

Abstract
The reactivities of a series of 1-substituted cyclobutene derivatives (carboxylate esters, carboxamides and carbinol esters) were investigated as substrates for ring-opening metathesis polymerization (ROMP) with [(H2IMes)(3-Br-pyridine)2(Cl)2Ru=CHPh]. Both the secondary amides of 1-cyclobutenecarboxylic acid and the esters of 1-cyclobutene-1-methanol undergo polymerization. The secondary amides provide translationally invariant polymers (E-olefins). Although the carbinol esters yield stereo- and regiochemically heterogeneous polymers, the 1-cyclobutenecarboxylic acid esters and tertiary amides undergo ring opening metathesis (ROM) but not ROMP. The regio- and stereochemical outcomes of these ROMP and ROM reactions were analyzed at the B3LYP/6-31G* and LANL2DZ levels of theory. Calculations suggest that the regiochemistry and stereochemistry of the addition to the propagating carbene to form the metallocyclobutane intermediate depend on both charge distribution and steric interactions.
Introduction
A variety of strained, cyclic monomers undergo ruthenium-catalyzed ring-opening metathesis polymerization (ROMP) to provide a range of polymeric materials.1–7 Development of N-heterocyclic carbene-substituted ruthenium catalysts with pyridyl ligands (3rd generation Grubbs catalysts),8 e.g., 1, provided entry into living ROMP reactions (Scheme 1). The fast initiation rate (ki ≫ kp), high thermal stability, and excellent functional group tolerance of catalyst 1 have enabled the pursuit of a multitude of applications.6,9–13 Norbornene, oxanorbornene, and oxanorbornene-dicarboximide derivatives are the most popular ROMP monomers because of their high ring strain, easy preparation, and the facility with which functional groups are attached.14 Living ROMP of these monomers generates linear polymers with accurate molecular weight control and low polydispersities (PDIs). However, internally heterogeneous mixtures are obtained because the polymerization reactions lack complete stereochemical and regiochemical control.
Scheme 1.

General mechanism of ring opening metathesis polymerization with catalyst 1 for the display of bioactive functional groups.
With the intention of obtaining polymers that are translationally invariant by virtue of a regioselective addition step and the absence of asymmetric centers on the backbone, we designed cyclobutenecarboxylic acid amide ROMP monomers, e.g. 1-(methyl ester)glycine cyclobutenecarboxylate 2a.10 We found that ROMP of substrate 2a with catalyst 1 provides regioregular polymers with strictly E-olefin geometry and excellent PDIs (Scheme 2).10
Scheme 2.
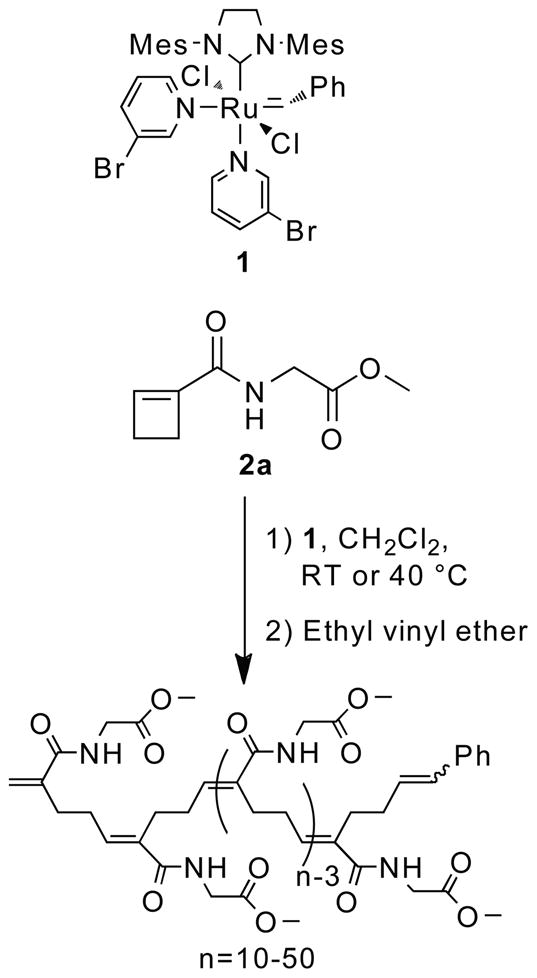
Ring opening metathesis polymerization (ROMP) of 2a.10
We have now investigated the reactivities of the readily available 1-substituted cyclobutenes 2b – 5 (Figure 1) under ROMP conditions. All secondary amides examined (2) provide polymers with translationally invariant backbones and excellent PDIs, whereas ROMP of the 1-cyclobutene-1-methanol esters (5) is neither regio- nor stereoselective. In contrast, both 1- cyclobutenecarboxylic acid tertiary amides (3) and 1-cyclobutenecarboxylic acid esters (4) undergo only a single ring-opening metathesis cycle (ROM) without polymerization. Here, we present the kinetic data for the metathesis transformations, the computed energies of key reaction intermediates and products, and the correlation between these energies and the observed reactivities.
Figure 1.
