

Abstract
Purpose of review
Tip links are thought to be an essential element of the mechanoelectrical transduction (MET) apparatus in sensory hair cells of the inner ear. The molecules that form tip links have recently been identified, and the analysis of their properties has not only changed our view of MET but also suggests that tip link defects can cause hearing loss.
Recent findings
Structural, histological and biochemical studies show that the extracellular domains of two deafness-associated cadherins, cadherin 23 (CDH23) and protocadherin 15 (PCDH15), interact in trans to form the upper and lower part of each tip link, respectively. High speed Ca2+ imaging suggests that MET channels are localized exclusively at the lower end of each tip link. Biochemical and genetic studies provide evidence that defects in tip links cause hearing impairment in humans.
Summary
The identification of the proteins that form tip links have shed new light on the molecular basis of MET and the mechanisms causing hereditary deafness, noise-induced hearing loss and presbycusis.
Keywords: Tip links, hair cells, hearing, deafness, mechanoelectrical transduction, cadherins, inherited deafness
Introduction
Tip links of sensory hair cells in the inner ear play a critical role in mechanoelectrical transduction (MET), the conversion of mechanical stimuli arising from sound and head movements into electrochemical signals. The dynamic properties and molecular composition of tip links has been the subject of intense debate over the past two decades. Two members of the cadherin family, cadherin 23 (CDH23) and protocadherin 15 (PCDH15), were recently identified as tip link constituents1-3. The identification CDH23 and PCDH15 as constituents of the tip link and the molecular asymmetry formed by heterophilic interaction of these molecules1 provides novel opportunities for understanding MET at the molecular level, and for determining the extent to which defects in MET cause hearing impairment. We summarize here recent advances in our understanding of the composition, function, and dysfunctions of tip links.
The current model of tip link function
MET in hair cells occurs in hair bundles, which are comprised of dozens of stereocilia organized in rows of decreasing heights. In the current model of MET, deflection of a hair bundle increases tension in a gating spring, which rapidly leads to increases in the open probability of MET channels4. MET channels are thought to be localized close to tip links5, the extracellular filaments connecting the tip of each stereocilium to the lateral membrane of its adjacent taller neighbor (Fig. 1). The molecular identity of the gating spring in hair cells is unknown, but tip links are thought to be the gating spring 5 or in series with it6. Following activation, mechanotransduction currents rapidly adapt, a process thought to be mediated at least in part by myosin motor proteins that reestablish tension in the gating spring4, 7, 8.
Figure 1.
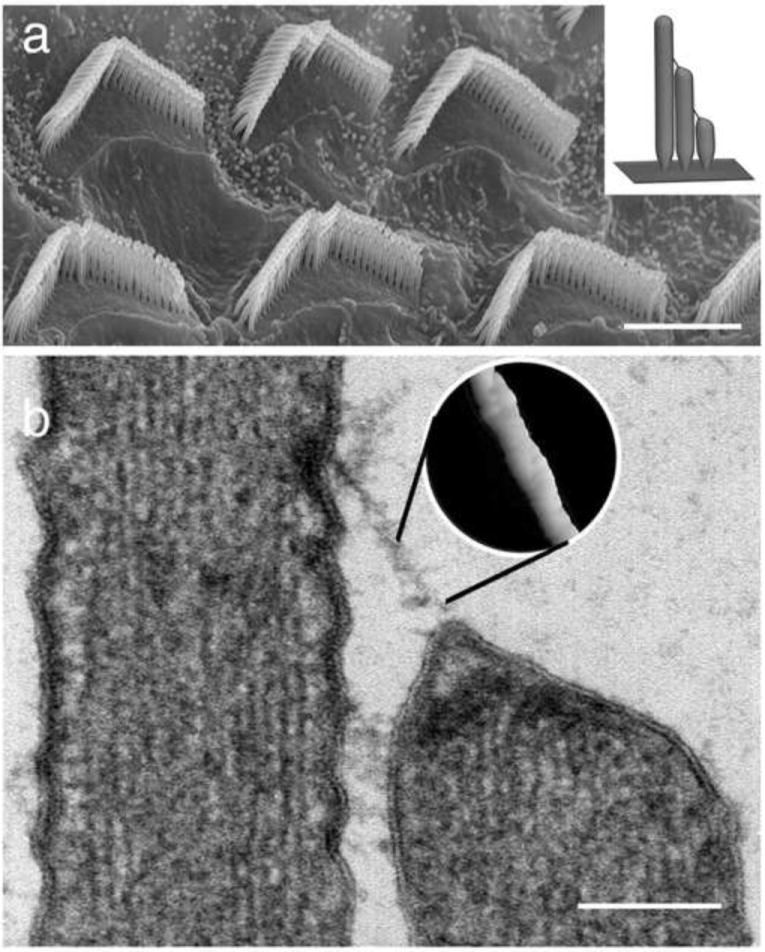
Stereocilia of the inner ear hair cells form organized bundles and are connected to each other by tip links. (a) Scanning electron micrograph showing stereocilia bundles on the apical surface of the outer hair cells of the rat organ of Corti. Bar = 5 μ m. (Inset in a) Model depicting a longitudinal section through a hair cell bundle. Note the staircase pattern and arrangement of tip links. (b) Thin section transmission electron micrograph showing a tip link connecting one stereocilium to an adjacent taller neighbor. Note the presence of electron dense material at the upper and lower insertion sites and that the tip of the shorter stereocilia is tented due to tip link tension (from Kachar et al., 2000). Bar = 150 nm. (Inset in b) The surface rendering of a freeze-etching image of the tip link provides a close up view of helical structure of the tip link. (Adapted from [6] : Kachar B, Parakkal M, Kurc M et al.: High-resolution structure of hair-cell tip links. Proc Natl Acad Sci U S A 2000, 97:13336-41)
