analysis of the immunoprecipitated proteins revealed 5-HT1A-R and Erk-dependent phosphorylation of PKCα at T638 (Fig. 3).
Figure 3. An PDK-1 site-deficient mutant of PKCα is phosphorylated at T638 in HN2-5 cells in an Erk-dependent manner upon stimulation of the 5-HT1A-R.
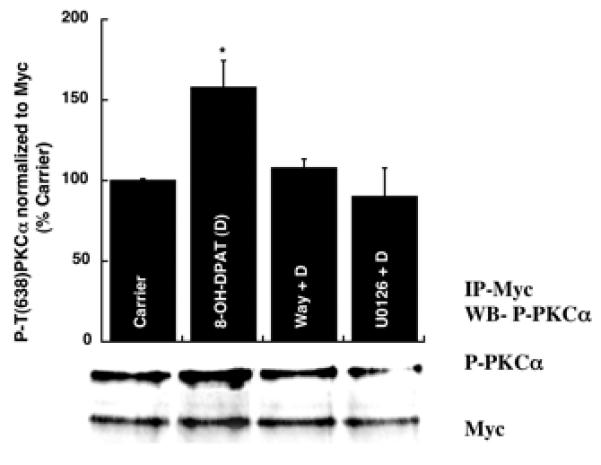
Transfected (empty vector or T497-mutant PKCα) and differentiated HN2-5 cells were treated with 1 μM D (1 h), in the absence and presence of 1 μM WAY or 10 μM U0126 (U). The Myc-tagged (T497→A) PKCα was immunoprecipitated with a Myc Ab. Western blotting analysis of the immunoprecipitate using a P-(T638/ 641)-PKCα/ßII antibody revealed phosphorylation of PKCα at T638, which was abrogated by WAY and U0126 (U). *p < 0.05 (versus all other sets).
PVLTP is a MAPK phosphorylation site and Erk1/2 causes phosphorylation of PKCα oligopeptide
All MAPK phosphorylates their substrates to a minimal consensus sequence Ser/Thr-Pro (S/TP) and many potential substrates contains this motif [19]. The T638 residue of PKCα is located within an authentic MAPK consensus phosphorylation site (PVXTP). A very similar phosphorylation site (PVATP) has been identified in the peroxiredoxin-6 protein [15]. In order to establish that PVXTP is an Erk1/2 phosphorylation site we performed an in vitro kinase assay using a mutant Erk construct in which a bulky amino acid residue in the ATP binding site (Q103) was replaced with glycine, enabling this mutant to utilize a bulky analog of ATP, cyclopentyl ATP. An 18-aa segment of PKCα protein sequence harboring the putative Erk target site (FFTRGQPVLTPPDQLVIA) was used as a substrate in an in vitro reaction. (4a) Flag-tagged mutant Erk vector or the empty vector were transfected into hippocampal neuron-derived HN2-5 cells. The transfected cells were differentiated and then treated with 1 μM 8-OH-DPAT for 30 min. The mutant Erk was immunoprecipitated using a Flag-antibody and the IP resuspended in PKC kinase buffer, supplemented with 32P-labeled cyclopentyl ATP and the 18-aa oligopeptide, followed by incubation at 30 °C for 20 min. HPTLC analysis of the product showed Erk-catalyzed phosphorylation of the PKCα peptide (Fig. 4b). Neither PKCα nor wild-type Erk can use cyclopentyl-[32P]ATP, therefore this phosphorylation must have been catalyzed by the mutant Erk molecule expressed in the differentiated HN2-5 cells.
Figure 4. In vitro analysis of Erk-catalyzed phosphorylation of PKCα.