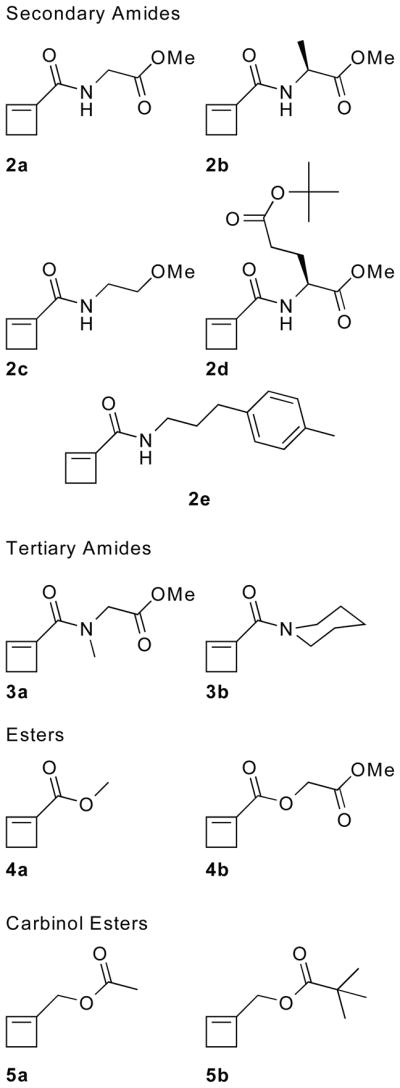
Cyclobutene monomers subjected to ROMP.
Results and Discussion
Preparation of Monomers
The known cyclobutene-1-carboxylic acid 6 was derivatized to provide all of the monomers (Scheme 3).10,15 It was converted to amides 2 by EDC coupling. Tertiary amide 3a was prepared in useable yield by a modified EDC procedure in which DMAP was added to the reaction mixture. Piperidine amide 3b was obtained by a procedure based on N,N′-bis(2-oxo-3-oxazolidinyl)phosphinic chloride (BOP-Cl). The known methyl ester 4a was prepared by treatment of acid 6 with N,N′-diisopropyl-O-methylisourea.16,17 Ester 4b was obtained by treating the acid with methyl bromoacetate in the presence of Hunig’s base. Reduction of ester 4a with DIBAL-H gave cyclobutene-1-carbinol, which was esterified with acetyl and pivaloyl chloride to provide 5a and 5b, respectively.
Scheme 3.

Synthesis of 1-substituted cyclobutenes. (i) 3a: EDC·HCl, DMAP, DIEA, CH2Cl2, rt; 3b: BOP-Cl, piperidine, DIEA, CH2Cl2, rt. (ii) EDC·HCl, DIEA, CH2Cl2, rt. (iii) 4a: N,N′-diisopropyl-O-methylisourea, Et2O, rt; 4b: BrCH2CO2CH3, KI, DIEA, rt. (iv) DIBAL-H, Et2O, −78 °C. (v) Pivaloyl chloride or acetyl chloride, DIEA, DMAP, CH2Cl2, 0 °C to rt.
Kinetics, Regiochemistry, and Stereochemistry of 1-Substituted Cyclobutene ROMP
When a 1-substituted cyclobutene coordinates to the ruthenium center of the catalyst, there are four Ru-cyclobutane intermediates that may be formed (cis- and trans-IM-1 and cis- and trans-IM-2, Scheme 4). In Pathway I, the Ru-cyclobutane ring is formed with a 1,3-relationship between the substituent-bearing carbons, whereas in Pathway II, there is a 1,2-relationship between them. Selectivity in formation of the metallocyclobutanes determines the regiochemistry and stereochemistry of chain extension (IM -> RP).
Scheme 4.

Two possible reaction pathways and their corresponding regio- and stereoisomeric intermediates and products in the ruthenium-catalyzed ring-opening reactions of 1-cyclobutene derivatives. For the ROM reactions and the ROMP initiating reaction, R2 = H, and R3 = Ph. For the ROMP chain extending reactions, R1 = R2 and R3 = the polymer chain.
ROMP of Secondary Amide Monomers
The ROMP kinetics of monomers 2a-2e were measured by monitoring the ROMP reactions in CD2Cl2 at 25 °C. We have previously shown that secondary amide cyclobutenes provide at least 50-mer polymers with excellent PDIs, if required.10 Here we studied the ROMP reaction in the context of the preparation of shorter polymers so that NMR analysis would be more informative.
The 1H-NMR spectra of the ROMP reaction of monomer 2b illustrate the method used to follow the kinetics of the reaction and the assignment of stereochemistry in the products (Figure 2). As a function of time, the integration of the peaks a, b and c (corresponding to protons in monomer 2b) decreased as the integration of peaks d and e (attributed to protons on the chain of the ring-opened ruthenium carbene) increased. During the course of the reaction of each monomer 2, a single broad peak appeared at 6.2 ppm in the 1H-NMR spectrum, indicating the formation of an internal trisubstituted olefin with E-configuration.10 There was no evidence of disubstituted olefin (signals in the 5 to 6 ppm region). In addition, the absence of a peak at 18.0–19.1 ppm indicated that the ruthenium alkylidene ([Ru]=CHPh or [Ru]=CH-CH2R) was not formed, and implied that an amide-substituted ruthenium carbene (RP-1 in Pathway I, Scheme 4) is the exclusive chain-lengthening species. The 10-mer polymers derived from secondary amides 2 are formed with high regioselectivity and stereoselectivity.
Figure 2.

Time course of ROMP of monomer 2b followed by 1H-NMR spectroscopy. Reaction conditions: CD2Cl2, [2b] = 0.1 M, [1] = 0.01 M, 25 °C. The peak at 5.33 ppm is CHDCl2.
The rates of consumption of monomers 2a–2e are very similar (t50 entries, Table 1, Figure S1). The polymerization rates of substrates 2 are approximately 4 times slower than those of 1,2-unsubstituted, 3-substituted cyclobutenes,18 in which the olefinic bond is disubstituted and the substituents are one atom removed from the carbons that undergo metathesis.
Table 1.