Ultrastructure of the tip link
Electron microscopy has revealed that tip links consist of right-handed coiled double strands with 20-25 nm periodicity and 4 nm diameter globular component6, 9. The upper part of the tip link splits close to the membrane into two filaments that terminate in an electron dense structure6. The lower tip-link part branches into two or three anchor filaments, which terminate in an electron dense structure at the tips of stereocilia6. Ca2+ chelators such as BAPTA disrupt tip links6, 10, 11 demonstrating that their structural integrity is Ca2+-dependent.
The tip link is an asymmetric filament formed by CDH23 and PCDH15
Two members of the cadherin family, CDH232, 12 and PCDH153, which have been linked to inherited forms of deafness in humans13-17, mice18, 19, and rats20, have recently been identified as integral components of tip links. CDH23 was proposed to be a component of the tip link because it localizes to the tips of stereocilia, and mutations in its gene lead to loss of tip links in zebrafish2, 12. Whether CDH23 was a true component of the tip link remained controversial due to several failed attempts to detect its expression in hair cells of adult animals21, 22. Meanwhile, it was shown that an antibody that stains the tips of stereocilia in avian hair cells recognizes PCDH1511, suggesting that PCDH15 was also a tip link component3.
A recent study1 proposed that each tip link is formed by CDH23 homodimers interacting in trans with PCDH15 homodimers, where CDH23 is localized to the upper part of the tip link and PCDH15 to the lower part (Fig. 2). Accordingly, antibodies that recognize specific epitopes along the CDH23 and PCDH15 extracellular cadherin domains (ECD) label the upper and lower part of a tip link, respectively1. Antibodies against the N-termini of CDH23 and PCDH15 stain the same position within the tip link, suggesting that the two cadherins interact via their N-termini1. Negative staining TEM shows that recombinant ECD of CDH23 and PCDH15 form coiled homodimers that interact at their N-termini. Furthermore, the membrane proximal C-terminus of CDH23 separates into two strands1, just as the upper end of tip links does6. In pull-down experiments, the CDH23 and PCDH15 ECDs interact via their N-termini in a Ca2+ dependent manner1.
Figure 2.
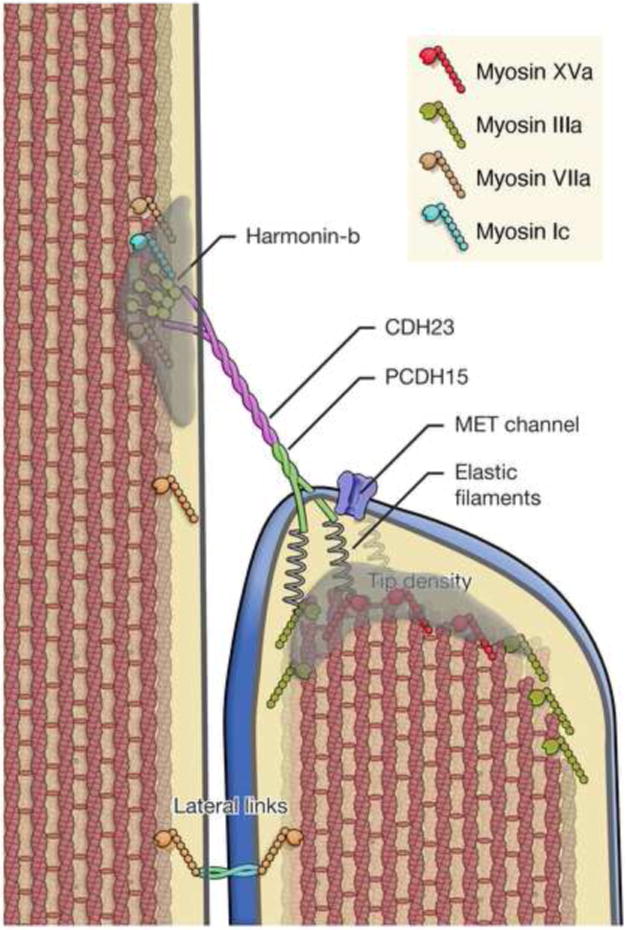
Schematic representation of the tip link complex illustrating structural features and key molecular components. CDH23 and PCDH15 comprise the tip link, which inserts into the stereocilia membrane at the sites of the upper and lower tip densities. Tip densities are presumed to contain scaffolding proteins, which bind to the cytoplasmic domain CDH23 and PCDH15 and anchor the tip link. In addition several myosins including myosin Ic7, myosin IIIa61, myosin VIIa47, and myosin XVa45 have been localized to the tip density region and proposed to participate in MET adaptation, stereocilia actin dynamics, localization of lateral links, and cargo transport. Tension on the tip link gates the mechanoelectrical transduction (MET) channel. The tip-link can exert force onto the channel either directly or indirectly by tenting the membrane4. The gating mechanism is presumed to involve a gating spring element through which force is applied to the channel gate. The channel gate opens in series with this spring, thus transiently reducing the force onto this spring and increasing compliance4. The coiled structure of the tip link and the properties expected of conventional cadherins suggest that the tip link is relatively stiff62. The spring element should then be in series with the tip link, possible as putative “elastic filaments” between the membrane and the dense actin core6. Original figure