(a) The scheme of the experiment, showing mutant Erk mediated phosphorylation of the PKCα oligopeptide harboring the turn motif and the T638 residue. (b) Transfected and differentiated HN2-5 cells were treated with 1 μM D (30 min), lysed, and the activated mutant Erk was immunoprecipitated (IP) using a Flag antibody (Ab) (upper panel). The IP pellet was used in a kinase assay (A) using cyclopentyl [γ32P] ATP and the PKCα peptide. The radiolabeled peptide moved faster than the free cyclopentyl [γ32P] ATP (smear) on an HPTLC plate. (c) PKCα harbors a putative phosphorylation site (PVLTP) and two docking site (KRGRIYL) and (KRGIIYRDLKL) for Erk1/2.
A kinase site mutant form of PKCα(K368→A) is also phosphorylated at T638 in an Erk-dependent manner
As an additional evidence beyond the observation that 5-HT1A-R stimulation in the HN2-5 cells causes Erk-dependent phosphorylation of the PDK-1 site mutant version of PKCα, we transfected the HN2-5 cells with the cDNA for a Myc-tagged kinase site mutant form of PKCα. As observed for the PDK-1 site mutant, 5-HT1A-R stimulation also caused increased T638 phosphorylation of the kinase site mutant in an Erk-dependent manner (Supplemental figure 1).
PKCα has two Erk1/2 docking sites
MAPKs recognize their substrates by initially binding to a motif called docking domain. The docking domain contains an amino acid sequence “K/R-K/R-(X)2–6-L-X-L”, which harbors a cluster of at least two positively charged amino acids followed by a spacer of 2-to-6 residues from a hydrophobic-X-hydrophobic sequence containing long-chain aliphatic amino acids such as Leu and Ile. Pro, Asn, and/or Gly, which form turns and break helices, occur at a high rate in the spacer and in the sequence immediately up- stream of the hydrophobic-X-hydrophobic element [20,21]. Another type of docking motif is a short peptide containing the sequence FXFP that is usually downstream of the phosphorylation site. This motif was originally found to bind the ERK subfamily but lately it has been shown to promote interactions with p38 alpha and p38 beta2 [22,23]. Substrates can contain the classical docking motif, or the FXFP sequence, or both [19]. Interestingly, the PKCα protein, like other MAPK substrates, also contains two putative docking domains such as RGRIYL and KRGIIYRDLKL (Fig. 4C). This provides further evidence that PKCα is a direct substrate of Erk1/2 in the 5-HT1A-R-mediated signaling cascade.
An earlier study by Kobe and coworkers used FRET to analyze and assess association between 5-HT1A-R and 5-HT7-R [17]. Both of these proteins mainly reside in the plasma membrane. Because of their restricted ability to move perpendicular to the plasma membrane, these two proteins will then be restricted to lateral movement, which may enhance the possibility of intermolecular interactions between these two molecules. This may have been the reason for a high FRET efficiency observed in this study. Even the FRET efficiency of non-specific interactions was relatively high (~5%). In contrast, P-Erk is distributed in the entire cell, whereas PKCα and P-PKCα remain in the cytosol and the cytoplasmic membranes. Consequently, we observed insignificant non-specific FRET efficiency and 7% FRET efficiency between Erk-CFP and PKCα-YFP in our experiments.
This short communication reports a fundamental discovery that the two pivotal kinases Erk and PKCα, which regulate myriad cellular processes, physically interact to regulate signaling receptor-mediated pathways that are central to synaptogenesis in the neonatal hippocampus [4]. Although this study delineates the mechanistic details in in vitro studies in a hippocampal neuron-derived cell line, our prior studies had demonstrated the profile of Erk-dependent, but PKC-independent phosphorylation of PKCα in cultured hippocampal slices [4]. Our current experiments employ intra-hippocampal injections of selective agonists and inhibitors of signaling intermediates to delineate these pathways in vivo [24]. These studies will verify the importance of Erk-mediated phosphorylation and stimulation of PKCα in neonatal development of the hippocampal circuitry. Identification of such synaptogenic signaling molecules may yield appropriate therapeutic targets, which can be manipulated in an early attempt to alleviate chances of later-life affective disorders that crucially depend on neonatal synaptogenesis in the hippocampus.