ROM and ROMP results.a
| Monomer | [monomer]/1 | Conversion Yield %b | Rxn Time/h | t50/minc | Mn (theo) | Mnd | Mwe | PDI |
|---|---|---|---|---|---|---|---|---|
| 2a | 10 | 93 | 3 | 3 | 1796 | 1835 | 2220 | 1.21 |
| 2b | 10 | 97 | 6 | 3 | 1936 | 1820 | 2522 | 1.39 |
| 2c | 10 | 85 | 3 | 4 | 1656 | 1349 | 1653 | 1.23 |
| 2d | 10 | 94 | 4 | 3 | 3077 | 3483 | 4796 | 1.38 |
| 2e | 10 | 96 | 6 | 3 | 2397 | 2222 | 3047 | 1.37 |
| 3a | 10 | 10 | 2 | ∞f | n/a | n/a | n/a | n/a |
| 3b | 10 | 10 | 2 | ∞f | n/a | n/a | n/a | n/a |
| 4a | 10 | 10 | 3 | ∞f | n/a | n/a | n/a | n/a |
| 4ag | 1 | 78 | 21 | 87 | n/a | n/a | n/a | n/a |
| 4b | 10 | 10 | 3 | ∞f | n/a | n/a | n/a | n/a |
| 4bg | 1 | 72 | 31 | 333 | n/a | n/a | n/a | n/a |
| 5a | 10 | 98 | 1.5 | <2 | 1366 | 2057 | 2740 | 1.33 |
| 5b | 10 | 99 | 1.5 | <2 | 1786 | 2067 | 2726 | 1.33 |
Reaction conditions: CD2Cl2, 25 °C, [monomer] = 0.1 M, [1] = 0.01 M.
Determined by 1H-NMR spectroscopy.
Reaction time for 50% consumption of monomer.
Number-averaged molecular weight by GPC using polystyrene standards.
Weight-averaged molecular weight by GPC using polystyrene standards.
The reaction stops after 10% consumption of monomer.
[1] = 0.1 M.
ROM of Tertiary Amide Monomers
Reaction of tertiary amides 3a and 3b with 10 mol% catalyst 1 resulted in the ring-opening metathesis (ROM) of approximately 10 mol% of the monomer. The ring-opening reaction requires 2 hours to reach this 10 % conversion; no polymerization is observed (Table 1, Figure S2).
ROM of 3a and 3b exhibits the same regioselectivity as that of the secondary amide monomers 2, and only resonances consistent with Pathway I were observed in the 1H-NMR spectra (Figure S3). Although secondary amides with γ-branching (i.e., 2b and 2d) undergo ROMP, tertiary amides, in which branching is at the β-position, yield only ring-opened monomer. We hypothesize that the increased steric bulk around the ruthenium in RP-1 hinders the binding of subsequent monomers to the tertiary amide-substituted carbene (see below).
ROM of Cyclobutenecarboxylic Acid Esters
We have reported that monomer 4a undergoes ROM but not ROMP with catalyst 1.19 Here, we describe the ROM of a second ester and the regiochemistry of the ROM reaction. As seen with monomer 4a, incubation of ester 4b with 10 mol% of catalyst 1 resulted in the ring opening metathesis (ROM) of approximately 10 mol% of the monomer with no polymerization (Table 1, Figure S2); that is, the remaining 90% of the monomer does not react.
An NMR time course of the reaction of 10 equivalents of monomer 4a with one equivalent of catalyst 1 is shown in Figure 3. The peak at 19.1 ppm corresponding to the carbene proton on the catalyst 1 disappears over time and no new carbene proton peak appears. At the same time, the styrenyl peaks from the ring-opened monomer appear (peak b, b′, c and c′). Therefore, RP-1 is formed preferentially to RP-2, as is observed for both secondary and tertiary amides. The 1-cyclobutene ester undergoes a single ring-opening reaction with the alkylidene catalyst to form an enoic carbene that does not react with remaining 1-cyclobutene ester. We conclude that the enoic ruthenium carbene, RP-1, cannot undergo further metathesis with an unsaturated ester.
Figure 3.

Time course of ROM of monomer 4a followed by 1H-NMR spectroscopy. Reaction conditions: CD2Cl2, [4a] = 0.1 M, [1] = 0.01 M, 25 °C. Resonances corresponding to H (d) and H (d′) are predicted to occur at 18.5 ppm. These resonances are not observed.
In order to determine the kinetics of the ring opening and to analyze the structures of the products in greater detail, we examined the reaction of esters 4a and 4b with one equivalent of catalyst (Figure S4). Again, the NMR spectra were monitored as a function of time. The ring-opening reactions of monomers 4a and 4b required more than 20 h to reach 75% consumption of monomer. The low reaction rates suggest that an ester further reduces the reactivity of the cyclobutene olefin relative to the amide-substituted cyclobutenes. In contrast to the secondary amides, these esters have no β- or γ-branching and are analogous in structure to primary amides 2. These comparisons suggest that the failure of esters 4a and 4b to polymerize is the result of an electronic factor rather than steric congestion at the catalytic center.
There are four possible ROM products that may be formed from monomer 4a or 4b (Figure 3). As noted above, ROM of 1-substituted esters was regioregular. The primary products [Ru]-9a and [Ru]-10a are formed as a 1/2 mixture of Z/E stryrenyl olefins (Figure 4a) through Pathway I (Scheme 4). The quenched reaction products were purified and characterized by 1H-NMR spectroscopy (Figure S6) and LC-MS (APCI) spectroscopy to establish their structures. A minor peak at 18.5 ppm in the 1H-NMR spectrum (Figure S5) was assigned to the carbene proton of species [Ru]-11a and [Ru]-12a (via Pathway II). It is reasonable to assume that [Ru]-11a and [Ru]-12a initiate ROM with monomer 4a; the resulting carbenes would be enoic and stable to further reaction prior to quenching. The time course (Figure 4a) revealed that the ruthenium carbenes [Ru]-11a and [Ru]-12a are formed in the initial reaction and are not secondary products from cross metatheses.