There are variations in the reported length of the tip link (90-200nm)23-25 compared to the molecular length of the CDH23-PCDH15 heterotetramer (180nm)1. A change in the conformation and configuration of the molecules may in part explain this variation. A recent report suggests that the Ca2+ concentration affects tip link length24. When in vitro Ca2+ concentration is similar to endolymph (50μM), tip link length is ~185 nm, while it is shortened by 20 nm when the Ca2+ concentration is increased to 1 mM, suggesting that Ca2+ may bridge Ca2+ binding sites inside the ECD and lead to a tightly folded conformation. The tension in the tip link could also affect the length by forcing unfolding of the CDH23 and PCDH15 extracellular domains. Molecular variability between tip links of different hair cells also cannot be excluded. In Cdh23-null waltzer mice, tip link-like structures are observed during hair cell development, which could potentially be PCDH15 homodimers interacting in trans26, though homophilic PCDH15 binding activity has so far not been detected in biochemical assays. Alternative splicing might also generate short isoforms of CDH2322 and PCDH153, 27, thereby leading to tip links of different lengths.
MET channels localize to the lower tip link insertion site
The intrinsic molecular asymmetry in the tip link suggests that the opposite ends of tip links might have distinct functions. Although MET channels were previously thought to be localized to both ends of tip links28, recent studies using high speed Ca2+ imaging demonstrate that upon mechanical stimulation, Ca2+ entry is ten-fold larger and faster in the middle and shortest rows of stereocilia than in the tallest row29. This finding strongly suggests that the MET channel is predominantly or exclusively localized to the lower tip link insertion point (Fig. 2). A structural study demonstrating the occasional tenting of the membrane at stereocilia tips in the shortest and middle row of stereocilia (Fig. 1B and Fig. 2), but not in the tallest row, supports this model as tenting indicates membrane tension, which might be favorable for activating the MET channel4, 6.
Developmental link and kinociliary link
In addition to the tip link, CDH23 and PCDH15 are implicated in forming transient links that connect the stereocilia to each other and to the single kinocilium of the hair bundle during development. Developmental links between stereocilia are initially broadly distributed along their length but are gradually removed as stereocilia mature30. Kinociliary links connect the lateral membrane of the kinocilium to the tallest stereocilia11. Like tip links, these links are sensitive to Ca2+ chelation11, 30. Moreover, antibodies against CDH23 and PCDH15 strongly label these links1, 11, 12, 21 and mutations in Cdh2319 and Pcdh1518 lead to defects in hair bundle morphology during embryogenesis, suggesting that these links regulate hair bundle development.
Proteins that bind to the cytoplasmic domains of CDH23 and PCDH15
Usher proteins are likely candidate components of the tip link/MET apparatus. Human Usher syndrome (USH) is characterized by congenital deafness with progressive visual deficit caused by retinitis pigmentosa31. Of the three known types of Usher syndrome, USH1 has the most severe phenotype causing congenital deafness and early onset retinitis pigmentosa. Mutations at seven loci have been linked to USH1 (USH1A-G). Two of the affected loci encode CDH23 (USH1D)15, 16 and PCDH15 (USH1F)13, 14. Other USH1 genes encode the adaptor proteins harmonin (USH1C)32 and SANS (USH1G)33, 34 as well as the unconventional myosin MYO7a (USH1B)35, 36. The similar phenotypes among USH1 patients suggest that USH1 proteins act in a common molecular network. This model is reinforced by biochemical evidence, which suggests that USH1 proteins assemble into larger protein complexes.
