

Abstract
Fibrodysplasia ossificans progressiva (FOP) is an autosomal dominant human disorder of bone formation that causes developmental skeletal defects and extensive debilitating bone formation within soft connective tissues (heterotopic ossification) during childhood. All patients with classic clinical features of FOP (great toe malformations and progressive heterotopic ossification) have previously been found to carry the same heterozygous mutation (c.617G>A; p.R206H) in the GS activation domain of activin A type I receptor/activin-like kinase 2 (ACVR1/ALK2), a bone morphogenetic protein (BMP) type I receptor. Among patients with FOP-like heterotopic ossification and/or toe malformations, we identified patients with clinical features unusual for FOP. These atypical FOP patients form two classes: FOP-plus (classic defining features of FOP plus one or more atypical features) and FOP variants (major variations in one or both of the two classic defining features of FOP). All patients examined have heterozygous ACVR1 missense mutations in conserved amino acids. While the recurrent c.617G>A; p.R206H mutation was found in all cases of classic FOP and most cases of FOP-plus, novel ACVR1 mutations occur in the FOP variants and two cases of FOP-plus. Protein structure homology modeling predicts that each of the amino acid substitutions activates the ACVR1 protein to enhance receptor signaling. We observed genotype-phenotype correlation between some ACVR1 mutations and the age of onset of heterotopic ossification or on embryonic skeletal development.
Keywords: ACVR1, ALK2, BMP type I receptor, fibrodysplasia ossificans progressiva, FOP, bone morphogenetic protein, clinical variants, heterotopic ossification
Introduction
The bone morphogenetic proteins (BMPs) are a family of highly conserved extracellular signaling proteins that regulate cell differentiation fates and that have critical functions in a wide variety of cells and tissues during embryonic development and postnatal life (Chen, et al., 2004; Gazzerro and Canalis, 2006; Massague, et al., 2005; Shi and Massague, 2003; Urist, 1965; Wagner, 2007; Wozney, et al., 1988). BMPs signal by binding to and activating transmembrane complexes of type I and type II BMP receptors. Both type I and II BMP receptors are serine/threonine kinases with similar functional domains. A single transmembrane domain links the extracellular N-terminal ligand-binding domain to the cytoplasmic C-terminal kinase domain. A unique feature of type I receptors is a cytoplasmic juxtamembrane region rich in glycine and serine residues (GS domain). Following ligand binding, serines and threonines in this region are phosphorylated by the BMP type II receptor, activating the BMP type I receptor to transmit BMP signals through SMAD and MAPK signaling pathways to regulate transcription of responsive target genes. BMP signaling is mediated through three known type I receptors: BMPRIA (ALK3), BMPRIB (ALK6), and ACVR1 (ALK2).
Mutations in ACVR1 (MIM# 102576) were recently identified as the genetic cause of the rare human disease fibrodysplasia ossificans progressiva (FOP; MIM# 135100) (Shore, et al., 2006). FOP is a severely disabling disease that causes endochondral bone formation at extra-skeletal (heterotopic) sites such as skeletal muscle, tendon, ligament, fascia, and aponeuroses (Cohen, et al., 1993; Kaplan, et al., 2005; Pignolo, et al., 2005; Rocke, et al., 1994; Roush, 1996). Heterotopic ossification begins in childhood and can be induced by trauma or occur without warning. Bone formation is episodic, progressive, and extensive, leading to flare-ups that form in a well-defined spatial pattern to cause extra-articular ankylosis of the joints of the axial and appendicular skeleton, immobilizing the patient in a “second skeleton” of heterotopic bone. In addition to this postnatal heterotopic bone formation, alterations during embryonic skeletal development also occur (Mahboubi, et al., 2001; Schaffer, et al., 2005).
ACVR1 DNA sequence analysis of FOP patients who have classic disease features (progressive heterotopic ossification and great toe malformations) revealed that the same recurrent single nucleotide change in ACVR1 occurs in each FOP patient. This mutation, c.617G>A, results in the substitution of arginine by histidine at codon 206 (p.R206H) within the GS domain of the receptor. Protein structural homology modeling predicted that this amino acid substitution results in a conformational change of the receptor that alters its sensitivity and activity (Groppe, et al., 2007; Shore, et al., 2006).
The goals of this study were to conduct detailed clinical evaluations of a large cohort of FOP patients in order to establish clinical homogeneity or sub-classes associated with FOP-like heterotopic ossification, and to determine whether the identified recurrent heterozygous ACVR1 mutation associated with classic FOP is present in all patients with FOP-like heterotopic ossification. We identified, in addition to patients with classic FOP, a small number of patients with unusual forms of FOP. Although these patients form FOP-like heterotopic ossification, they have additional features that are not commonly associated with FOP (FOP-plus) or have major variations in the classic defining features of FOP (FOP variants). We determined that all patients with any form of FOP carry heterozygous ACVR1 mutations. However, in addition to the recurrent ACVR1 c.617G>A mutation found in all cases of classic FOP and most cases of FOP-plus, previously undescribed mutations in the ACVR1 gene are associated with the wider range of FOP variable expressivity that is clinically observed in FOP variants.
Materials and Methods
Subjects and Clinical Evaluation
We evaluated 112 FOP (classic and atypical) cases from five continents (104 sporadic and 8 families) who were referred to one or more of us because of progressive heterotopic ossification and/or great toe malformations. Of the 104 with sporadic FOP [32 classically affected patients from our initial study (Shore, et al., 2006) plus 72 additional patients], 84 individuals had classic FOP (classic defining features of FOP). We additionally identified 20 patients with additional atypical features and/or variation of the classic FOP phenotype: 8 had FOP-plus (classic defining features of FOP plus one or more atypical features), and 12 had FOP variants (major variations in one or both of the classic defining features of FOP). One family had variant FOP in three members. We previously reported 7 families with inheritance of classic FOP. Routine medical history, physical examination, and clinically-relevant photographic and radiographic studies were obtained on each patient. [See Table 1 for detailed clinical features and natural history of all three FOP forms.] FOP heterotopic ossification is considered “early onset” before 2 years of age and “late onset” after 10 years. The classic course of progression and severity of heterotopic ossification has been previously defined in detail (Cohen, et al., 1993; Rocke, et al., 1994). Under approval by the Investigational Review Board of the University of Pennsylvania, peripheral blood samples were obtained following informed consent from patients and unaffected individuals. Cell lines were established by Epstein Barr Virus (EBV) transformation of peripheral blood mononuclear cells. To generate haploid chromosome 2 cell lines for patient #11, cells were fused with the E2 mouse cell line and hybrid cell lines obtained (GMP Companies).
Table 1. Clinical Features of Classic FOP, FOP-plus, and Variant FOP Patients.