Figure 4.

Rate of formation of ring-opened monomer products formed from monomers 4a and 4b. Reaction conditions: [4a] or [4b] = 0.1 M, [1] = 0.1 M, 25 °C.
High ROM regiospecificity was also observed with monomer 4b. The ROM products of 4b formed through the enoic carbene (Pathway I, Scheme 4) are likewise a mixture of Z and E styrene olefins ([Ru]-9b and [Ru]-10b) with a Z/E ratio around 2/3 (Figure 4b). At 75% consumption of monomer, the combined yield of [Ru]-11b and [Ru]-12b generated through formation of the alkylidene carbene (Pathway II, Scheme 4) is less than 4% (data not shown). Thus, Pathway I (Scheme 4) to form the enoic carbene is the energetically favorable pathway.
ROMP and ROM of Carbinol Esters 5
Metathesis reactions with 1 equivalent of catalyst 1 and 10 equivalents of monomer 5a or 5b were monitored by 1H-NMR spectroscopy at room temperature for 1.5 hours (Table 1, Figure S7). Greater than 98% of each monomer was consumed in 1 hour. The t50 values are at least 5 times smaller than the t50 values for monomers 2–4; the carbinol esters 5 undergo ROMP much faster than secondary amides 2.
In the 1H-NMR spectrum of the [Ru]-13 polymer, the methylene protons adjacent to the ester oxygen are observed as three broad signals centered at 4.64, 4.57 and 4.46 ppm of approximately equal intensity (Figure 5, peak d and Figure S8, peaks f, g and h, Table S1). The assignment of these resonances to the allylic oxygen-bearing carbon was corroborated by 13C-NMR, 13C-APT, gCOSY and gHMQC spectroscopy of the purified 10-mer products 13 and 14 (Table S1, Figure S8). The presence of three signals indicates that at least three, and perhaps all four (if peaks are overlapping) of the possible substitution patterns on (-CH2OR)-bearing double bonds are present (Figure 6). Furthermore, the 1H-NMR spectrum of the [Ru]-13 polymer exhibits multiple broad resonances for the olefinic protons in the polymer backbone at 5.35–5.62 ppm (Figure 5, peak c), indicating that the polymer backbone is composed of different types of olefins (Figure 6). Therefore, the polymerization is neither regio- nor stereoselective.
Figure 5.
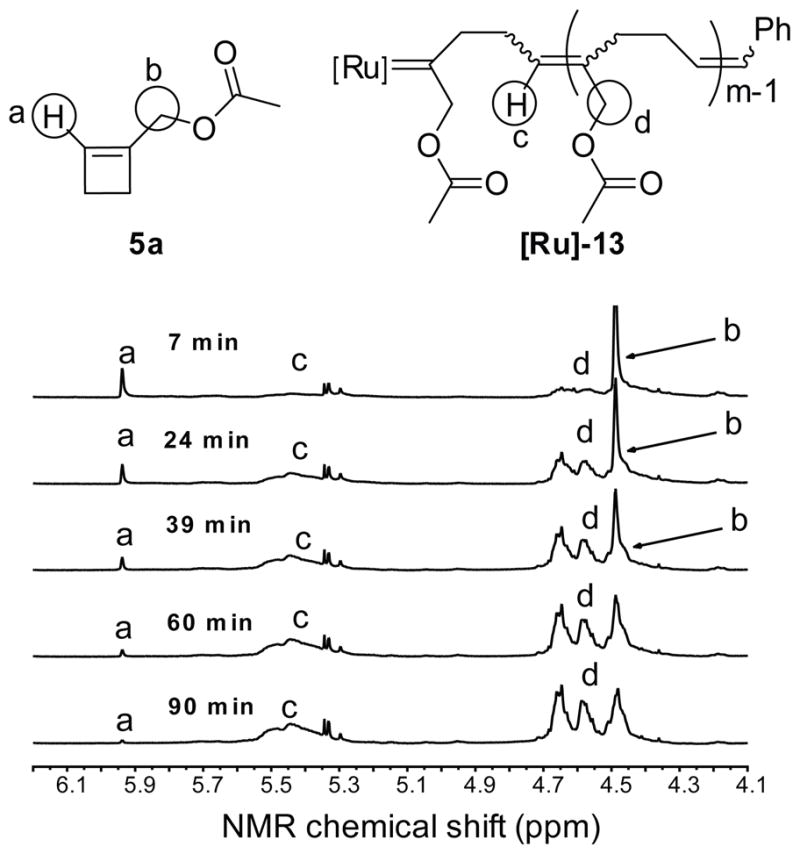
Time course of ROMP of monomer 5a followed by 1H-NMR spectroscopy. Reaction conditions: CD2Cl2, [5a] = 0.1 M, [1] = 0.01 M, 25 °C.
Figure 6.