The cytoplasmic domain of CDH23 has two alternative splicing isoforms, CDH23+68 and CDH23-6837, while three prominent cytoplasmic splice variants, CD1, 2, and 3, have been described for PCDH153. The N-termini of CDH23+68, CDH23-68 and PCDH15 fit the consensus sequence that mediates protein-protein interactions with PDZ motifs. Interestingly, the USH1C protein harmonin contains three PDZ domains, localizes to stereocilia, and binds in vitro to CDH23+68, CDH23-68 and PCDH15-CD138-41. Binding to PCDH15-CD1 is mediated by interactions of its extreme C-terminus with PDZ2 of harmonin38. Interactions between CDH23 and harmonin are more complex. The extreme C-terminus of CDH23+68 and CDH23-68 can bind to PDZ2 of harmonin, while a more membrane proximal domain in the two CDH23 isoforms bind to the N-terminal fragment of harmonin39-41. Additionally, CDH23-68 but not CDH23+68 can bind to harmonin PDZ139.
Recent findings show that harmonin is broadly distributed in stereocilia of developing hair bundles but becomes strongly concentrated at the upper tip link insertion site as hair cells mature40. Harmonin therefore shows a similar redistribution as CDH23 during hair bundle maturation42. Mutations in CDH2343 or in the harmonin PDZ2 domain44 prevent harmonin localization to the upper tip link insertion site, suggesting that PDZ2-mediated interactions with CDH23 are critical for its localization. Interestingly, harmonin is also mislocalized in deaf circler (dfcr) mice, which carry a mutation in harmonin deleting its actin-binding domain44. While the electron dense plaque at the upper tip link insertion site is lost in dfcr mice, tip links are maintained and MET currents can be evoked44. However, the properties of the MET currents, such as channel activation and adaptation, are affected. These findings suggest that harmonin is an essential component of the upper tip link plaque (Fig. 2) and thus is important for MET.
The USH1B protein myosin VIIa or MYO7A also localizes to stereocilia35, 36, 45, 46 and can bind to the USH1 proteins PCDH15-CD1, harmonin, and SANS. Interactions with PCDH15-CD1 are mediated by the SH3 domain in the MYO7A tail domain, while the MyTh4-FERM domain binds to harmonin PDZ1 and SANS34, 38. MYO7A is a plus-end directed actin based molecular motor and has been proposed to transport cargo towards the tips of stereocilia47. Consistent with this model, localization of PCDH15 and harmonin is disturbed in MYO7A-deficient mice40, 48.
Myosin Ic (MYO1C) is thought to participate in MET channel adaptation and has been localized to the upper and lower insertion points of the tip link49, 50. MYO1C and CDH23 co-immunoprecipitated when expressed in heterologous cells2 and recombinant MYO1C localizes to the stereocilia tips of wild type but not CDH23-deficient mice51. These findings suggest that CDH23 is in series with the adaptation motor complex, but the mechanisms that mediate interactions between the two proteins are currently unclear.
Deafness associated with tip link dysfunction
Genetic mouse models have been instrumental in providing insight into the function of tip link proteins. These findings also reinforce the concept that CDH23, PCDH15 and their associated proteins are multifunctional. Previously identified mouse models such as shaker-1 (Myosin VIIa)36, deaf circler (harmonin)32, waltzer (CDH23)19, Ames waltzer (PCDH15)18 and Jackson circler (SANS)33, exhibit disrupted stereociliary bundles, which indicate the importance of these proteins in hair bundle development. The mutations in the mouse models resemble similar mutations in USH1D patients, suggesting that defects in hair bundle development cause USH1. However, missense mutations in some USH1 genes cause recessive, late onset deafness without visual impairment. For example, while a deletion in CDH23 resulting from nonsense, splice-site or frame shift mutations cause human USH1D, missense mutations in CDH23 cause the less severe DFNB12 phenotype52, 53. This phenotypic heterogeneity suggests that some mutations might affect developmental linkages in hair cells, while others specifically affect tip links. Recently the first mouse model for recessive forms of deafness caused by missense mutations in any USH1 gene was described. This mouse, named salsa, was identified in an ENU mutagenesis screen and carries a missense mutation in the extracellular domain of CDH2354. The salsa mutation leads to amino acid substitution in the highly conserved Ca2+ binding motif in the 7th ECD of CDH23. Organization of salsa hair bundles is intact, but tip links are progressively lost from the basal to apical turn of the cochlea after the development of functional hearing 54. Hearing loss in salsa is similarly progressive, suggesting that it is caused by tip link loss54. Interestingly, many mutations in DFNB12 patients also affect Ca2+ binding sites of CDH23. Biochemical assay demonstrated that the salsa and DFNB12 mutations affect adhesive interactions between CDH23 and PCDH15, even though the mutated sites are distant from the ligand-binding domain (ECD1). Ca2+ binding stabilizes the structure of classical cadherins55, suggesting that structural defects caused by the salsa/DFNB12 mutations secondarily affect adhesive function. The phenotype of salsa suggests that DFNB12 is a new class of deafness caused by the loss of the tip links due to an unstable interaction between CDH23 and PCDH15. A DFNB23-causative missense mutation in PCDH15-ECD is also known to affect the interaction between CDH23 and PCDH151, suggesting a common pathogenesis underlying both DFNB12 and DFNB23. Establishing mouse models for DFNB23 will be important for testing this hypothesis.