| Classic FOP | Atypical Features of FOP-plus or FOP Variant Patients | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient #: | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | |
| ACVR1 mutation: | |||||||||||||||||||||
| Codon change | R206H | R206H | R206H | R206H | R206H | R206H | R206H | Q207E | G328R | G328R | G328R | G328W | G328W | G328E | G328E | G356D | G356D | G356D | G356D | R375P | P197_F198 del insL |
| Nucleotide change (cDNA position) | c.617G>A | c.617G>A | c.617G>A | c.617G>A | c.617G>A | c.617G>A | c.617G>A | c.619C>G | c.982G>A | c.982G>A | c.982G>C | c.982G>T | c.982G>T | c.983G>A | c.983G>A | c.1067G>A | c.1067G>A | c.1067G>A | c.1067G>A | c.1124G>C | c.590_592delCTT |
| Gender | M, F | M | F | F | M | M | F | M | M | 3F | M | F | F | F | F | F | M | M | F | F | F |
| Age of HO onset (years) | 1-10 | 3 | 14 | 0.3 | 2 | 9 | 1.5 | 0.5 | 26 | 13; 22; none | 21 | 2 | 8 | 2 | 2 | 2 | 0.3 | ? | 15 | 14 | 11.5 |
| High resolution karyotype | - | normal | normal | normal | normal | normal | normal | - | - | - | - | normal | normal | normal | normal | - | normal | normal | - | - | - |
| Classic/Defining FOP Features: | |||||||||||||||||||||
| Characteristic malformations of great toe (hallux valgus, malformed 1st metatarsal, and/or monophalangism) | 100% | X | X | X | X | X | X | X | X | X# (right) | |||||||||||
| Progressive heterotopic ossification in characteristic anatomic patterns | 100% | X | X | X | X | X | X | X | X | X## | X | X | X | X | X | X | X | ? | X | X | X |
| Common Variable FOP Features: | |||||||||||||||||||||
| Conductive hearing impairment | >50% | X | X | X | X | X | X | X | X | X | X | ||||||||||
| Cervical spine malformations* | >80% | X | X | X | X | X | X | X | X** | X | X | X | X | ||||||||
| Proximal medial tibial osteochondromas | >90% | X | X | X | X | X | X | X | ‡ | X | X | X | X | X | X | ||||||
| Short broad femoral necks | >70% | X | X | X | X | X | X | X | X | X | X | X | X | X | X | ||||||
| Thumb malformations (short 1st metacarpal, +/- monophalangism) | ∼50% | X | X | X | X | X | X | X | X | X | X | X | X | X | |||||||
| Atypical FOP Clinical Features: | |||||||||||||||||||||
| Severe variable reduction deficits of digits | 0% | X | X | X | X | X# (left) | X | X | |||||||||||||
| Absent finger/toe nails in digits with severe reduction deficits | 0% | X | X | X | X | X# (left) | X | X | |||||||||||||
| Normal or minimal changes in great toes | 0% | X† | X†† | X††† | X | X | |||||||||||||||
| Intra-articular synovial osteochondromatosis of hips and DJD of hips | 0% | X | X | X | |||||||||||||||||
| Sparse, thin scalp hair (more prominent in 2nd decade) | 0% | X | X | X | X | X | X | ||||||||||||||
| Mild cognitive impairment | 0% | X | X | X | X | X | X | ||||||||||||||
| Severe growth retardation | 0% | X | X | ||||||||||||||||||
| Cataracts | 0% | X | |||||||||||||||||||
| Retinal detachment | 0% | X | |||||||||||||||||||
| Childhood glaucoma | 0% | X | X | ||||||||||||||||||
| Craniopharygioma | 0% | X | |||||||||||||||||||
| Persistence of primary teeth in adulthood | 0% | X | NA (3 yo) | ||||||||||||||||||
| Anatomic abnormalities of cerebellum | 0% | X | X | ||||||||||||||||||
| Diffuse cerebral dysfunction w seizures | 0% | X | |||||||||||||||||||
| Polyostotic fibrous dysplasia | 0% | X | |||||||||||||||||||
| Primary amenorrhea | 0% | X | X | ||||||||||||||||||
| Aplastic anemia | 0% | X | |||||||||||||||||||
| Hypospadias | 0% | X | |||||||||||||||||||
| Cerebral cavernous malformations | 0% | X | |||||||||||||||||||
This patient has asymmetric malformations of his great toes: right = classic, left = more severe deficit.
See Table 2.
Intra-articular ankylosis of facet joints and early degenerative changes of cervical spine.
the mother has mild orthotopic changes in c-spine; the daughters do not.
Distal femoral osteochondroma only.
Right great toe is normal; minor changes in 1st left metatarsal.
Two members of this family, the mother and one daughter, have normal toes; one daughter has minor changes of both 1st metatarsals.
Characteristic short monophalangic great toe, but additionally the patient is missing middle phalanges of 4th and 5th toes bilaterally.
No detected H.O at the time of this report (patient is 2.3 years of age; typical age of onset is 2-10 years).
Note: The DNA mutation numbering is based on cDNA sequence, with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence (GenBank NM_001105.4) according to nomenclature guidelines (www.hgvs.org/mutnomen). The initiation codon is codon 1.
Mutation Analysis
We screened genomic DNA from buccal swabs, blood, or lymphoblastoid cell lines for mutations in ACVR1 by PCR-amplification of the 9 exons containing protein-coding sequences (ACVR1 Transcript Report Ensembl v35, accession number ENST00000263640; GenBank RefSeq NM_001105.4 and NP_001096.1) using exon-flanking primers (Shore, et al., 2006). DNA sequence analysis of genomic DNA used an ABI 3730XL sequencer (University of Pennsylvania School of Medicine DNA Sequencing Facility). The complete ACVR1 coding region was sequenced for all atypical and variant FOP patients. Sequence data were analyzed using 4Peaks software v.1.6 (http://www.mekentosj.com/4peaks/). (Electropherograms were reviewed but are not shown.) For each identified mutation, we verified the absence of a mutation in at least 98 individuals (196 alleles). Mutations are identified by nucleotide numbering that reflects the cDNA sequence, with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence, according to journal guidelines (www.hgvs.org/mutnomen). The protein initiation codon is codon 1. Differences in restriction endonuclease recognition sites were identified (MacVector v.7.2 software); genomic DNA (0.1 ug) was PCR-amplified and purified PCR products were digested with the appropriate restriction enzymes.
Protein Structure Homology Modeling
TβRI crystal structure coordinates were downloaded from the Protein Data Bank (http://www.rscb.org). Structure-based homology models of mutant ACVR1 cytoplasmic domains were calculated through the automated SwissModel routines (Biozentrum, Basel; http://swissmodel.expasy.org//SWISS-MODEL.html) with the FKBP12-bound crystal structure of TβRI (PDB #1B6C) as a three-dimensional template. Molecular models were analyzed and figures prepared with PyMOL (DeLano Scientific, Palo Alto; http://pymol.sourceforge.net/).
Results
Individuals with Classic FOP
Patients with classic FOP have two defining clinical features (Figure 1A-1D): characteristic congenital malformations of the great toes and progressive heterotopic ossification in characteristic anatomic patterns (Shore, et al., 2006). In addition, common but variable features are seen in most individuals with FOP (Table 1) including proximal medial tibial osteochondromas (greater than 90% prevalence), cervical spine malformations (greater than 80% prevalence), short, broad femoral necks (greater than 70% prevalence), conductive hearing impairment (greater than 50% prevalence), and malformations of the thumb – specifically short first metacarpals and/or monophalangism of the thumbs (greater than 50% prevalence; Figure 1E-1H) (Kaplan, et al., 2005). To date, we examined 84 sporadic cases and 7 families with classic FOP, and all affected individuals were heterozygous for the canonical (c.617G>A; p.R206H) mutation in ACVR1.
Figure 1. Characteristic and Variable Features of Classic FOP.
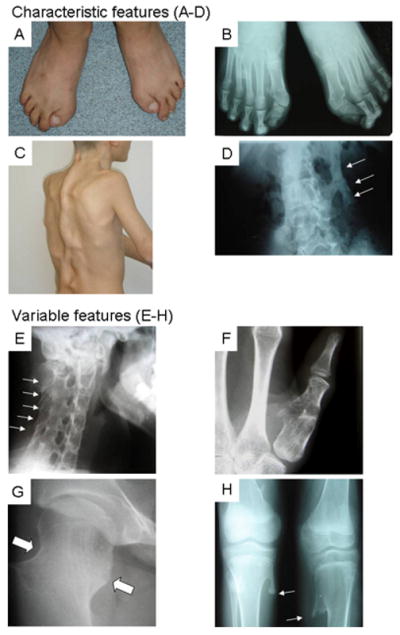
Composite of characteristic (A-D) and common variable (E-H) features of classic FOP. A photograph (A) and radiograph (B) of the feet in a classically-affected 15 year-old boy shows short, malformed monophalangic great toes. A photograph of his back (C) and a radiograph of his lumbar spine (D) reveal ribbons, sheets, and plates of heterotopic bone (D, arrows). Lower panel collage (E-H) from several affected individuals depicts common variable features of classic FOP including orthotopic fusion of sub-axial facet joints of the cervical spine (E, arrows) prior to the onset of heterotopic ossification, short monophalangic malformed thumb (F), short broad femoral neck (G, arrows), and proximal medial tibial osteochondromas (H, arrows).
Individuals with Classic FOP Plus Atypical Clinical Features (“FOP-plus”)
Eight individuals in our study had FOP-plus (patients #1-7 and 15; Table 1): the classic clinical features of FOP plus one or more atypical features. Six of these individuals (patients #1-6) had the canonical c.617G>A (p.R206H) mutation in ACVR1. One individual (patient #7) had a unique missense mutation (p.Q207E) in the GS domain of ACVR1 and one patient (patient #15) had a missense mutation (p.G356D) in the protein kinase domain of ACVR1. Atypical features will be highlighted in the following brief summaries of patients with FOP-plus.
