

Abstract
Peptide amphiphiles are a class of molecules that combine the structural features of amphiphilic surfactants with the functions of bioactive peptides and are known to assemble into a variety of nanostructures. A specific type of peptide amphiphiles are known to self-assemble into one-dimensional (1D) nanostructures under physiological conditions, predominantly nanofibers with a cylindrical geometry. The resultant nanostructures could be highly bioactive and are of great interest in many biomedical applications, including tissue engineering, regenerative medicine and drug delivery. In this context, we highlight our strategies for using molecular self-assembly as a toolbox to produce peptide amphiphile nanostructures and materials and efforts to translate this technology into applications as therapeutics. We also review our recent progress in using these materials for treating spinal cord injury, inducing angiogenesis, and for hard tissue regeneration and replacement.
Keywords: peptide amphiphiles, self-assembly, peptides, nanofibers, one-dimensional nanostructures, bionanotechnology, regenerative medicine
1. Introduction
The cultural and economic pressures from humans seeking high quality of life and long lifespan have prompted the development of new strategies for the practice of medicine. This has coincided with, and likely promoted, the emergence of bionanotechnology. The current question is not whether bionanotechnology will have an impact on our quality of life but rather how to best harness this technique for a wide range of medical applications and when the resulting products will be brought to the clinic. Over the past decade, the focus of nanoscience has shifted from the development of novel nanostructures and the investigation of their formation mechanisms to the exploration of their potential applications. A broad range of nanostructures, from carbon nanotubes, buckyballs, inorganic nanoparticles, and dendrimers to self-assembled micelles and nano-objects of natural or synthetic molecules, have been evaluated in disease diagnostics, drug delivery, tissue engineering and regenerative medicine. The approval of Abraxane by the Food and Drug Administration (FDA) in 2005, the first medicine that employs nanotechnology, recognized these efforts. In this particular nanomedicine, an anticancer drug, taxol, was loaded into protein nanoparticles, appearing to be less toxic and more effective when used for metastatic breast cancer treatments. With a number of nanomedicine or nano-enhanced drugs in clinical use and many others entering preclinical and clinical trials, bionanotechnology has begun to revolutionize traditional medical therapies.
The rapidly growing needs for new medical therapies continue to challenge the field of nanotechnology and nanoscience. The earliest examples of nanomedicines are essentially reformulations of existing drugs using nanostructured materials; however, the development of future medicines that could be more predictive, preemptive, personalized and regenerative, demands customized fabrication of nanostructures with properties specifically tailored for the desired medical purpose. This consequently requires a better structural and functional control of materials and drugs at the molecular level. This is where self-assembly can make a major contribution, allowing for the necessary control of bioactive architectures with adjustable sizes, shapes, and more importantly, surface chemistries, that can emulate three-dimensional structures of biological proteins.
Molecular self-assembly presents a very attractive strategy to construct nanoscale materials due to its simplicity in application1–3. Furthermore, it is still the only practical approach for producing a variety of nanostructures4. This free energy driven process spontaneously organizes molecules into ordered structures at multiple length scales. The greatest merit of this method for fabricating nanoscale objects is that structural features of the final assemblies can be readily and finely tuned by molecular chemistry, assembling environment (pH, solvents, co-assembling molecules, and temperature), and assembly kinetics. For applications of these self-assembled materials, the ultimate goal is not necessarily to produce thermodynamically stable structures but for the material to achieve its desired function. Thus, kinetically trapped, or metastable, structures could enhance the diversity of these materials offering structures that can maintain function within the desired timescale or novel properties that vary with time. For example, materials used for artificial cell scaffolds should support and signal transplanted or native cells and then degrade once they are no longer needed. In order to achieve maximal function in a minimally invasive way, self-assembling networks or hydrogels that can degrade within a controlled timescale have great potential for cell delivery.
Peptides have been recognized as a very useful building block for creating self-assembling nanostructures for medical applications due to their inherent biocompatibility and biodegradability. In particular, β-sheet forming peptides demonstrate the extraordinary ability to assemble into one-dimensional (1D) nanostructures through intermolecular hydrogen bonding5. Interactions among 1D peptide-based nanostructures can lead to the formation of three-dimensional networks. The chemical design versatility of peptides, in combination with their ability to adopt specific secondary structures, provides a unique platform for the design of materials with controllable structural features at the nanoscale. Additionally, the biodegradability, further tunable by incorporation of specific amino acid sequences or control over their self-assembled structures, allows for the construction of bioactive hydrogels that can mimic the structure and function of native extracellular matrix. Though the large-scale synthesis of proteins comprising hundreds of residues still remains a formidable challenge, oligopeptides can be produced rather easily using standard solid-phase synthesis. These oligopeptides could serve as an effective, low-cost alternative for functional mimicry of large proteins. The relationship of function and structure in natural proteins is pervasive, with the spatial organization provided by specific folding and residue interactions directly contributing to the functional activity. Through self-assembly, we hope to precisely combine functional peptide sequences with the versatile structure of self-assembling synthetic molecules to produce customized nanoscale engineered biomaterials for various medical applications.