Intriguingly, a polymorphism in the mouse genome that maps to the CDH23 extracellular domain is associated with noise induced hearing loss (NIHL) and presbycusis. This polymorphism changes the splicing of the primary CDH23 transcript leading to an in-frame deletion, which likely affects CDH23 structure and function56. In humans, several non-synonymous polymorphisms in CDH23 also increase the risk of NIHL57. These findings suggest that tip link fragility is also associated with NIHL and presbycusis.
Conclusions and future directions
Based on recent advances, inner ear research is now at a new horizon. In the following section we discuss two important future areas of research.
1. Identification of the MET channel
The asymmetric molecular composition of the tip link together with localization of the MET channels at its lower end sets the stage for understanding MET at the molecular level. For example, of the three prominent PCDH15 cytoplasmic domains, PCDH15-CD3 is has been specifically localized at the tip of stereocilia and is a good candidate to bind to the MET channel or channel-associated molecules3. Recently, a study using yeast two-hybrid screening identified HCN1 (hyperpolarization-activated, cyclic nucleotide-gated nonselective cation channel isoform-1) as a binding partner of PCDH15-CD358. The role of HCN1 in MET has yet to be determined. Earlier studies reported channel properties that are significantly different than that expected for the MET channel itself59, 60. Further exploration of the molecular dynamics, protein-protein interactions, and mechanical properties of the tip link complex will further clarify our understanding of the molecular identity and intricacies of the MET machinery.
2. Presbycusis and tip link dysfunction
In mammals, hair cells are terminally differentiated and do not regenerate, yet need to maintain the highly organized MET machinery over decades. This is awe-inspiring as we consider that hair bundles deflect thousands of times per second when stimulated and that the load-bearing molecules in the MET apparatus must restore and retain their structural and functional integrity with each deflection. Imperfections in hair cells accumulate with age and might eventually overload the ability of the cell to maintain a functional MET apparatus. A further understanding of the mechanisms controlling maintenance and turnover of these extraordinary structures will hopefully lead to a better understanding of presbycusis and provide new therapeutic approaches to sensorineural hearing loss.
Acknowledgments
This work was supported by the Intramural Program of NIDCD, NIH.
References
- 1.Kazmierczak P, Sakaguchi H, Tokita J, et al. Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature. 2007;449:87–91. doi: 10.1038/nature06091. [DOI] [PubMed] [Google Scholar]
- 2.Siemens J, Lillo C, Dumont RA, et al. Cadherin 23 is a component of the tip link in hair-cell stereocilia. Nature. 2004;428:950–5. doi: 10.1038/nature02483. [DOI] [PubMed] [Google Scholar]
- 3.Ahmed ZM, Goodyear R, Riazuddin S, et al. The tip-link antigen, a protein associated with the transduction complex of sensory hair cells, is protocadherin-15. J Neurosci. 2006;26:7022–34. doi: 10.1523/JNEUROSCI.1163-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ricci AJ, Kachar B, Gale J, et al. Mechano-electrical transduction: new insights into old ideas. J Membr Biol. 2006;209:71–88. doi: 10.1007/s00232-005-0834-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pickles JO, Comis SD, Osborne MP. Cross-links between stereocilia in the guinea pig organ of Corti, and their possible relation to sensory transduction. Hear Res. 1984;15:103–12. doi: 10.1016/0378-5955(84)90041-8. [DOI] [PubMed] [Google Scholar]
- 6.Kachar B, Parakkal M, Kurc M, et al. High-resolution structure of hair-cell tip links. Proc Natl Acad Sci U S A. 2000;97:13336–41. doi: 10.1073/pnas.97.24.13336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gillespie PG, Cyr JL. Myosin-1c, the hair cell’s adaptation motor. Annu Rev Physiol. 2004;66:521–45. doi: 10.1146/annurev.physiol.66.032102.112842. [DOI] [PubMed] [Google Scholar]
- 8.Stauffer EA, Scarborough JD, Hirono M, et al. Fast adaptation in vestibular hair cells requires myosin-1c activity. Neuron. 2005;47:541–53. doi: 10.1016/j.neuron.2005.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsuprun V, Santi P. Helical structure of hair cell stereocilia tip links in the chinchilla cochlea. J Assoc Res Otolaryngol. 2000;1:224–31. doi: 10.1007/s101620010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Assad JA, Shepherd GM, Corey DP. Tip-link integrity and mechanical transduction in vertebrate hair cells. Neuron. 1991;7:985–94. doi: 10.1016/0896-6273(91)90343-x. [DOI] [PubMed] [Google Scholar]