Supplementary Material
A kinase site mutant form (K368→A) of PKCα is phosphorylated at T638 in Erk1/2 dependent manner upon stimulation of the 5-HT1A-R in HN2-5 cells.
Acknowledgment
This project was supported by a grant from the NIH (MH071376). We express our gratitude to Dr. Yalin Wang for his help in FRET analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Adayev T, Ranasinghe B, Banerjee P. Transmembrane signaling in the brain by serotonin, a key regulator of physiology and emotion. Bioscience Reports. 2005;25:363–385. doi: 10.1007/s10540-005-2896-3. [DOI] [PubMed] [Google Scholar]
- 2.Banerjee P, Mehta M, Kanjilal B. The 5-HT1A Receptor: A Signaling Hub linked to Health and Emotional Balance. In: Chattopadhyay A, editor. Frontiers in Neuroscience. CRC Press; 2007. pp. 133–155. [PubMed] [Google Scholar]
- 3.Adayev T, Ray I, Sondhi R, Sobocki T, Banerjee P. The G protein-coupled 5-HT1A receptor causes suppression of caspase-3 through MAPK and protein kinase Calpha. Biochim Biophys Acta. 2003;1640:85–96. doi: 10.1016/s0167-4889(03)00023-5. [DOI] [PubMed] [Google Scholar]
- 4.Mehta M, Ahmed Z, Fernando SS, Cano-Sanchez P, Adayev T, Ziemnicka D, Wieraszko A, Banerjee P. Plasticity of 5-HT1A receptor-mediated signaling during early postnatal brain development. J Neurochem. 2007;101:918–28. doi: 10.1111/j.1471-4159.2007.04448.x. [DOI] [PubMed] [Google Scholar]
- 5.Khundmiri SJ, Dean WL, McLeish KR, Lederer ED. Parathyroid hormone-mediated regulation of Na+-K+-ATPase requires ERK-dependent translocation of protein kinase Calpha. J Biol Chem. 2005;280:8705–13. doi: 10.1074/jbc.M408606200. [DOI] [PubMed] [Google Scholar]
- 6.Mauro A, De Cesaris P, Scoglio A, Bouche M, Molinaro M, Aquino A, Zani BM. PKCalpha-mediated ERK, JNK and p38 activation regulates the myogenic program in human rhabdomyosarcoma cells. J. Cell Sci. 2002;115:3587. doi: 10.1242/jcs.00037. C. C. [DOI] [PubMed] [Google Scholar]
- 7.Rucci N, DiGiacinto C, Orru L, Millimaggi D, Baron R, Teti A. A novel protein kinase C alpha-dependent signal to ERK1/2 activated by alphaVbeta3 integrin in osteoclasts and in Chinese hamster ovary (CHO) cells. J. Cell Sci. 2005;118:3263. doi: 10.1242/jcs.02436. [DOI] [PubMed] [Google Scholar]
- 8.Wen-sheng W. rotein kinase C α trigger Ras and Raf-independent MEK/ERK activation for TPA-induced growth inhibition of human hepatoma cell HepG2. Cancer Lett. 2006;239 doi: 10.1016/j.canlet.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 9.Toker A, Newton AC. Cellular Signaling: Pivoting around PDK-1. Cell. 2000;103:185–188. doi: 10.1016/s0092-8674(00)00110-0. [DOI] [PubMed] [Google Scholar]
- 10.Seki T, Matsubayashi H, Amano T, Shirai Y, Saito N, Sakai N. Phosphorylation of PKC activation loop plays an important role in receptor-mediated translocation of PKC. Genes to Cells. 2005;10:225–239. doi: 10.1111/j.1365-2443.2005.00830.x. [DOI] [PubMed] [Google Scholar]
- 11.Keranen LM, Dutil EM, Newton AC. Protein kinase C is regulated in vivo by three functionally distinct phosphorylations. Curr Biol. 1995;5:1394–1403. doi: 10.1016/s0960-9822(95)00277-6. [DOI] [PubMed] [Google Scholar]