Patient #1 (c.617G>A; p.R206H) had intercurrent aplastic anemia that developed at 10 years of age and was treated successfully with an HLA-matched bone marrow transplantation from an unaffected sibling (Kaplan, et al., 2007b). The bone marrow transplantation cured the aplastic anemia but did not affect the progression of the FOP. At 35 years of age, the patient had 100 percent donor lineage in all cell lines of hematopoietic origin. DNA obtained from a buccal swab confirmed the classic ACVR1 mutation while DNA from peripheral blood of both patient and his unaffected sibling donor did not, confirming bone marrow engraftment in the patient.
Patient #2 (c.617G>A; p.R206H) had intercurrent polyostotic fibrous dysplasia (Frame, et al., 1972), a condition typically caused by somatic cell activating mutations of the GNAS gene (MIM# 139320). DNA was not available from affected somatic tissues and GNAS sequence analysis was not possible. Genomic DNA from peripheral blood was negative, as expected, for GNAS mutations (data not shown) but ACVR1 mutation screening revealed the c.617G>A (p.R206H) mutation.
Patient #3 (c.617G>A; p.R206H) was diagnosed at 10 years of age with a craniopharyngioma which was successfully resected although panhypopituitarism required daily replacement of adrenal steroids, thyroid hormone, and vasopressin (DDAVP).
Patient #4 (c.617G>A; p.R206H) was diagnosed with severe childhood glaucoma at the age of two, had a bifid uvula, thin hair, and a history of mild developmental delay.
Patient #5 (c.617G>A; p.R206H) suffered from severe retinopathy of prematurity in addition to bilateral cataracts, and glaucoma of the left eye. At four months of age, he was diagnosed with an inguinal hernia which was surgically repaired without the formation of heterotopic bone. He has severe motor and cognitive developmental delays with myoclonic seizures and diffuse cerebral dysfunction. He had a successful HLA-matched liver transplantation due to liver failure, attributed to cytomegalovirus infection and takes antiepleptic medications as well as sirolimus and prednisolone as chronic immunosupression. Progressive heterotopic ossification was first noted at age 9 following the liver transplantation and flare-ups of heterotopic ossification continued in characteristic anatomic patterns despite the immunosupression. A high resolution karyotype was normal.
Patient #6 (c.617G>A; p.R206H) had growth retardation with height and weight persistently less than fifth percentile for age. At eighteenth months of age, resections of large soft tissue swellings on her back exacerbated heterotopic bone formation. She rapidly developed severe scoliosis and thoracic lordosis with death at eight years of age from heart failure that complicated thoracic insufficiency syndrome (Kaplan and Glaser, 2005).
Patient #7 had a clinical appearance nearly identical to patient #6, but he had not undergone invasive diagnostic biopsies. Nevertheless, he had failure to thrive with height and weight persistently below the fifth percentile. A unique heterozygous mutation (c.619C>G; p.Q207E) in the GS domain of ACVR1 was detected in his genomic DNA.
Patient #15 had persistence of primary teeth into adulthood, and primary amenorrhea. Sequence analysis of genomic DNA revealed a heterozygous mutation (c.1067G>A; p.G356D) in the protein kinase domain of ACVR1.
Individuals with Variant FOP Features (“FOP Variants”)
Twelve cases (patients #8-14; 16-20) are variants of classic FOP, with major variations in one or both of the classic defining features of FOP (Table 1). Variations from the classic FOP features will be highlighted in the following brief summaries.
Patient #8 had short thumbs at birth, although his toes appeared normal. He had no soft tissue lesions or heterotopic ossification during childhood. At 26 years of age, he tore the right anterior cruciate ligament, underwent a surgical repair, and formed heterotopic bone at the operative site. Surgical resection of mature heterotopic bone was attempted but failed. By age 30, he had a stooped posture, a painless reciprocal gait, 80 percent residual movement of the cervical spine, 150 degrees abduction of the shoulders, and full range of motion of the jaw, elbows, wrists, and ankles. Radiographs revealed heterotopic ossification in the neck, back, and left knee. Both hips showed mild degenerative changes. Radiographs of the right great toe were normal, however there was a small boney irregularity of the left distal first metatarsal (Figure 2A). A CT scan of the head and neck revealed a hypoplastic cerebellum. Sequence analysis of genomic DNA revealed a heterozygous mutation (c.982G>A; p.G328R) in the protein kinase domain of ACVR1.
Figure 2. Digital Malformations in FOP Variants.

All FOP variants patients whose images are depicted here (A-H) had heterozygous missense mutations at either codon 328 (A-E) or codon 536 (F-H) in the kinase domain of ACVR1 in contrast to all classically-affected individuals with FOP who had a recurrent mutation in ACVR1 at codon 206 (R206H) (Figure 1); protein RefSeq NP_001096.1. All three variants with the G328R mutation (A, B, plus members of family #9, not shown) had either normal great toes or minimal malformations, while the hands were normal. All four variants with either the G328W mutation (C; plus patient #12, not shown) or the G328E mutation (D, E) had severe truncation deformities of multiple digits (C-E) and/or syndactyly (C, D). Patients #13 and #14 (D, E; G328E mutation) had slightly different malformation patterns from each other: patient #13 (D) had severe truncation of the great toes, whereas patient #14 (E) had more severe reduction deficits of the posterior digits. The hand malformations were similar in each, although patient #14 (E) was missing a post-axial digit on both hands. Both patients lacked nails in all severely affected digits. Three variants with the G356D mutation (F, G, H) had severe truncation malformations of the thumbs and great toes, although variable degrees of terminal symphalangism were noted and the digital truncations of patient #16 (F) were asymmetric in the hands and feet.
Patient/family #9 included three individuals, a mother (10.II.2) and two daughters (10.III.1 and 10.III.2; Table 2) who were reported previously (as family #2) (Virdi, et al., 1999) with a mild FOP phenotype characterized by either normal toes or mildly-affected toes and no or late onset of heterotopic ossification (Table 1). No substantial progression of FOP has occurred in any of the individuals since the previously published report. Sequence analysis of genomic DNA revealed a heterozygous mutation (c.982G>A; p.G328R) in the protein kinase domain of ACVR1 in all three affected individuals, but not in the unaffected father (10.II.1) or maternal grandmother (10.I.2).
Table 2. Phenotypes of FOP variant patient/family #9*.
| 10.II.2 | 10.III.1 | 10.III.2 | |
|---|---|---|---|
| Congenital malformation of great toe | none | minor | none |
| Age of onset of HO | 22 | no HO | 13 |
| Severity of HO | mild | - | mild |
| Exacerbation of HO following trauma | + | - | + |
| Intra-articular synovial osteochondromatosis of the hip | - | - | + |
| Radiographic anomalies of the cervical spine | + | - | - |
| ACVR1 mutation | c.982G>A | c.982G>A | c.982G>A |
Key:
Affected mother (10.II.2) and two affected daughters (10.III.1 and 10.III.2)
Absent (-); Present (+); heterotopic ossification (HO)
ACVR1 mutation numbering reflects the cDNA sequence with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence (NM_001105.4).
Patient #10 had normal hands but short malformed great toes at birth. Surgical correction was attempted and pre-operative films were not available, however post-surgical radiographs showed monophalangism of the great toes, with surgical correction of hallux valgus, and absence of the middle phalanges of the fourth and fifth toes bilaterally (Figure 2B). Heterotopic ossification began at 21 years of age in the characteristic anatomic patterns. At 32 years, he developed headaches and a computed tomographic scan of the brain revealed multiple cerebral cavernous malformations (CCMs). No genetic workup was undertaken for the CCMs. Sequence analysis of genomic DNA revealed a heterozygous mutation (c.982G>C; p.G328R) in the protein kinase domain of ACVR1.
Patient #11 was noted at birth to have severe reduction deficits of the great toes, with lack of toenails in the affected digits, and severe malformations of the thumbs (Figure 2C). During the second decade of life, her scalp hair became thin, and its growth rate slowed dramatically. Her eyebrows were sparse. She had mild cognitive impairment with difficulty in abstract thinking but no difficulty in attention. A computed tomographic brain scan revealed anatomic abnormalities of the cerebellum without associated impairment in movement. An MRI of the brain was not performed. Sequence analysis of genomic DNA revealed a heterozygous mutation (c.982G>T; p.G328W) in the protein kinase domain of ACVR1.