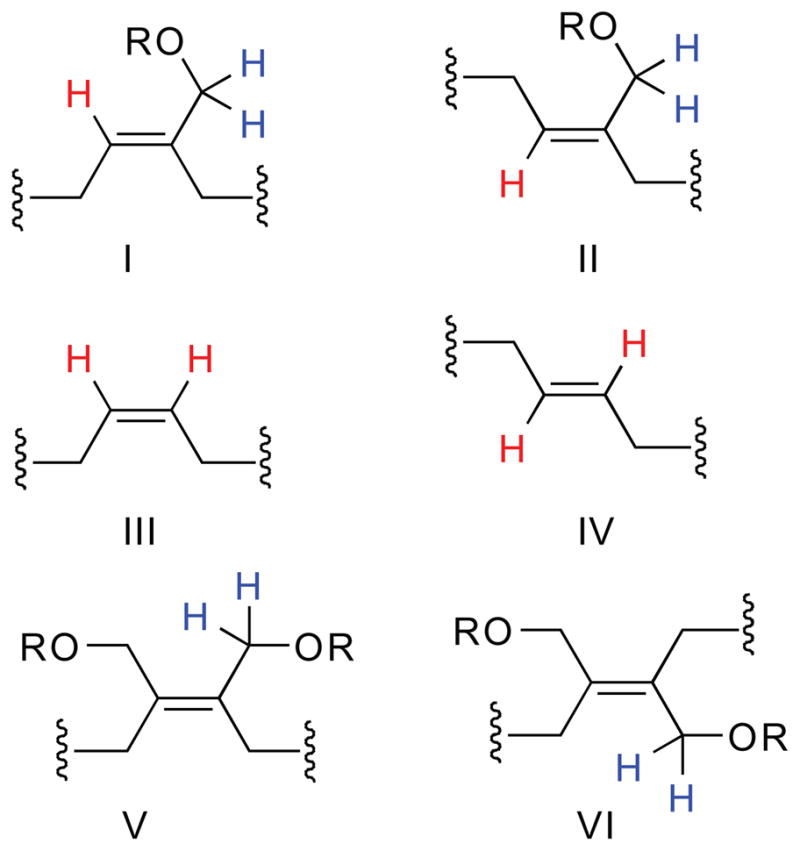
Six possible alkene configurations in the polymer backbone upon ROMP of monomers 5.
Computational Analysis of Regio- and Stereoselectivity
In order to understand the effects of structure on reactivity in each of the ring opening reactions, we analyzed the energies of the stable and metastable species along the two polymerization pathways by ab initio methods. The isomer formed depends on which step along the reaction coordinate is rate-determining. The rate of formation of the 14-electron ruthenium complex upon dissociation of bromopyridine from 1 is the same regardless of cyclobutene structure and will not contribute to the regio or stereoselectivities. Likewise, during the catalytic cycle, competition between the cyclobutene and the bromopyridine does not alter the regio- or stereoselectivity.
Therefore, two kinetic steps after dissociation were considered: (a) formation of the metallocyclobutane intermediate, and (b) formation of ring-opened product. In order to understand the regioselectivity of π-complex (π-SM) formation, we calculated the natural bond orbital charge populations for the cyclobutene monomers and their respective propagating carbenes. For the polymerization of cyclobutene amide 2a, we considered reaction with the amide substituted carbene, which is the species formed in the reaction; for the polymerization of the carbinol ester 5b, we considered only the reaction with the substituted carbene that is the analogous isomer (Figure 7a). For the ROM monomers, 3 and 4, we analyzed the initial reaction with the benzylidene carbene 1 (Figure 7a). In order to predict the relative activation energies for metallocyclobutane formation and subsequent ring opening, we calculated the energies of the reaction intermediates (IM) and products (RP).
Figure 7.

(a) NBO charge populations of 1-substituted cyclobutene derivatives 2a, 3a, 4a and 5b, the corresponding ring-opened ruthenium carbenes of 2a and 3a, and ruthenium benzylidene. Hartree-Fock calculations were performed with the 6-311G++* basis set (for cyclobutene monomers) and the LANL2DZ basis set (for ruthenium carbenes) in Gaussian 03W. (b) AIM electron densities (atomic units) for the olefin bonds in 1-substituted cyclobutene derivatives 2a and 4a were calculated with AIMPAC.
Calculation Methods
For optimization of the geometries of the cyclobutene monomers (2a, 3a, 4a and 5b), we used B3LYP/6-31G*. This method is frequently employed for simple organic compounds20–23 because it provides accurate structural predictions with economical calculations. In order to limit the number of atoms included in the propagating ruthenium carbenes and thereby facilitate the calculations, we modeled the substituents as (CH2CH2CH=CHPh); i.e., we used structures [Ru]-15 and [Ru]-16. For these carbenes, and for the ruthenium benzylidene [Ru]-17 (Figure 7a), we used B3LYP/LANL2DZ. This method has been used extensively for the geometry optimization of ruthenium carbenes. The LANL2DZ basis set has superior properties with respect to effective core potentials (ECPs) and polarization functions on the ruthenium24–37 and calculated results agree very well with experimental data, e.g., X-ray crystal structures,32,33 IR and NMR spectra,36 thermal parameters,25,26,37 or metathesis reactivities.24,25,28–31,34 For natural bond orbital (NBO) charge populations (Figure 7a), we used Hartree-Fock with the 6-31G++* basis set (for cyclobutene monomers) and the LANL2DZ basis set (for ruthenium carbenes) in Gaussian 03W.
The geometry optimizations and energy calculations of the intermediates (IM) and products (RP) were also performed with B3LYP/LANL2DZ in Gaussian 03W. To simplify the computation, the polymer chain and the 1-carbonyl amide group were modeled as a methyl group and a CONHCH3 substituent, respectively (Figure S9). To prevent ring opening and to find the local minimum of the intermediate structures, only one bond in the metallocyclobutane ring was optimized while the other three bond lengths were kept constant; the partial optimization was rotated to optimize the other three bonds, until the optimized structure changed little in energy (ΔE < 6.3×10−4 kcal/mol). Vibrational frequency calculations using B3LYP/LANL2DZ were performed for all model compounds (Figure S9); there were no imaginary vibrational frequencies indicating that local minima had been found for each structure. Free energies computed in Gaussian 03W for structures (Figure 8) in solvent (CH2Cl2) include the electronic energy plus the solvation free energy from the conductor-like polarizable continuum model (CPCM) based on the United Atom Kohn-Sham (UAKS) radii.