- 11.Goodyear RJ, Richardson GP. A novel antigen sensitive to calcium chelation that is associated with the tip links and kinocilial links of sensory hair bundles. J Neurosci. 2003;23:4878–87. doi: 10.1523/JNEUROSCI.23-12-04878.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sollner C, Rauch GJ, Siemens J, et al. Mutations in cadherin 23 affect tip links in zebrafish sensory hair cells. Nature. 2004;428:955–9. doi: 10.1038/nature02484. [DOI] [PubMed] [Google Scholar]
- 13.Alagramam KN, Yuan H, Kuehn MH, et al. Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum Mol Genet. 2001;10:1709–18. doi: 10.1093/hmg/10.16.1709. [DOI] [PubMed] [Google Scholar]
- 14.Ahmed ZM, Riazuddin S, Bernstein SL, et al. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet. 2001;69:25–34. doi: 10.1086/321277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bork JM, Peters LM, Riazuddin S, et al. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet. 2001;68:26–37. doi: 10.1086/316954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bolz H, von Brederlow B, Ramirez A, et al. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat Genet. 2001;27:108–12. doi: 10.1038/83667. [DOI] [PubMed] [Google Scholar]
- 17.Ahmed ZM, Riazuddin S, Ahmad J, et al. PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet. 2003;12:3215–23. doi: 10.1093/hmg/ddg358. [DOI] [PubMed] [Google Scholar]
- 18.Alagramam KN, Murcia CL, Kwon HY, et al. The mouse Ames waltzer hearing-loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nat Genet. 2001;27:99–102. doi: 10.1038/83837. [DOI] [PubMed] [Google Scholar]
- 19.Di Palma F, Holme RH, Bryda EC, et al. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat Genet. 2001;27:103–7. doi: 10.1038/83660. [DOI] [PubMed] [Google Scholar]
- 20.Naoi K, Kuramoto T, Kuwamura Y, et al. Characterization of the Kyoto Circling (KCI) Rat Carrying a Spontaneous Nonsense Mutation in the Protocadherin 15 (Pcdh15) Gene. Exp Anim. 2009;58:1–10. doi: 10.1538/expanim.58.1. [DOI] [PubMed] [Google Scholar]
- 21.Michel V, Goodyear RJ, Weil D, et al. Cadherin 23 is a component of the transient lateral links in the developing hair bundles of cochlear sensory cells. Dev Biol. 2005;280:281–94. doi: 10.1016/j.ydbio.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 22.Lagziel A, Ahmed ZM, Schultz JM, et al. Spatiotemporal pattern and isoforms of cadherin 23 in wild type and waltzer mice during inner ear hair cell development. Dev Biol. 2005;280:295–306. doi: 10.1016/j.ydbio.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 23.Tsuprun V, Goodyear RJ, Richardson GP. The structure of tip links and kinocilial links in avian sensory hair bundles. Biophys J. 2004;87:4106–12. doi: 10.1529/biophysj.104.049031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24*.Furness DN, Katori Y, Nirmal Kumar B, et al. The dimensions and structural attachments of tip links in mammalian cochlear hair cells and the effects of exposure to different levels of extracellular calcium. Neuroscience. 2008;154:10–21. doi: 10.1016/j.neuroscience.2008.02.010. This article provides a possible explanation for variations in tip link length measured in previous experiments by demonstrating changes in tip link length as a function of extracellular calcium concentration and between different species.
- 25.Auer M, Koster AJ, Ziese U, et al. Three-dimensional architecture of hair-bundle linkages revealed by electron-microscopic tomography. J Assoc Res Otolaryngol. 2008;9:215–24. doi: 10.1007/s10162-008-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rzadzinska AK, Steel KP. Presence of interstereocilial links in waltzer mutants suggests Cdh23 is not essential for tip link formation. Neuroscience. 2009;158:365–8. doi: 10.1016/j.neuroscience.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alagramam KN, Miller ND, Adappa ND, et al. Promoter, alternative splice forms, and genomic structure of protocadherin 15. Genomics. 2007;90:482–92. doi: 10.1016/j.ygeno.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Denk W, Holt JR, Shepherd GM, et al. Calcium imaging of single stereocilia in hair cells: localization of transduction channels at both ends of tip links. Neuron. 1995;15:1311–21. doi: 10.1016/0896-6273(95)90010-1. [DOI] [PubMed] [Google Scholar]
- 29**.Beurg M, Fettiplace R, Nam JH, et al. Localization of inner hair cell mechanotransducer channels using high-speed calcium imaging. Nat Neurosci. 2009;12(5):553–558. doi: 10.1038/nn.2295. High speed calcium imaging demonstrated a much higher calcium influx in stereocilia of the second and third row compared to first row implying that the channels themselves are present only at the lower insertion sites of the tip links suggesting that the mechanoelectric transduction apparatus is not symmertric as previously proposed.