Examination of polymorphic markers in the FOP linkage region on chromosome 2 also revealed a loss of heterozygosity (LOH) region in patient #11. We determined (data not shown) that this LOH is a de novo 1.8 kb region that is not present in either of the patient's parents. Genomic DNA database analysis and comparison of the deleted region across species revealed that the deletion contains no identified gene coding regions and that the sequence is not conserved in mouse genomic DNA. No other patients examined (including patients #12, 13, 14 reported here) contained this deletion.
Patient #12 showed severe reduction deficits of the great toes and thumbs at birth, with absent toenails in the affected digits (Connor and Evans, 1982). Progressive heterotopic ossification in characteristic anatomic patterns began at age 8. She had mild conductive hearing impairment and short broad femoral necks. During the second decade of life, she developed sparse thin hair on her scalp and mild cognitive impairment without any difficulty in attention. Sequence analysis of genomic DNA revealed a heterozygous mutation (c.982G>T; p.G328W) in the protein kinase domain of ACVR1, the same mutation identified in patient #11.
Patient #13 (Connor and Evans, 1982) had severe reduction deficits of the great toes and thumbs at birth, with absence of toenails in the affected digits (Figure 2D). During the second decade of life, she developed sparse thin hair on her scalp and had mild cognitive impairment without any difficulty in attention. Sequence analysis of genomic DNA revealed a heterozygous mutation (c.983G>A; p.G328E) in the protein kinase domain of ACVR1.
Patient #14 was noted at birth to have severe reduction deficits of the great toes and thumbs as well as absent toenails of the affected digits (Figure 2E). During the second decade of life, she was noted to have sparse, thin hair on her scalp and sparse eyebrows. She had mild cognitive impairment without any deficit in attention. DNA sequence analysis of genomic DNA revealed a heterozygous mutation (c.983G>A; p.G328E) in the protein kinase domain of ACVR1, the same mutation that was found in patient #13.
Patient #16 had asymmetrical malformed great toes and thumbs with missing nails on the severely affected digits at birth. The distal interphalangeal joints in his index fingers were absent (Figure 2F) and he had hypospadias. Sequence analysis of genomic DNA revealed a heterozygous mutation (c.1067G>A; p.G356D) in the protein kinase domain of ACVR1.
Patient #17 had bilateral absence of both great toes and hypoplastic thumbs with severe shortening of the first metacarpals (Figure 2G). His eyebrows and eyelashes were sparse. He was 28 months of age at last examination and had no manifestations of soft tissue swellings or heterotopic ossification; typical age of onset is 2-10 years. Since his toe and thumb malformations were suggestive of FOP, the ACVR1 gene was examined. Sequence analysis of genomic DNA revealed a heterozygous mutation (c.1067G>A; p.G356D) in the protein kinase domain of ACVR1.
Patient #18 had severe reduction deficits of the great toes and thumbs at birth, with absent or hypoplastic nails of the affected digits (Figure 2H). Heterotopic ossification began at 15 years of age causing mobility restriction of the neck and back; a flare-up of the right hip occurred at 16 years old. Severe alopecia, and primary amenorrhea were noted. Sequence analysis of genomic DNA revealed a heterozygous mutation (c.1067G>A; p.G356D) in the protein kinase domain of ACVR1. This is the same mutation identified in patients #15, 16, and 17.
Patient #19 had clinically and radiographically normal toes. FOP flare-ups initiated at age 14 and progression of disease was slow and evanescent. At 40 years of age, she had limited motion of the cervical spine and shoulders with heterotopic ossification in the neck, back, chest, and right hip, but was still ambulatory. Sequence analyses of genomic DNA revealed a heterozygous mutation (c.1124G>C; p.R375P) in the protein kinase domain of ACVR1.
Patient #20 had clinically and radiographically normal toes. At 11 years of age, painful flexion contracture of the left hip prompted imaging studies that showed a poorly defined soft tissue mass of the left iliopsoas muscle. Following a biopsy that was diagnosed as “aggressive fibromatosis,” she developed heterotopic ossification with ankylosis of the left hip. Within six months she had ankylosis of all major joints of the axial and appendicular skeleton. Sequence analysis of genomic DNA revealed a unique in-frame three basepair heterozygous mutation (c.590_592delCTT; P197_F198delinsL) in the GS domain of ACVR1 that replaces amino acids proline (codon 197) and phenylalanine (codon 198) with leucine.
ACVR1 Mutations
All mutations identified in classic FOP, FOP-plus, and variant FOP patients are single nucleotide substitutions that cause missense mutations, with the exception of a 3-nucleotide deletion (patient #20) that replaces two amino acids with a single amino acid. In families with inherited FOP (such as #9 in this study), all affected members have a mutation and no unaffected members carry the mutation. For most cases of sporadic FOP-plus and FOP variants, both parental samples were unavailable for analysis, however, de novo mutations were confirmed for patients #11 and #18. Absence of each of the identified mutations was verified in at least 98 unaffected individuals. None of these ACVR1 sequence variants are reported in SNP databases (http://www.ncbi.nlm.nih.gov/SNP; http://www.ensembl.org/Homo_sapiens/genesnpview). Unlike the recurrent c.617G>A mutation, none of the rarer mutations in FOP-plus and variant FOP patients alter a CpG dinucleotide.
In addition to direct DNA sequence analysis, most of the identified ACVR1 mutations were verified experimentally by differential restriction endonuclease digestion (data not shown). New enzyme digestion sites are formed by the c.619C>G (NruI), and c.1067G>A (DrdI) nucleotide substitutions. Each of the single nucleotide substitutions identified in codon 328 eliminates a StyI digestion site.
ACVR1 is a protein that has been highly conserved during vertebrate evolution. Comparison of the human ACVR1 protein sequence (509 amino acids) to ACVR1 in other species shows high degrees of similarity (for example, 99.8% in chimp, 98.4% in mouse, 97.9% in chick). Each of the mutated amino acids in the FOP variant and FOP-plus patients is conserved across species (Figure 3). Unlike the classic FOP mutation that changes codon 206, an amino acid that may contribute to receptor specificity [ACVR1 receptors encode arginine (R) while BMPRIA and BMPRIB have lysine (K) at the analogous position; see suppl. Fig 2 in Shore, et al. (2006)], each FOP variant mutation and FOP-plus mutation is invariant among all three human type I BMP receptors: ACVR1 (ALK2), BMPRIA (ALK3), and BMPRIB (ALK6).
Figure 3. Position and Conservation of ACVR1 Amino Acid Changes.

ACVR1 encodes a 509 amino acid protein that contains a ligand binding region, a transmembrane (TM) domain, a glycine-serine rich region (GS), and a protein kinase domain. The numbers below the protein representation indicate the amino acid codons included in each identified domain; the protein initiation codon is codon 1 (RefSeq NP_001096.1). The relative positions of all identified mutations are shown with the altered amino acids in bold with light shading. Each mutation in the ACVR1 gene occurs in an identical amino acid at the corresponding position of ACVR1/ALK2 across species (darker shading). Clustal W was used for multiple protein sequence alignment. The schematics are drawn approximately to scale.
No frameshift or nonsense mutations were identified in the ACVR1 sequence, suggesting that in each case a mutant receptor protein with altered function is produced. All of the identified mutations occur in either the GS or protein kinase domains, regions of the ACVR1 receptor that are important in conferring downstream signal transduction.
Protein Modeling
Although the family of activin-like kinases (ALKs) includes seven different receptors, only the structure of the intracellular domain of TβRI (ALK5; PDB 1IAS) has been experimentally determined (Huse, et al., 1999). However, the extensive cytoplasmic domain homology within the ALK family allows reliable structure-based homology modeling of wild-type and mutant ACVR1 receptor kinases (Figure 4). Our previous modeling predicted that the highly conserved p.R206H substitution, found in all classic FOP and in six patients (#1-6) with FOP-plus, results in an aberrant ion pair or salt bridge that acts as a pH-sensitive switch that leads to ligand-independent activation of the receptor (Groppe, et al., 2007).
Figure 4. Sites of FOP mutations in a structured-based homology model of the ACVR1 receptor kinase domain.

Panels show models of the wild type ACVR1 protein with specific amino acids that are implicated in structural changes as a result of mutations are indicated. The protein initiation codon is codon 1 (RefSeq NP_001096.1).
A. Mutation sites within the GS regulatory region. Arginine 206, which forms an ion pair with aspartate 269 (dashed lines), is substituted with histidine in all patients with classic FOP and six of eight patients (#1-6) with FOP-plus. The adjacent residue, glutamine 207, is substituted with glutamate in FOP-plus patient #7. A three-nucleotide deletion replaces proline 197 and phenylalanine 198 with a leucine residue in variant FOP patient #20. The surface of the FKBP12 binding protein at the binding protein-receptor interface is depicted in grey.