Figure 8.

(a) Cyclobutene ROMP reaction scheme and relative free energy profiles for the reactions of (b) monomers 2, R2 = CONHMe, and (c) monomers 5, R2 = CH2OAc. Structures were optimized with B3LYP/LANL2DZ in Gaussian 03W program. Free energies computed for structures in solvent (CH2Cl2) at 298.15 K include the electronic energy plus the solvation free energy from the CPCM solvation model based on the UAKS radii using Gaussian 03W. SM: starting material; π-SM: π-complex of starting material; TS: transition state; IM: intermediate; π-RP: π-complex of reaction product; RP: reaction product. In the potential minima and maxima, dashed lines represent energy estimates and solid lines represent calculated energies.
NBO Charge Calculations
For the ester and amide cyclobutene monomers, the calculations showed that the electron density is higher on C-1 than on C-2 as expected for olefins bearing conjugated electron withdrawing groups (monomers 2a, 3a, 4a). In contrast to the carbonyl substituents, the C-1 methanol ester substituent (monomer 5b) is electron donating, making C-1 more electropositive than C-2. In all of the ruthenium carbenes, the metal atom is more electropositive than the adjacent carbon atom.
Relative Energies of Intermediates and Products
The NBO calculations allowed us to rationalize the regiochemical results of the ROM and ROMP processes. In the case of monomers 2 – 4, alignment of the cyclobutene and the electron-deficient Ru to pair C-1 and the Ru to form the π-complex (π-SM) is favored by the electronic factors; however, in the case of monomers 5, electronic factors favor a cycloaddition in which C-2 becomes bonded to the ruthenium (Figure 7). Assuming then that steric effects between the coordinating cyclobutene and the carbene are relatively unimportant (because of the long distance between the cyclobutene and Ru in the π-complex), we ordered the energies of the regioisomeric cyclobutene π-complexes (π-SM) according to the charge distribution of the cyclobutenes.
Noting energy values from the literature for the ring strain of cyclobutenes and for π-stabilization of ruthenium carbene complexes, we developed a comprehensive picture (Figure 8) of the ROMP processes of 2 and 5. On the basis of the calculations of Goddard and coworkers,38 we estimated the strain energy of 1-substituted cyclobutene to be ~26 kcal/mol. We used the strain energy to plot the difference in energy between the starting 14 electron ruthenium carbene (SM) energy and the mean product carbene (RP) energy. The calculated energy differences between the metallocyclobutane intermediates (IM) and the product carbenes (RP) were used to order the isomers on the free energy profile. For the ROMP of 5, we plotted the energies for the reaction that begins with the more substituted of the two regioisomeric ruthenium carbenes. Further, we assumed that the most favorable π-stabilized 16-electron ruthenium carbene (π-SM) is about 14 kcal/mol more stable than the uncomplexed carbene (SM) and that the most favorable π-RP is 14 kcal/mol more stable than the corresponding RP. An 18 kcal/mol stabilization energy was calculated by Zhao and Truhlar for ethylene39 and measured by Chen and coworkers for norbornene;40 however, there are additional steric interactions present in cyclobutene π-SM complexes (Figure 8). The TSa energies were ordered according to the relative stability of their corresponding metallocyclobutane intermediates (IM) to account for steric effects that develop in the transition state. All of the species were placed along the reaction coordinates of monomers 2 and 5 in their respective free energy profiles (Figure 8).
The cis-Pathway I Is Favored for Secondary Amide Monomers
In the case of 1-substituted cyclobutene amides, starting (π-SM), intermediate (IM) and reaction product (RP) complexes on the cis-pathway I are always the lowest energy isomers. By extension, the transition states are expected to be the lowest energy transition states and the corresponding activation energies for their formation the most favorable. This picture is consistent with the high selectivity for ring-opening metathesis of amides 2 (>99%) of the more substituted ruthenium carbene and (E)-olefin),10 and by extension for ROM of monomers 3 and 4.
Activation Energies in Pathways I and II Are Nearly Equivalent for Carbinol Ester Monomers 5
In the case of the carbinol esters 5, if we consider the partitioning of metallocyclobutane intermediate forward to ring-opened product versus backward to the coordinated cyclobutene, the forward reaction is favored due to the release of ring strain upon formation of π-RP, and π-SM is not in equilibrium with IM. Likewise by the Hammond postulate, we surmise that TSb is not rate limiting. Thus, the energy of TSa determines the regio and stereoselectivities. The NBO charge and energy calculations suggest that trans-π-SM-2 and cis-π-SM-2 are lower in energy than the π-SM-1 regioisomers. However, upon formation of the metallocyclobutane intermediate, steric interactions destabilize IM-2 relative to IM-1 counteracting electronic effects. As these steric interactions develop in the transition state, the difference in TSa energies between isomers is much smaller than in either the π-SM or IM species. The consequence is that trans and cis-π-SM-1 react at a comparable rate to trans and cis-π-SM-2. Thus, poor regio- and stereoselectivity are observed. In addition, the overall estimated activation energies for carbinol esters 5 are smaller than for those of the secondary amides 2; this result is consistent with the higher rates of propagation observed for monomers 5 (Table 1).
Geometry of ROM Intermediate from Tertiary Amide Monomers 3
Kinetic studies with monomers 3a and 3b revealed that only ROM reactions occurred. We hypothesized that the second N-substituent may block the approach and binding of an incoming monomer at the ruthenium carbene center. To test our hypothesis, geometry optimization of the N,N-disubstituted carbonyl ruthenium carbene formed from monomer 3a was performed with B3LYP/LANL2DZ in Gaussian 03W. In the optimized structure of the ruthenium carbene, the N-methyl group blocks the face of the metal carbene to which the incoming cyclobutene must bind (Figure 9a). Therefore, we conclude that severe steric crowding is responsible for a lack of propagation after the initial ring opening occurs.