- 30.Goodyear RJ, Marcotti W, Kros CJ, et al. Development and properties of stereociliary link types in hair cells of the mouse cochlea. J Comp Neurol. 2005;485:75–85. doi: 10.1002/cne.20513. [DOI] [PubMed] [Google Scholar]
- 31.Kremer H, van Wijk E, Marker T, et al. Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum Mol Genet. 2006;15(Spec No 2):R262–70. doi: 10.1093/hmg/ddl205. [DOI] [PubMed] [Google Scholar]
- 32.Verpy E, Leibovici M, Zwaenepoel I, et al. A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat Genet. 2000;26:51–5. doi: 10.1038/79171. [DOI] [PubMed] [Google Scholar]
- 33.Kikkawa Y, Shitara H, Wakana S, et al. Mutations in a new scaffold protein Sans cause deafness in Jackson shaker mice. Hum Mol Genet. 2003;12:453–61. doi: 10.1093/hmg/ddg042. [DOI] [PubMed] [Google Scholar]
- 34.Weil D, El-Amraoui A, Masmoudi S, et al. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet. 2003;12:463–71. doi: 10.1093/hmg/ddg051. [DOI] [PubMed] [Google Scholar]
- 35.Weil D, Blanchard S, Kaplan J, et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature. 1995;374:60–1. doi: 10.1038/374060a0. [DOI] [PubMed] [Google Scholar]
- 36.Gibson F, Walsh J, Mburu P, et al. A type VII myosin encoded by the mouse deafness gene shaker-1. Nature. 1995;374:62–4. doi: 10.1038/374062a0. [DOI] [PubMed] [Google Scholar]
- 37.Di Palma F, Pellegrino R, Noben-Trauth K. Genomic structure, alternative splice forms and normal and mutant alleles of cadherin 23 (Cdh23) Gene. 2001;281:31–41. doi: 10.1016/s0378-1119(01)00761-2. [DOI] [PubMed] [Google Scholar]
- 38.Adato A, Michel V, Kikkawa Y, et al. Interactions in the network of Usher syndrome type 1 proteins. Hum Mol Genet. 2005;14:347–56. doi: 10.1093/hmg/ddi031. [DOI] [PubMed] [Google Scholar]
- 39.Siemens J, Kazmierczak P, Reynolds A, et al. The Usher syndrome proteins cadherin 23 and harmonin form a complex by means of PDZ-domain interactions. Proc Natl Acad Sci U S A. 2002;99:14946–51. doi: 10.1073/pnas.232579599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boeda B, El-Amraoui A, Bahloul A, et al. Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 2002;21:6689–99. doi: 10.1093/emboj/cdf689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41*.Pan L, Yan J, Wu L, et al. Assembling stable hair cell tip link complex via multidentate interactions between harmonin and cadherin 23. Proc Natl Acad Sci U S A. 2009;106:5575–80. doi: 10.1073/pnas.0901819106. A novel binding mode between CDH23 and harmonin was demonstrated by biochemical study and the detailed structure of the binding site by NMR.
- 42.Rzadzinska AK, Derr A, Kachar B, et al. Sustained cadherin 23 expression in young and adult cochlea of normal and hearing-impaired mice. Hear Res. 2005;208:114–21. doi: 10.1016/j.heares.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 43*.Lefevre G, Michel V, Weil D, et al. A core cochlear phenotype in USH1 mouse mutants implicates fibrous links of the hair bundle in its cohesion, orientation and differential growth. Development. 2008;135:1427–37. doi: 10.1242/dev.012922. This article defined the role of USH1 proteins in maintaing the fibrous structure including developmental and kinociliary links during hair cell development.