B, C. Multiple Glycine 328 missense mutations. Seven FOP variants (#8-14) had substitutions of glycine 328. Codon 328, in the protein kinase domain, resides in a loop at the bottom of a surface cavity bordering the GS loop and the N-terminal end of the αC helix. The surface of the kinase domain is depicted in green. For clarity, FKBP12 is not shown. The view in C is similar to panel B, with the FKBP12 binding site and GS loop rolled slightly forward toward the viewer.
D. Mutation sites within the receptor kinase active site. Glycine 356 is substituted with aspartate in patients #15-18 (one with FOP-plus and three FOP variants) and arginine 375 with proline in FOP variant patient #19. The ion pair between arginine 375 and aspartate 354 blocks a cation binding site required for ATP hydrolysis by the enzyme. The lysine 235-glutamate 248 ion pair is conserved in all protein kinases and modulates enzyme activity by altering active site conformation.
In addition to the p.R206H mutation, we identified two other GS domain mutations. Protein modeling predicts that introduction of an acidic residue such as p.Q207D, an engineered constitutive activating TβRI mutation (Wieser, et al., 1995), or p.Q207E (patient #7; FOP-plus) would disrupt an ion pair formed between the neighboring basic residue (ACVR1 Arg206) and an invariant acidic residue (ACVR1 Asp269). Formation of the non-native ion pair (in cis), even transiently, is predicted to sterically hinder the binding of the inhibitory FKBP12 binding protein (Figure 4A), a protein which is required to maintain the receptor kinase in an auto-inhibited state until activated by ligand-induced assembly of the heterotetrameric signaling complex (Huse, et al., 2001). The three-nucleotide deletion in FOP variant patient #20 (c.590_192delCTT), replaces proline 197 and phenylalanine 198 with a single leucine residue, and removes one of the two residues (Phe 198, Leu199) comprising the FKBP12 binding site (Figure 4A); this loss of the entire FKBP12-ACVR1 binding interface would abolish all interaction with the inhibitory protein. Thus all three mutations in the GS region are predicted to share the common effect to perturb, diminish or abolish interactions with FKBP12.
ACVR1 mutations within the protein kinase domain were also found. Glycine 328, a site of multiple mutations in FOP variants, is replaced with arginine (patients #8-10), tryptophan (patients #11-12), or glutamate (patients #13-14). Glycine 328 is at the bottom of a cavity formed by a flanking surface loop, by the GS loop, and by the N-terminal end of the αC helix (Figure 4B and 4C). The structural basis for the G328 mutations are not clear since none of the substitutions significantly alters the conformation of the polypeptide chain as determined by homology modeling (not shown). Introduction of bulky, charged or hydrogen-bonding sidechains into the cavity could impair receptor function by affecting GS domain interaction with FKBP12 or SMAD proteins. Alternatively, the cleft could be a substrate-binding site for phosphorylations in the MAP kinase pathway with the substituted residues causing enhanced interactions with target substrates. A third possibility is that introduction of these new sidechains results in displacement of the αC helix, a key regulatory element of all protein kinases (see below).
Two additional protein kinase domain FOP mutations, p.G356D (patients #15-18) and p.R375P (patient #19), occur within the receptor kinase active site (Figure 4D). Glycine 356 substitution with aspartate has no detectable effect on the backbone conformation of the receptor (not shown). However, an ion pair between lysine 235 and glutamate 248 (Figure 4D), found in all protein kinases that modulate enzyme activity by αC helix conformation changes (Huse and Kuriyan, 2002), may be altered by introducing an ion pair-forming partner (aspartate 356) that causes loss of auto-inhibition of the kinase. Arginine 375 forms a conserved ion pair with aspartate 354, blocking a cation-binding site required for ATP hydrolysis. Loss of this ion pair due to substitution of arginine with proline allows cation binding and promotes phosphorylation by the receptor (a similar view for TβRI interactions has been reported (Huse, et al., 1999)).
Discussion
Small variations in genotype can give rise to large variations in phenotype that provide important insight into the mechanisms of human disease. Among patients identified with FOP-like heterotopic ossification and/or great toe malformations, we identified twenty patients who showed notable variation in the clinical presentation commonly observed in patients with FOP. All patients with “classic FOP” features have a recurrent c.617G>A (p.R206H) mutation in the ACVR1 gene. In this study, we expanded our analysis of classic FOP patients and examined DNA from the atypical FOP patients for ACVR1 gene mutations. This analysis has led to several significant conclusions: All patients with FOP-type heterotopic ossification have mutations in the protein-coding region of ACVR1, and all are missense mutations or in-frame deletions. While all patients with classic FOP have the identical single nucleotide and resulting amino acid change, some patients with atypical clinical presentation of FOP have alternate mutations in the ACVR1 gene. The classic FOP mutation occurs in most patients with “FOP-plus” (classic features plus additional unusual features); their additional features may be coincidental or may be due to genetic modifiers. However, some patients with FOP-plus have novel mutations, which may explain their additional features. “FOP variant” patients (variations in the classic defining features of FOP) have novel mutations that can provide insights into the effect of altered ACVR1 function during developmental, cognitive, and homeostatic processes. Additionally, understanding the specific effect of a missense mutation on ACVR1 function could help guide the design of pharmacologic agents that will modify or prevent the cause of the disease.
The consequences of specific ACVR1 mutations during postnatal life are likely different from those during embryogenesis. Studies of identical twin pairs with FOP (Hebela, et al., 2005) have shown that although environmental influences have major effects on the course of heterotopic ossification, genetic factors are the major influence on developmental defects (such as embryonic skeletal formation) in FOP. Among patients with classic FOP, FOP-plus, and FOP variants, a wide range of variability in great toe malformations is observed, and our data suggest that genotype-phenotype correlations may explain at least some of this variation. In some patients, different mutations (for example, p.R206H and p.Q207E, both in the GS domain) show similar toe malformations. We also observed that similar phenotypes are associated with mutations in different domains of the ACVR1 receptor. For example, mutations in the protein kinase domain or the GS activation domain occur in FOP variants with normal or mildly malformed great toes (patients #8, 9, 10, 19, 20; Table 1). By contrast, the same mutation (p.G356D) that was identified in four patients (#15-18), and in an additional recent case report (Furuya, et al., 2008), is associated with a wide range of phenotypic consequences, possibly due to differences in genetic backgrounds of the individuals (Shore, et al., 2005).
Other developmental phenotypes may only be caused by specific ACVR1 mutations, such as the severely shortened thumbs and great toes, alopecia, nail dysplasia, and learning disorders (Botchkarev, 2003; Botchkarev and Sharov, 2004; Hens, et al., 2007; Kobielak, et al., 2007; Wang, et al., 2004) that occur in four FOP variants (#11-14) with glycine to tryptophan or glutamic acid codon 328 mutations. These codon 328 mutations are distinct from the glycine to arginine mutations in less severe FOP variants (#8-10) who had normal/minimally affected great toes and late onset heterotopic ossification. ACVR1 therefore appears to be particularly sensitive to codon 328 mutations, suggesting importance in regulating receptor function and BMP signaling during embryonic development.
Progressive postnatal heterotopic ossification is the common feature shared by all patients with classic FOP, FOP-plus, and FOP variants. Although the rate of progression and the severity of heterotopic ossification varies among individuals with classic FOP, we found correlation between the severity of heterotopic ossification and specific mutations among the ACVR1 mutations identified in FOP variant patients. These data support that all of the identified ACVR1 missense mutations influence the promiscuous post-natal induction of cartilage and bone cell differentiation, however, as supported by our protein modeling data, the proposed molecular mechanisms may vary.
All of the mutations in ACVR1 associated with classic, FOP-plus and variant forms of FOP reside in or adjacent to the GS regulatory region or active site of the kinase. None were mapped to the larger C-terminal domain or lobe, which serves only as a scaffold to stabilize the receptor. Furthermore, the structural basis for loss of autoinhibition by the mutant receptors is predicted by structural modeling, with the exception of the multiple substitutions at glycine 328 that could perturb the receptor through one of several plausible mechanisms.