- 12.Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochemical Journal. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis RJ. The mitogen-activated protein kinase signal transduction pathway. J Biol Chem. 1993;268:14553–6. [PubMed] [Google Scholar]
- 14.Fantz DA, Jacobs D, Glossip D, Kornfeld K. Docking sites on substrate proteins direct extracellular signal-regulated kinase to phosphorylate specific residues. J Biol Chem. 2001;276 doi: 10.1074/jbc.M102512200. [DOI] [PubMed] [Google Scholar]
- 15.Wu Y, Feinstein SI, Manevich Y, Chowdhury I, Pak JH, Kazi A, Dodia C, Speicher DW, Fisher AB. Mitogen-activated protein kinase-mediated phosphorylation of peroxiredoxin 6 regulates its phospholipase A(2) activity. Biochem J. 2009;419:669–79. doi: 10.1042/BJ20082061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eblen ST, Kumar NV, Shah K, Henderson MJ, Watts CK, Shokat KM, Weber MJ. Identification of novel ERK2 substrates through use of an engineered kinase and ATP analogs. J Biol Chem. 2003;278:14926–35. doi: 10.1074/jbc.M300485200. [DOI] [PubMed] [Google Scholar]
- 17.Kobe F, Renner U, Woehler A, Wlodarczuk J, Zeug A, Richter DW, Neher E, Ponimaskin EG. Stimulation- and palmitoylation-dependent changes in oligomeric conformation of 5-HT1A receptors. BBA: Molecular Cell Research. 2008;1783:1503–1516. doi: 10.1016/j.bbamcr.2008.02.021. [DOI] [PubMed] [Google Scholar]
- 18.Gao T, Toker A, Newton AC. The carboxyl terminus of protein kinase c provides a switch to regulate its interaction with the phosphoinositide-dependent kinase, PDK-1. J Biol Chem. 2001;276:19588–96. doi: 10.1074/jbc.M101357200. [DOI] [PubMed] [Google Scholar]
- 19.Sharrocks AD, Yang SH, Galanis A. Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem Sci. 2000;25:448–53. doi: 10.1016/s0968-0004(00)01627-3. [DOI] [PubMed] [Google Scholar]
- 20.Yang SH, Yates PR, Whitmarsh AJ, Davis RJ, Sharrocks AD. The Elk-1 ETS-domain transcription factor contains a mitogen-activated protein kinase targeting motif. Mol. Cell. Biol. 1998;18:710–720. doi: 10.1128/mcb.18.2.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bardwell AJ, Flatauerm LJ, Matsukuma K, Thorner J, Bardwell L. A conserved docking site in MEKs mediates high-affinity binding to MAP kinases and cooperates with a scaffold protein to enhance signal transmission. J. Biol. Chem. 2001;276:10374–10386. doi: 10.1074/jbc.M010271200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobs D, Glossip D, Xing H, Muslin AJ, Kornfeld K. Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 1999;13:163–75. [PMC free article] [PubMed] [Google Scholar]
- 23.Galanis A, Yang S-H, Sharrocks AD. Selective Targeting of MAPKs to the ETS Domain Transcription Factor SAP-1. Journal of Biological Chemistry. 2001;276:965–973. doi: 10.1074/jbc.M007697200. [DOI] [PubMed] [Google Scholar]
- 24.Mogha A, Debata PR, Banerjee P. Signaling by the 5-HT1A receptor causes postnatal synaptogenesis in the hippocampus; 41st Annual Meeting of The American Society for Neurochemistry; Santa Fe, NM. 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A kinase site mutant form (K368→A) of PKCα is phosphorylated at T638 in Erk1/2 dependent manner upon stimulation of the 5-HT1A-R in HN2-5 cells.