Figure 9.

(a) Schematic model based on the B3LYP/LANL2DZ optimized structure (Gaussian 03W) of the new N,N-disubstituted carbonyl ruthenium carbene for monomer 3a illustrating how the second N-substituent blocks access to the ruthenium carbene of a second monomer. (b) Model of the ester coordination observed in the B3LYP/LANL2DZ optimized structure (Gaussian 03W) of the enoic carbene formed from monomer 4a.
Inability of Ester Monomers 4 to form Homopolymer
Our initial hypothesis for the lack of polymerization of enoic carbene was that the electron withdrawing nature of the ester deactivated the cyclobutene olefin to further metathesis reaction. However, the NBO calculations followed by AIM electron density analysis reveal that the charge density on the ester-substituted cyclobutene is not significantly different than the charge density on the secondary amide-substituted cyclobutene, which does undergo ROMP (Figure 7). Ab initio calculations for the enoic carbene reaction product of 4a revealed that the ester oxygen forms a chelate at the open coordination site of the 14-electron ruthenium center (Figure 9b). This chelation stabilizes the ester-substituted carbene by approximately 7 kcal/mol relative to the amide-substituted carbene. Similar ester coordination was observed in calculations by which Fomine and coworkers investigated the reactivity of the enoic carbene35 and in the reaction of enoates in cross metathesis.41 Although ring-opening metathesis with the relatively electron-rich olefin of cyclohexene19,42,43 is still feasible, the chelated enoic carbene is kinetically trapped from further reaction with the sterically hindered 1-substituted cyclobutene ester, 4.
Conclusion
In summary, we have found that cyclobutenes undergo stereo- and regioregular ring opening metathesis when substituted with an electron withdrawing carbonyl at the 1-position. Regioregular addition to the catalyst carbene is consistent with both the calculated charge distributions for the carbene and the cyclobutene and the minimization of steric interactions. In the case of more electron-rich 1-substituents, the inverse rank order of energies between the intermediates and the π-complexed starting materials strongly suggests that the corresponding activation energies to reach the transition states may be close enough in magnitude to make ROMP of carbinol ester monomers 5 neither regio- nor stereoselective.
Neither cyclohexene carboxylic acid esters nor tertiary amides ROMP. When an enoic carbene is formed from ester monomers 4, the formation of a ruthenium chelate with the ester oxygen traps the enoic carbene in a stabilized state that precludes the reaction of these esters with 1-substituted cyclobutene esters, e.g., in homopolymerizations. This reactivity opens an avenue for preparing alternating polymers with unique functionality.19 Steric crowding prevents propagation for tertiary amide substituents. In conclusion, of the functionalized cyclobutenes studied, the secondary amides 2 exhibit the optimal level of reactivity and stereo- and regio- control for the generation of translationally invariant polymers.
Supplementary Material
Acknowledgments
This research was supported by NIH grants R01HD38519 (N.S.), S10RR021008 (N.S.), and GM074776 (K.A.P.), NYSTAR award (FDP C040076, NS) and NSF grant CHE0131146 (NMR). We thank Dr. James Marecek for his assistance with NMR spectroscopy and Dr. Derek Middlemiss for his consultation on calculations.
Footnotes
Supporting Information Available: Additional figures, experimental methods and results including spectroscopic data and coordinates of calculated structures. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Komiya Z, Pugh C, Schrock RR. Macromolecules. 1992;25:6586–6592. [Google Scholar]
- 2.Lynn DM, Mohr B, Grubbs RH. J Am Chem Soc. 1998;120:1627–1628. [Google Scholar]
- 3.Royappa AT, Saunders RS, Rubner MF, Cohen RE. Langmuir. 1998;14:6207–6214. [Google Scholar]
- 4.Gordon EJ, Gestwicki JE, Strong LE, Kiessling LL. Chem Biol. 2000;7:9–16. doi: 10.1016/s1074-5521(00)00060-0. [DOI] [PubMed] [Google Scholar]
- 5.Cairo CW, Gestwicki JE, Kanai M, Kiessling LL. J Am Chem Soc. 2002;124:1615–1619. doi: 10.1021/ja016727k. [DOI] [PubMed] [Google Scholar]
- 6.Baessler KA, Lee Y, Roberts KS, Facompre N, Sampson NS. Chem Biol. 2006;13:251–259. doi: 10.1016/j.chembiol.2005.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts KS, Konkar S, Sampson NS. ChemBioChem. 2003;4:1229–1231. doi: 10.1002/cbic.200300672. [DOI] [PubMed] [Google Scholar]