- 44.Grillet N, Xiong W, Reynolds A, et al. Harmonin Mutations Cause Mechanotransduction Defects in Cochlear Hair Cells. Neuron. 2009 doi: 10.1016/j.neuron.2009.04.006. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rzadzinska AK, Schneider ME, Davies C, et al. An actin molecular treadmill and myosins maintain stereocilia functional architecture and self-renewal. J Cell Biol. 2004;164:887–97. doi: 10.1083/jcb.200310055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hasson T, Heintzelman MB, Santos-Sacchi J, et al. Expression in cochlea and retina of myosin VIIa, the gene product defective in Usher syndrome type 1B. Proc Natl Acad Sci U S A. 1995;92:9815–9. doi: 10.1073/pnas.92.21.9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michalski N, Michel V, Bahloul A, et al. Molecular characterization of the ankle-link complex in cochlear hair cells and its role in the hair bundle functioning. J Neurosci. 2007;27:6478–88. doi: 10.1523/JNEUROSCI.0342-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Senften M, Schwander M, Kazmierczak P, et al. Physical and functional interaction between protocadherin 15 and myosin VIIa in mechanosensory hair cells. J Neurosci. 2006;26:2060–71. doi: 10.1523/JNEUROSCI.4251-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holt JR, Gillespie SK, Provance DW, et al. A chemical-genetic strategy implicates myosin-1c in adaptation by hair cells. Cell. 2002;108:371–81. doi: 10.1016/s0092-8674(02)00629-3. [DOI] [PubMed] [Google Scholar]
- 50.Garcia JA, Yee AG, Gillespie PG, et al. Localization of myosin-Ibeta near both ends of tip links in frog saccular hair cells. J Neurosci. 1998;18:8637–47. doi: 10.1523/JNEUROSCI.18-21-08637.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Phillips KR, Tong S, Goodyear R, et al. Stereociliary myosin-1c receptors are sensitive to calcium chelation and absent from cadherin 23 mutant mice. J Neurosci. 2006;26:10777–88. doi: 10.1523/JNEUROSCI.1847-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Astuto LM, Bork JM, Weston MD, et al. CDH23 mutation and phenotype heterogeneity: a profile of 107 diverse families with Usher syndrome and nonsyndromic deafness. Am J Hum Genet. 2002;71:262–75. doi: 10.1086/341558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pennings RJ, Topsakal V, Astuto L, et al. Variable clinical features in patients with CDH23 mutations (USH1D-DFNB12) Otol Neurotol. 2004;25:699–706. doi: 10.1097/00129492-200409000-00009. [DOI] [PubMed] [Google Scholar]
- 54**.Schwander M, Xiong W, Tokita J, et al. A mouse model for nonsyndromic deafness (DFNB12) links hearing loss to defects in tip links of mechanosensory hair cells. Proc Natl Acad Sci U S A. 2009;106:5252–7. doi: 10.1073/pnas.0900691106. Experiments carried out in this work suggest that CDH23 plays a distinct role in mechanotransduction. It also identifies a mouse model for DFNB12, a new class of hearing disorder related to defects in the tip link.
- 55.Leckband D, Prakasam A. Mechanism and dynamics of cadherin adhesion. Annu Rev Biomed Eng. 2006;8:259–87. doi: 10.1146/annurev.bioeng.8.061505.095753. [DOI] [PubMed] [Google Scholar]
- 56.Noben-Trauth K, Zheng QY, Johnson KR. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat Genet. 2003;35:21–3. doi: 10.1038/ng1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57**.Sliwinska-Kowalska M, Noben-Trauth K, Pawelczyk M, et al. Single nucleotide polymorphisms in the cadherin 23 (CDH23) gene in Polish workers exposed to industrial noise. Am J Hum Biol. 2008;20:481–3. doi: 10.1002/ajhb.20744. Identification of an isoform of CDH23 carried by some humans who are particularly susceptible to noise-induced hearing loss and provides a framework for studies on the genetic basis of susceptibility to noise-induced hearing loss.
- 58.Ramakrishnan NA, Drescher MJ, Barretto RL, et al. Calcium-dependent Binding of HCN1 Channel Protein to Hair Cell Stereociliary Tip Link Protein Protocadherin 15 CD3. J Biol Chem. 2009;284:3227–38. doi: 10.1074/jbc.M806177200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao X, Bucchi A, Oren RV, et al. In vitro characterization of HCN channel kinetics and frequency dependence in myocytes predicts biological pacemaker functionality. J Physiol. 2009;587:1513–25. doi: 10.1113/jphysiol.2008.163444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Corey DP, Hudspeth AJ. Response latency of vertebrate hair cells. Biophys J. 1979;26:499–506. doi: 10.1016/S0006-3495(79)85267-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Salles FT, Merritt RC, Jr, Manor U, et al. Myosin IIIa boosts elongation of stereocilia by transporting espin 1 to the plus ends of actin filaments. Nat Cell Biol. 2009;11:443–50. doi: 10.1038/ncb1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sotomayor M, Corey DP, Schulten K. In search of the hair-cell gating spring elastic properties of ankyrin and cadherin repeats. Structure. 2005;13:669–82. doi: 10.1016/j.str.2005.03.001. [DOI] [PubMed] [Google Scholar]