All of the reported mutations in FOP and its variants are predicted by protein structure homology modeling to activate the ACVR1 protein and enhance receptor signaling. Constitutive ACVR1 expression in embryonic chick limbs induces expansion of chondrogenic anlagen, strongly suggesting that ACVR1 signaling alters cell fate and induces undifferentiated mesenchyme to form cartilage (Zhang, et al., 2003). In the chick however, the heterotopic bone was seen at birth, whereas in humans with FOP, the effects were not seen at birth, and occurred only in the postnatal period. These findings suggest the intriguing possibility that the FOP mutations are mildly constitutively active and hyper-responsive to receptor stimulation. Recent data support these findings (Billings, et al., 2008; Shen, et al., 2007).
Enhanced expression of BMP transcriptional targets is observed in FOP cells (Fiori, et al., 2006; Kaplan, et al., 2006; Serrano de la Pena, et al., 2005). Overactive BMP signaling in FOP cells may lead paradoxically to orthotopic ankylosis of the joints and early degenerative joint disease as seen in FOP patients and in animal models of promiscuous BMP signaling (Ahn, et al., 2003; Fiori, et al., 2006; Gannon, et al., 1997; Glaser, et al., 2003; Kan, et al., 2004; Kaplan, et al., 2006; Kaplan, et al., 2007c; Kaplan, et al., 1990; O'Connell, et al., 2007; Serrano de la Pena, et al., 2005; Shafritz, et al., 1996). Aberrant ACVR1 signaling may also be relevant to the pathogenesis of degenerative joint disease (Oshin and Stewart, 2007), as seen in early orthotopic degenerative changes of the great toe, thumb, cervical spine, and in the costovertebral joints before the appearance of FOP flare-ups and subsequent heterotopic ossification.
Animal and human studies of mutations in BMPRIB (ALK6; another BMP type I receptor associated with brachydactyly type A2), as well as GDF5 and NOGGIN, suggest that mutations in BMP type I receptors affect cartilage formation in a dominant-negative manner (Baur, et al., 2000; Dawson, et al., 2006; Lehmann, et al., 2006; Lehmann, et al., 2007; Lehmann, et al., 2003; Seemann, et al., 2005; Yi, et al., 2000). We cannot yet rule out the possibility that a similar effect is present during embryonic development with the mutations in classic FOP, FOP-plus, and FOP variants that lead to malformations of the great toes. Mutant ACVR1 may oligomerize with other type I receptors such as BMPRIA and BMPRIB, accounting for some of the in vitro and in vivo effects seen in individuals with FOP and its variants (Gilboa, et al., 2000; Nohe, et al., 2002).
All of the classic and common variable features of FOP as well as many, if not all, of the atypical features evaluated in our study could plausibly be ascribed to dysregulation of the BMP signaling pathway. A recent report that mutations in BMP4 cause eye, brain, and digit abnormalities suggests that BMP4 signaling through ACVR1 could lead to at least some of the atypical features found in some FOP patients (Bakrania, et al., 2008). However, it is not yet known if atypical FOP features such as aplastic anemia (Kaplan, et al., 2007b), polyostotic fibrous dysplasia (Frame, et al., 1972), craniopharyngioma (Davis and Camper, 2007), cerebellar abnormalities (Angley, et al., 2003; Qin, et al., 2006), childhood glaucoma (Plikus, et al., 2008; Wordinger, et al., 2002; Wordinger, et al., 2007), cataracts (Andreev, et al., 2006; Wordinger and Clark, 2007), persistence of primary teeth (Thesleff, 2006), primary amenorrhea (Knight and Glister, 2006), hypospadias (Morgan, et al., 2003), or severe growth retardation (Lee, et al., 2007) (each seen in only one or two individuals) were intercurrent findings coincidentally associated with FOP or whether they were causally related to the underlying mutations in ACVR1 and unmasked by polymorphisms in the BMP or other signaling pathways in the affected individuals. Further studies of BMP signaling in animal models of classic and variant FOP will be critical to address these questions.
Identification of disease-causing mutations in ACVR1 has important diagnostic and therapeutic implications. Presently, there is no definitive treatment for patients with FOP or its variants (Glaser and Kaplan, 2005) and the identification of heterozygous missense mutations in ACVR1 reveals a pharmaceutical target for the development of signal transduction inhibitors (STIs) such as dorsomorphin or its derivatives (Kaplan, et al., 2007a; Yu, et al., 2008) as well as other therapeutic strategies(Kaplan, et al., 2007a). However, in addition to treating FOP, postnatal inhibition of ACVR1 could have a significant role in treating common acquired disorders of orthotopic and heterotopic ossification and, conversely, the mutation(s) of FOP and its variants could be harnessed for tissue engineering to form new bone for therapeutic applications. Genotype-phenotype correlations of the FOP ACVR1 mutations will help elucidate ACVR1 signaling mechanisms and in vivo functions to further these goals.
Acknowledgments
We thank members of our research laboratory for their contributions, including George Feldman for his work related to patient #11 and Bob Caron for figure preparation. We also thank Kathryn Ewens for helpful discussions. This work was supported in part by the Center for Research in FOP and Related Disorders, the International FOP Association, the Ian Cali Endowment, the Weldon Family Endowment, the Isaac and Rose Nassau Professorship of Orthopaedic Molecular Medicine, and by grants from the Rita Allen Foundation and the National Institutes of Health (R01-AR41916).
References
- Ahn J, de la Pena LS, Shore EM, Kaplan FS. Paresis of a bone morphogenetic protein-antagonist response in a genetic disorder of heterotopic skeletogenesis. J Bone Joint Surg (Am) 2003;85A:667–674. doi: 10.2106/00004623-200304000-00013. [DOI] [PubMed] [Google Scholar]
- Andreev K, Zenkel M, Kruse F, Junemann A, Schlotzer-Schrehardt U. Expression of bone morphogenetic proteins (BMPs), their receptors, and activins in normal and scarred conjunctiva: role of BMP-6 and activin-A in conjunctival scarring? Exp Eye Res. 2006;83:1162–1170. doi: 10.1016/j.exer.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Angley C, Kumar M, Dinsio KJ, Hall AK, Siegel RE. Signaling by bone morphogenetic proteins and Smad1 modulates the postnatal differentiation of cerebellar cells. J Neurosci. 2003;23:260–268. doi: 10.1523/JNEUROSCI.23-01-00260.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakrania P, Efthymiou M, Klein JC, Salt A, Bunyan DJ, Wyatt A, Ponting CP, Mmartin A, Williams S, Lindley V, Gilmore J, Restori M, Robson AG, Neveu MM, Holder GE, Collin JRO, Robinson DO, Fardon P, Johansen-Berg H, Gerrelli D, Ragge NK. Mutations in BMP4 cause eye, brain, and digit developmental anomalies: Overlap between the BMP4 and Hedgehog signaling pathways. Am J Hum Genet. 2008;82:304–319. doi: 10.1016/j.ajhg.2007.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur ST, Mai JJ, Dymecki SM. Combinatorial signaling through BMP receptor IB and GDF5: shaping of the distal mouse limb and the genetics of distal limb diversity. Development. 2000;127:605–619. doi: 10.1242/dev.127.3.605. [DOI] [PubMed] [Google Scholar]
- Billings PC, Fiori JL, Bentwood JL, O'Connell MP, Jiao X, Nussbaum B, Caron RJ, Shore EM, Kaplan FS. Dysregulated BMP signaling and enhanced osteogenic differentiation of connective tissue progenitor cells from patients with fibrodysplasia ossificans progressiva (FOP) J Bone Miner Res. 2008;23:305–313. doi: 10.1359/JBMR.071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botchkarev VA. Bone morphogenetic proteins and their antagonists in skin and hair follicle biology. J Invest Dermatol. 2003;120:36–47. doi: 10.1046/j.1523-1747.2003.12002.x. [DOI] [PubMed] [Google Scholar]