Over the past decade, our laboratory has designed and synthesized a broad range of amphiphilic molecules to create self-assembling biomaterials6–10. A broad class of amphiphiles known as peptide amphiphiles (PAs) incorporate a short hydrophobic block, in most cases an alkyl chain, onto one end of a short peptide sequence with overall hydrophilicity relative to the alkyl chain1,2,5,9,10. These molecules combine the structural features of amphiphilic surfactants with the functions of bioactive peptides. The amphiphilic nature of the molecules allows the specific presentation of hydrophilic peptide signals on the surfaces of the assembled nanostructures. The peptide amphiphiles developed in our laboratory self assemble into high-aspect-ratio nanofibers under specific solution conditions (pH, ionic strength, and temperature). From the standpoint of medical applications, we are interested in using individual assemblies to encapsulate and deliver small hydrophobic drugs, and employing three-dimensional networks of these high-aspect-ratio nanofibers as bioactive scaffolds to support, deliver, and signal cells. In this paper, we will focus on the work from our laboratory to synthesize PA nanostructures and translate this technology into therapies for regenerative medicine. We highlight our strategies for using molecular self-assembly as a toolbox to produce functional materials. Additionally we will discuss our recent work interfacing these materials with biology, specifically treating spinal cord injury, inducing angiogenesis, and constructing useful materials for hard tissue regeneration and replacement. Interesting work from other research groups regarding the self-assembly of peptide amphiphiles can be found elsewhere11,12 and references therein.
2. Self-assembly of Peptide Amphiphiles
2.1 Supramolecular Design
Figure 1 shows the chemical structure of a representative PA molecule, which is composed of four key structural features.9,13 Region 1, the hydrophobic domain, typically consists of a long alkyl tail. Region 2 consists of a short peptide sequence capable of forming intermolecular hydrogen bonding. Region 3 contains charged amino acids for enhanced solubility in water and for the design of pH and salt responsive nanostructures and networks. Region 4 typically is used for the presentation of bioactive signals, usually through epitopes that can interact with cells or proteins. The first generation of PAs in our laboratory was designed specifically to form cylindrical nanofibers under physiological conditions9,10. The internal packing of these PA molecules into high-aspect-ratio nanofibers is illustrated in Figure 1B.
Figure 1.

(A) Molecular Structure of a representative peptide amphiphile with four rationally designed chemical entities. (B) Molecular graphics illustration of an IKVAV-containing peptide amphiphile molecule and its self-assembly into nanofibers; (C) Scanning electron micrograph of the IKVAV nanofiber network formed by adding cell media (DMEM) to the peptide amphiphile aqueous solution; (D) Transmission electron micrograph of the IKVAV nanofibers13,27.
The first and arguably the most important design element of the PA is the strongly amphiphilic nature of the molecule. The amphiphilicity that results from the incorporation of the hydrophobic alkyl tail allows the self-assembled nanostructures to specifically present peptide signals on the nanofiber periphery. This enables the engineering of different types of bioactive epitopes on the nanofiber surfaces. The hydrophobic tails are tunable by using different alkyl chain lengths or by using different hydrophobic components10. In most of our examples, the hydrophobic block is introduced by coupling palmitic acid with the N-terminal amine of a short peptide on resin.
The second design element is the short peptide sequence immediately adjacent to the hydrophobic segment. This short peptide is typically composed of hydrophobic amino acids that have strong propensity to form intermolecular hydrogen bonding, typically in the form of β-sheets. This unique feature leads to the one dimensional nature of the self-assembled nanostructures. These 1D nanostructures can further entangle into networks. In addition, the molecular packing within a cylindrical geometry allows for the presentation of biological signals at very high density on the fiber surface. Recently, we have found that this region of the PA molecule can also be modified to control the mechanical properties of the resulting nanofiber networks14, as well as provide control over the shape of these one-dimensional nanostructures15.