- 8.Love JA, Morgan JP, Trnka TM, Grubbs RH. Angew Chem Int Ed. 2002;41:4035–4037. doi: 10.1002/1521-3773(20021104)41:21<4035::AID-ANIE4035>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 9.Bielawski CW, Grubbs RH. Prog Polym Sci. 2007;32:1–29. [Google Scholar]
- 10.Lee JC, Parker KA, Sampson NS. J Am Chem Soc. 2006;128:4578–4579. doi: 10.1021/ja058801v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walker R, Conrad RM, Grubbs RH. Macromolecules. 2009;42:599–605. doi: 10.1021/ma801693q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baessler K, Lee Y, Sampson NS. ACS Chem Biol. 2009;4:357–366. doi: 10.1021/cb900013d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee Y, Sampson NS. ChemBioChem. 2009;10:929–937. doi: 10.1002/cbic.200800791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith D, Pentzer EB, Nguyen ST. Polym Rev. 2007;47:419–459. [Google Scholar]
- 15.Campbell A, Rydon HN. J Chem Soc. 1953:3002–3008. [Google Scholar]
- 16.Griffin RJ, Arris CE, Bleasdale C, Boyle FT, Calvert AH, Curtin NJ, Dalby C, Kanugula S, Lembicz NK, Newell DR, Pegg AE, Golding BT. J Med Chem. 2000;43:4071–4083. doi: 10.1021/jm000961o. [DOI] [PubMed] [Google Scholar]
- 17.Mathias LJ. Synthesis-Stuttgart. 1979:561–576. [Google Scholar]
- 18.Lapinte V, de Fremont P, Montembault VR, Fontaine L. Macromol Chem Phys. 2004;205:1238–1245. [Google Scholar]
- 19.Song A, Parker KA, Sampson NS. J Am Chem Soc. 2009;131:3444–5. doi: 10.1021/ja809661k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujitsuka M, Cho DW, Tojo S, Yamashiro S, Shinmyozu T, Majima T. J Phys Chem A. 2006;110:5735–5739. doi: 10.1021/jp060709d. [DOI] [PubMed] [Google Scholar]
- 21.Shambayati S, Blake JF, Wierschke SG, Jorgensen WL, Schreiber SL. J Am Chem Soc. 1990;112:697–703. [Google Scholar]
- 22.Aleman C, Casanovas J. J Phys Chem A. 2005;109:8049–8054. doi: 10.1021/jp053404s. [DOI] [PubMed] [Google Scholar]
- 23.Keating AE, GarciaGaribay MA, Houk KN. J Am Chem Soc. 1997;119:10805–10809. [Google Scholar]
- 24.Adlhart C, Hinderling C, Baumann H, Chen P. J Am Chem Soc. 2000;122:8204–8214. [Google Scholar]
- 25.Fomine S, Ortega JV, Tlenkopatchev MA. J Mol Catal A-Chem. 2007;263:121–127. [Google Scholar]
- 26.Hofmann P, Volland MAO, Hansen SM, Eisentrager F, Gross JH, Stengel K. J Organomet Chem. 2000;606:88–92. [Google Scholar]
- 27.Hoffmann M, Marciniec B. J Mol Model. 2007;13:477–83. doi: 10.1007/s00894-006-0166-3. [DOI] [PubMed] [Google Scholar]
- 28.Cornejo A, Fraile JM, Garcia JI, Gil MJ, Martinez-Merino V, Mayoral JA, Salvatella L. Angew Chem Int Ed. 2005;44:458–461. doi: 10.1002/anie.200461418. [DOI] [PubMed] [Google Scholar]
- 29.Fomine S, Tlenkopatchev MA. Appl Catal A-Gen. 2009;355:148–155. [Google Scholar]
- 30.Buchmeiser MR, Wang DR, Zhang Y, Naumov S, Wurst K. Eur J Inorg Chem. 2007:3988–4000. [Google Scholar]
- 31.Straub BF. Adv Synth Catal. 2007;349:204–214. [Google Scholar]
- 32.Chou HH, Lin YC, Huang SL, Liu YH, Wang Y. Organometallics. 2008;27:5212–5220. [Google Scholar]
- 33.Cantat T, Demange M, Mezailles N, Ricard L, Jean Y, Le Floch P. Organometallics. 2005;24:4838–4841. [Google Scholar]
- 34.Occhipinti G, Bjorsvik HR, Jensen VR. J Am Chem Soc. 2006;128:6952–6964. doi: 10.1021/ja060832i. [DOI] [PubMed] [Google Scholar]
- 35.Fomine S, Tlenkopatchev MA. Organometallics. 2007;26:4491–4497. [Google Scholar]
- 36.Cabeza JA, Del Rio I, Miguel D, Perez-Carreno E, Sanchez-Vega MG. Dalton Trans. 2008:1937–42. doi: 10.1039/b718770j. [DOI] [PubMed] [Google Scholar]
- 37.Wang D, Wurst K, Knolle W, Decker U, Prager L, Naumov S, Buchmeiser MR. Angew Chem Int Ed. 2008;47:3267–3270. doi: 10.1002/anie.200705220. [DOI] [PubMed] [Google Scholar]
- 38.Khoury PR, Goddard JD, Tam W. Tetrahedron. 2004;60:8103–8112. [Google Scholar]
- 39.Zhao Y, Truhlar DG. Org Lett. 2007;9:1967–1970. doi: 10.1021/ol0705548. [DOI] [PubMed] [Google Scholar]
- 40.Torker S, Merki D, Chen P. J Am Chem Soc. 2008;130:4808–4814. doi: 10.1021/ja078149z. [DOI] [PubMed] [Google Scholar]
- 41.Giudici RE, Hoveyda AH. J Am Chem Soc. 2007;129:3824–3825. doi: 10.1021/ja070187v. [DOI] [PubMed] [Google Scholar]
- 42.Choi TL, Lee CW, Chatterjee AK, Grubbs RH. J Am Chem Soc. 2001;123:10417–10418. doi: 10.1021/ja016386a. [DOI] [PubMed] [Google Scholar]
- 43.Ulman M, Belderrain TR, Grubbs RH. Tetrahedron Lett. 2000;41:4689–4693. [Google Scholar]
- 44.Becke AD. J Chem Phys. 1996;104:1040–1046. [Google Scholar]
- 45.Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 46.Lee CT, Yang WT, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 47.http://www.chemistry.mcmaster.ca/aimpac/.
- 48.Sanford MS, Love JA, Grubbs RH. Organometallics. 2001;20:5314–5318. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.