- Botchkarev VA, Sharov AA. BMP signaling in the control of skin development and hair follicle growth. Differentiation. 2004;72:512–526. doi: 10.1111/j.1432-0436.2004.07209005.x. [DOI] [PubMed] [Google Scholar]
- Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth Factors. 2004;22:233–241. doi: 10.1080/08977190412331279890. [DOI] [PubMed] [Google Scholar]
- Cohen RB, Hahn GV, Tabas JA, Peeper J, Levitz CL, Sando A, Sando N, Zasloff M, Kaplan FS. The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. A study of forty-four patients. J Bone Joint Surg (Am) 1993;75:215–9. doi: 10.2106/00004623-199302000-00008. [DOI] [PubMed] [Google Scholar]
- Connor JM, Evans DA. Fibrodysplasia ossificans progressiva. The clinical features and natural history of 34 patients. J Bone Joint Surg (Br) 1982;64:76–83. doi: 10.1302/0301-620X.64B1.7068725. [DOI] [PubMed] [Google Scholar]
- Davis SW, Camper SA. Noggin regulates Bmp4 activity during pituitary induction. Dev Biol. 2007;305:145–160. doi: 10.1016/j.ydbio.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson K, Seeman P, Sebald E, King L, Edwards M, Williams J, Mundlos S, Krakow D. GDF5 is a second locus for multiple-synostosis syndrome. Am J Hum Genet. 2006;78:708–712. doi: 10.1086/503204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiori JL, Billings PC, Serrano de la Pena LS, Kaplan FS, Shore EM. Dysregulation of the BMP-p38 MAPK signaling pathway in cells from patients with fibrodysplasia ossificans progressiva (FOP) J Bone Miner Res. 2006;21:902–909. doi: 10.1359/jbmr.060215. [DOI] [PubMed] [Google Scholar]
- Frame B, Azad N, Reynolds WA, Saeed SM. Polyostotic fibrous dysplasia and myositis ossificans progressiva. A report of coexistence. Am J Dis Child. 1972;124:120–122. doi: 10.1001/archpedi.1972.02110130122020. [DOI] [PubMed] [Google Scholar]
- Furuya H, Ikezoe K, Wang L, Ohyagi Y, Motomura K, Fujii N, Kira JI, Fukumaki Y. A unique case of fibrodysplasia ossificans progressiva with an ACVR1 mutation, G356D, other than the common mutation (R206H) Am j Med Genet Part A. 2008;146:459–463. doi: 10.1002/ajmg.a.32151. [DOI] [PubMed] [Google Scholar]
- Gannon FH, Kaplan FS, Olmsted E, Finkel GC, Zasloff MA, Shore E. Bone morphogenetic protein 2/4 in early fibromatous lesions of fibrodysplasia ossificans progressiva. Hum Pathol. 1997;28:339–43. doi: 10.1016/s0046-8177(97)90133-7. [DOI] [PubMed] [Google Scholar]
- Gazzerro E, Canalis E. Bone morphogenetic proteins and their antagonists. Rev Endocr Metab Disorders. 2006;7:51–65. doi: 10.1007/s11154-006-9000-6. [DOI] [PubMed] [Google Scholar]
- Gilboa L, Nohe A, Geissendorfer T, Sebald W, Henis YI, Knaus P. Bone morphogenetic protein receptor complexes on the surface of live cells: a new oligomerization mode for serine/threonine kinase receptors. Mol Biol Cell. 2000;11:1023–1035. doi: 10.1091/mbc.11.3.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser DL, Economides AN, Wang LL, Liu X, Kimble RD, Fandl JP, Wilson JM, Stahl N, Kaplan FS, Shore EM. In vivo somatic cell gene transfer of an engineered noggin mutein prevents BMP4-induced heterotopic ossification. J Bone Joint Surg (Am) 2003;85A:2332–2342. doi: 10.2106/00004623-200312000-00010. [DOI] [PubMed] [Google Scholar]
- Glaser DL, Kaplan FS. Treatment considerations for the management of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:243–250. [Google Scholar]
- Groppe JC, Shore EM, Kaplan FS. Functional Modeling of the ACVR1 (R206H) mutation in FOP. Clin Orthop Rel Res. 2007;462:87–92. doi: 10.1097/BLO.0b013e318126c049. [DOI] [PubMed] [Google Scholar]
- Hebela N, Shore EM, Kaplan FS. Three pairs of monozygotic twins with fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:205–208. [Google Scholar]
- Hens JR, Dann P, Zhang JP, Harris S, Robinson GW, Wysolmerski J. BMP4 and PTHrP interact to stimulate ductal outgrowth during embryonic mammary development and t inhibit hair follicle induction. Development. 2007;134:1221–1230. doi: 10.1242/dev.000182. [DOI] [PubMed] [Google Scholar]
- Huse M, Chen YG, Massague J, Kuriyan J. Crystal structure of the cytoplasmic domain of the type I TGF beta receptor in complex with FKBP12. Cell. 1999;96:425–436. doi: 10.1016/s0092-8674(00)80555-3. [DOI] [PubMed] [Google Scholar]
- Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- Huse M, Muir TW, Xu L, Chen YG, Kuriyan J, Massague J. The TGF beta receptor activation process: an inhibitor- to substrate-binding switch. Mol Cell. 2001;8:671–682. doi: 10.1016/s1097-2765(01)00332-x. [DOI] [PubMed] [Google Scholar]
- Kan L, Hu M, Gomes WA, Kessler JA. Transgenic mice overexpressing BMP4 develop a fibrodysplasia ossificans progressiva (FOP)-like phenotype. Amer J Pathol. 2004;165:1107–1115. doi: 10.1016/S0002-9440(10)63372-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan FS, Fiori J, De La Pena LS, Ahn J, Billings PC, Shore EM. Dysregulation of the BMP-4 signaling pathway in fibrodysplasia ossificans progressiva. Ann NY Acad Sci. 2006;1068:54–65. doi: 10.1196/annals.1346.008. [DOI] [PubMed] [Google Scholar]
- Kaplan FS, Glaser DL. Thoracic insufficiency syndrome in patients wiith fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:213–216. [Google Scholar]
- Kaplan FS, Glaser DL, Pignolo RJ, Shore EM. A new era for fibrodysplasia ossificans progressiva: a druggable target for the second skeleton. Expert Opin Biol Ther. 2007a;7:705–712. doi: 10.1517/14712598.7.5.705. [DOI] [PubMed] [Google Scholar]
- Kaplan FS, Glaser DL, Shore EM, Deirmengian G, Gupta R, Delai P, Morhart R, Smith R, Le Merrer M, Rogers JG, Connor JM, Kitterman JA. The phenotype of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:183–188. [Google Scholar]
- Kaplan FS, Glaser DL, Shore EM, Pignolo RJ, Xu MQ, Zhang Y, Senitzer D, Forman SJ, Emerson SG. Hematopoietic stem-cell contribution to ectopic skeletogenesis. J Bone Joint Surg (Am) 2007b;89A:347–357. doi: 10.2106/JBJS.F.00472. [DOI] [PubMed] [Google Scholar]
- Kaplan FS, Groppe JC, Pignolo RJ, Shore EM. Ann NY Acad Sci. 2007c. Sep 13, Morphogen receptor genes and metamorphogenes: skeleton keys to the metamorphosis. epub. [DOI] [PubMed] [Google Scholar]
- Kaplan FS, Tabas JA, Zasloff MA. Fibrodysplasia ossificans progressiva: a clue from the fly? Calcif Tissue Int. 1990;47:117–25. doi: 10.1007/BF02555995. [DOI] [PubMed] [Google Scholar]
- Knight PG, Glister C. TGF-beta superfamily members and ovarian follicle development. Reproduction. 2006;132:191–206. doi: 10.1530/rep.1.01074. [DOI] [PubMed] [Google Scholar]
- Kobielak K, Stokes N, de la Cruz J, Polak L, Fuchs E. Loss of a quiescent niche but not follicle stem cells in the absence of bone morphogenetic protein signaling. Proc Natl Acad Sci USA. 2007;104:10063–10068. doi: 10.1073/pnas.0703004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, Dacquin R, Mee PJ, McKee MD, Jung DY, Zhang Z, Kim JK, Mauvais-Jarvis F, Ducy P, Karsenty G. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130:456–469. doi: 10.1016/j.cell.2007.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann K, Seemann P, Boergermann J, Morin G, Reif S, Knaus P, Mundlos S. A novel R486Q mutation in BMPR1B resulting in either a brachydactyly type C/symphalangism-like phenotype or brachydactyly type A2. Eur J Hum Genet. 2006;14:1248–1254. doi: 10.1038/sj.ejhg.5201708. [DOI] [PubMed] [Google Scholar]