The third element incorporated into the design of PAs is the addition of charged amino acids following the hydrophobic peptide sequence. The number of charged amino acids added should be enough to guarantee good solubility and assist with peptide purification but not too many as to interfere with the self-assembly of PAs into 1D nanostructures under physiological conditions. Since these charged amino acids are relatively weak acids and weak bases, nanofiber growth can be triggered by changing solution pH or simply raising the concentration of screening electrolytes in the solution. Depending on the amount and type of added salts, the resulting PA nanofibers can consequently bundle and form a robust network. This salt or pH-responsive design element is critical for developing injectable therapies for minimally invasive percutaneous delivery. This allows unassembled PA molecules to be combined with bioactive entities, such as growth factors, DNA or glycosaminoglycans, or with cells to form a liquid cocktail with very low viscosity. Upon injection into the tissue, electrolytes in the physiological environment can immediately induce the self-assembly of PAs into nanofibers and subsequent formation of gelled networks encapsulating the desired payload.
The last element in the design of PAs is the incorporation of different peptide epitopes that can be tailored for various purposes without changing the cylindrical geometry. For use in biological studies, epitopes that promote cell adhesion are frequently incorporated into PAs. It is known that cells adhere to the extracellular matrix (ECM) through the formation of a focal adhesion complex between cell membrane integrins and ECM proteins16,17. The incorporation of such cell adhesion sequences into networks of one-dimensional PA assemblies allows for both structural and functional mimicry of native ECM. One commonly used short peptide epitope for biological adhesion is RGDS, often in an abbreviated RGD form. This epitope exists on many ECM proteins, notably fibronectin, and has been shown to be specifically responsible for fostering cell adhesion to these proteins18,19. As a functional component in biological studies, this epitope has been frequently incorporated into PAs9,10,20–31. Another common epitope is the laminin-derived IKVAV, a pentapeptide sequence crucial for cell attachment, migration, and neurite outgrowth32,33. The IKVAV epitope has been specifically incorporated into PAs as a bioactive sequence for neural applications13,27,34. In order to limit the negative effects epitope incorporation may have on the assembly and gelation behavior of PA molecules, and further to allow the epitope to access its bioactive structural conformation, a short spacer (e.g. one or two glycines) is often used between the charged groups and the epitope.
2.2 Assembly Mechanisms
The driving forces that govern self-assembly of PAs in water arise from at least three major energy contributions: hydrophobic interactions of the alkyl tails, hydrogen bonding among the middle peptide segments, and electrostatic repulsions between the charged amino acids. The first two are attractive forces that tend to promote the aggregation of PA molecules, whereas electrostatic repulsions from the charged components favor disassociation of PA molecules. The final assemblies, including size, shape and interfacial curvature, reflect a delicate balance of each of these energy contributions. In water, individual PA molecules are expected to assume a tapered shape due to the hydrophobicity of the alkyl tail and the hydration of the incorporated charged groups. Simulations by Tsonchev and coworkers predict that tapered objects prefer a spherical geometry that allows molecules to pack more effectively35. However, studies of PA assemblies using microcopy techniques, such as transmission electron microscopy (TEM), scanning electron microscopy (SEM) and atomic force microcopy (AFM) predominantly reveal cylindrical nanofibers, though frequently with different lengths.
For the assembly of PAs in water, it is not the tapered shape of a single PA molecule but rather the sheet-like structure of a group of PAs linked together through intermolecular hydrogen bonding that determines the interfacial curvature of the final assemblies. To evaluate this claim, we have used molecular simulations to study the self-assembly behavior of PAs in water36. This simple model takes into account only hydrophobic interaction and intermolecular hydrogen bonding (Figure 2). Without considering any effect from hydrogen bonding, the “pure” hydrophobic interaction of these molecules produces micelles of finite size. This micellization follows a closed association pathway that involves a dominant nucleation mechanism above a critical micellization temperature and/or concentration. In contrast, the system with only “pure” hydrogen bonding follows an open association scenario that is characterized by a broad distribution of one-dimensional β-sheets resulting from step-by-step aggregation of molecules. Within a system in which both hydrophobic interaction and hydrogen bonding coexist, the phase behavior and assembly kinetics differ depending on the strength of intermolecular bonding. In a relatively weak hydrogen bonding system, spherical micelles may form with random β-sheets interspersed throughout the corona. The ordering of β-sheets is compromised in order to fit into the spherical interfacial curvature. Increasing the hydrogen bonding energy breaks the spherical symmetry and eventually results in long one-dimensional cylindrical fibers where β-sheets grow along the long axis of the nanofiber. Thus, it is this combined effect of intermolecular hydrogen bonding among the peptide segments and the hydrophobic collapse of alkyl tails that leads to the formation of cylindrical nanofibers in dilute aqueous solutions. Hartgerink and coworkers investigated the role of hydrogen bonding in the self-assembly of peptide amphiphiles and found that the four amino acids closest to the core of the nanofiber determine the ability of a PA to form the cylindrical nanostructure.37 Spherical micelles are formed when the hydrogen bonding associated with these four amino acids is disrupted.