- Lehmann K, Seemann P, Silan F, Goecke TO, Irgang S, Kjaer KW, Kjaergaard S, Mahoney MJ, Morlot S, Reissner C, Kerr B, Wilkie AOM, Mundlos S. A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN. Am J Hum Genet. 2007;81:388–396. doi: 10.1086/519697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann K, Seemann P, Stricker S, Sammar M, Meyer B, Suring K, Majewski F, Tinschert S, Grzeschik KH, Muller D, Knaus P, Nurnberg P, Mundlos S. Mutations in bone morphogenetic protein receptor 1B cause brachydactyly type A2. Proc Natl Acad Sci USA. 2003;100:12277–12282. doi: 10.1073/pnas.2133476100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahboubi S, Glaser DL, Shore EM, Kaplan FS. Fibrodysplasia ossificans progressiva. Pediatr Radiol. 2001;31:307–314. doi: 10.1007/s002470100447. [DOI] [PubMed] [Google Scholar]
- Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- Morgan EA, Nguyen SB, Scott V, Stadler HS. Loss of Bmp7 and Fgf8 signaling in Hoxa13-mutant mice causes hypospadia. Development. 2003;130:3095–3109. doi: 10.1242/dev.00530. [DOI] [PubMed] [Google Scholar]
- Nohe A, Hassel S, Ehrlich M, Neubauer F, Sebald W, Henis YI, Knaus P. The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J Biol Chem. 2002;277:5330–5338. doi: 10.1074/jbc.M102750200. [DOI] [PubMed] [Google Scholar]
- O'Connell MP, Billings PC, Fiori JL, Deirmengian G, Roach HI, Shore EM, Kaplan FS. HSPG modulation of BMP signaling in fibrodysplasia ossificans progressiva cells. J Cell Biochem. 2007;102:1493–1503. doi: 10.1002/jcb.21370. [DOI] [PubMed] [Google Scholar]
- Oshin AO, Stewart MC. The role of bone morphogenetic proteins in articular cartilage development, homeostasis and repair. Vet Comp Orthop Traum. 2007;20:151–158. doi: 10.1160/vcot-07-02-0018. [DOI] [PubMed] [Google Scholar]
- Pignolo RJ, Suda RK, Kaplan FS. The fibrodysplasia ossificans progressiva lesion. Clin Rev Bone Miner Metab. 2005;3:195–200. [Google Scholar]
- Plikus MV, Mayer JA, de la Cruz D, Baker RE, Maini PK, Maxson R, Chuong CM. Cyclic dermal BMP signaling regulates stem cell activation during hair regeneration. Nature. 2008;451:340–344. doi: 10.1038/nature06457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Wine-Lee L, Ahn KJ, Crenshaw EB., 3rd Genetic analyses demonstrate that bone morphogenetic protein signaling is required for embryonic cerebellar development. J Neurosci. 2006;26:1896–1905. doi: 10.1523/JNEUROSCI.3202-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocke DM, Zasloff M, Peeper J, Cohen RB, Kaplan FS. Age- and joint-specific risk of initial heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. Clin Orthop Rel Res. 1994;301:243–248. [PubMed] [Google Scholar]
- Roush W. Protein builds second skeleton. Science. 1996;273:1170. doi: 10.1126/science.273.5279.1170. [DOI] [PubMed] [Google Scholar]
- Schaffer AA, Kaplan FS, Tracy MR, O'Brien ML, Dormans JP, Shore EM, Harland RM, Kusumi K. Developmental anomalies of the cervical spine in patients with fibrodysplasia ossificans progressiva are distinctly different from those in patients with Klippel-Feil syndrome - Clues from the BMP signaling pathway. Spine. 2005;30:1379–1385. doi: 10.1097/01.brs.0000166619.22832.2c. [DOI] [PubMed] [Google Scholar]
- Seemann P, Schwappacher R, Kjaer KW, Krakow D, Lehmann K, Dawson K, Stricker S, Pohl J, Ploger F, Staub E, Nickel J, Sebald W, Knaus P, Mundlos S. Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2. J Clin Invest. 2005;115:2373–2381. doi: 10.1172/JCI25118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano de la Pena LS, Billings PC, Fiori JL, Ahn J, Kaplan FS, Shore EM. Fibrodysplasia ossificans progressiva (FOP), a disorder of ectopic osteogenesis, misregulates cell surface expression and trafficking of BMPRIA. J Bone Miner Res. 2005;20:1168–1176. doi: 10.1359/JBMR.050305. [DOI] [PubMed] [Google Scholar]
- Shafritz AB, Shore EM, Gannon FH, Zasloff MA, Taub R, Muenke M, Kaplan FS. Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. N Eng J Med. 1996;335:555–561. doi: 10.1056/NEJM199608223350804. [DOI] [PubMed] [Google Scholar]
- Shen Q, Xu M, Little SC, Kaplan FS, Mullins MC, Shore EM. Activation of BMP signaling by the FOP ACVR1 R206H mutation. J Bone Miner Res. 2007;22:S43–S43. [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Shore EM, Feldman GJ, Xu M, Kaplan FS. The genetics of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:201–204. [Google Scholar]
- Shore EM, Xu MQ, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, Le Merrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- Thesleff I. The genetic basis of tooth development and dental defects. Am j Med Genet Part A. 2006;140:2530–2535. doi: 10.1002/ajmg.a.31360. [DOI] [PubMed] [Google Scholar]
- Urist MR. Bone: formation by autoinduction. Science. 1965;150:893–899. doi: 10.1126/science.150.3698.893. [DOI] [PubMed] [Google Scholar]
- Virdi AS, Shore EM, Oreffo ROC, Li M, Connor JM, Smith R, Kaplan FS, Triffitt JT. Phenotypic and molecular heterogeneity in fibrodysplasia ossificans progressiva. Calcif Tissue Int. 1999;65:250–255. doi: 10.1007/s002239900693. [DOI] [PubMed] [Google Scholar]
- Wagner TU. Bone morphogenetic protein signaling in stem cells--one signal, many consequences. FEBS J. 2007;274:2968–2976. doi: 10.1111/j.1742-4658.2007.05839.x. [DOI] [PubMed] [Google Scholar]
- Wang CKL, Omi M, Ferrari D, Cheng HC, Lizarraga G, Chin HJ, Upholt WB, Dealy CN, Kosher RA. Function of BMPs in the apical ectoderm of the developing mouse limb. Dev Biol. 2004;269:109–122. doi: 10.1016/j.ydbio.2004.01.016. [DOI] [PubMed] [Google Scholar]
- Wieser R, Wrana JL, Massague J. GS domain mutations that constitutively activate T beta R-I, the downstream signaling component in the TGF-beta receptor complex. EMBO J. 1995;14:2199–2208. doi: 10.1002/j.1460-2075.1995.tb07214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wordinger RJ, Agarwal R, Talati M, Fuller J, Lambert W, Clark AF. Expression of bone morphogenetic proteins (BMP), BMP receptors, and BMP associated proteins in human trabecular meshwork and optic nerve head cells and tissues. Mol Vision. 2002;8:241–250. [PubMed] [Google Scholar]
- Wordinger RJ, Clark AF. Bone morphogenetic proteins and their receptors in the eye. Exp Biol Med. 2007;232:979–992. doi: 10.3181/0510-MR-345. [DOI] [PubMed] [Google Scholar]
- Wordinger RJ, Fleenor DL, Hellberg PE, Pang IH, Tovar TO, Zode GS, Fuller JA, Clark AF. Effects of TGF-beta2, BMP-4, and gremlin in the trabecular meshwork: implications for glaucoma. Invest Opthalmol Visual Sci. 2007;48:1191–1200. doi: 10.1167/iovs.06-0296. [DOI] [PubMed] [Google Scholar]
- Wozney JM, Rosen V, Celeste AJ, Mitsock LM, Whitters MJ, Kriz RW, Hewick RM, Wang EA. Novel regulators of bone formation: molecular clones and activities. Science. 1988;242:1528–1534. doi: 10.1126/science.3201241. [DOI] [PubMed] [Google Scholar]
- Yi SE, Daluiski A, Pederson R, Rosen V, Lyons KM. The type I BMP receptor BMPRIB is required for chondrogenesis in the mouse limb. Development. 2000;127:621–630. doi: 10.1242/dev.127.3.621. [DOI] [PubMed] [Google Scholar]
- Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, Lin HY, Bloch KD, Peterson RT. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nature Chemical Biology. 2008;4:33–41. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Schwarz EM, Rosier RN, Zuscik MJ, Puzas JE, O'Keefe RJ. ALK2 functions as a BMP type I receptor and induces Indian hedgehog in chondrocytes during skeletal development. J Bone Miner Res. 2003;18:1593–1604. doi: 10.1359/jbmr.2003.18.9.1593. [DOI] [PubMed] [Google Scholar]