Figure 2.

Schematic phase diagram of peptide amphiphile assemblies. The phase diagram includes regions with (a) free molecules, (b) spherical micelles, (c) micelles with β-sheets on the outside forming the corona, (d) long cylindrical fibers, (e) stacks of parallel sheets, (f) single β-sheets, and (g) the amorphous aggregate phase36.
2.3 Nanofiber Internal Structures
The molecular level packing within PA nanofibers determines their morphological and structural as well as functional properties, including the persistence length, the packing density and order of epitopes presented on the surfaces. As a result, the internal packing within PA nanofibers can greatly affect molecular dynamics and bio-signaling abilities of these nanostructures and therefore their performance as biomaterials. Our studies have shown that the ability of avidin binding to a biotin functionalized nanofiber surface was highly dependent on the architecture and therefore molecular packing of PA molecules within the nanostructures25.
We have studied the internal molecular packing of nanofibers using spectroscopic techniques, such as circular dichroism (CD), nuclear magnetic resonance (NMR), transmission infrared spectroscopy (IR), polarization modulation-infrared reflection-absorption spectroscopy (PM-IRRAS), and UV-vis absorption and photoluminescence measurements. These studies reveal that PA cylindrical nanofibers are composed of β-sheets that are oriented parallel to the long axis of the fibers38. Stern-Volmer quenching studies on PAs with covalently attached tryptophan and pyrene fluorophores have shown that the shell of PA nanofibers remains well-solvated in the assembled/gel states39. Chromophores buried deeper within the aggregates demonstrated a weaker but still significant response to aqueous quenchers in comparison to those located closer to the nanofiber surface. In contrast, various degrees of internal order of the hydrophobic tails were detected using infrared techniques, depending on the molecular architecture and the peptide sequence38. These results also suggest that order of the hydrophobic segment correlates, to some extent, with order within the peptidic domains. PAs with a branched peptide domain yielded nanofibers with the lowest degree of internal order. Our studies on PAs derivatized with diacetylene-containing hydrophobic tails support this conclusion that a higher degree of internal order exists in the nanofibers formed by the linear PA40. In this example, we polymerized diacetylene groups present in the hydrophobic component of PA molecules and were able to create polydiacetylene backbones. Since this reaction is well known to be a topotactic reaction, polymerization within the nanofiber demonstrated a high degree of internal order. Interestingly, it was also found that the chirality established within the β-sheet region can induce chirality in hydrophobic tails40. These findings provide insight for the rational design of PA molecules with the well-controlled epitope presentation. For example, a lower degree of internal order may be beneficial to present epitopes on the nanofiber surface that can be easily accessed by protein receptors, whereas a high degree of internal order might be crucial for templated mineralization on the nanofiber surface.
2.4 Strategies to Program Self-Assembly
Control of PA nanostructures and their subsequent gelation behavior can be achieved through the manipulation of the molecular forces that contribute to the self-assembly process. This can be implemented through molecular design, variation of the assembly environment, and introduction of co-assembling molecules. First, the chemical structure of the PA defines its functionality at the smallest length scale. The dominant molecular interactions that drive the assembly process can be varied depending on the chemical conformation, allowing for control over the assemblies of these molecules. Accordingly, we have investigated the effects of altering the length of the hydrophobic tail and the number of charged groups10. While the length and the diameter of the resulting nanofibers, as well as the conditions required for their self-assembly and gelation, are influenced by these modifications, the cylindrical shape of nanofibers remains the same regardless of the number of charged residues or the hydrophobic tail lengths when the peptide segment consists of a sufficient number of β-sheet forming hydrophobic amino acids.
The shape of assembled nanostructures can be controlled through the design of the middle peptide segment. Initially, we found that the self-assembly of PAs is remarkably tolerant to changes within this middle peptide segment, always forming cylindrical nanofibers. However, recent work has found that by introducing hydrophilic amino acids in this domain, specific sequences of peptide amphiphiles self-assemble in such a way as to eliminate all interfacial curvature and generate completely flat nanobelts (Figure 3)15. The key structural feature required for this nanobelt formation appears to be an alternating hydrophobic and hydrophilic amino acid sequence within this middle peptide segment. Peptides with alternating hydrophobic and hydrophilic amino acids are known to have a strong propensity to form β-sheet structures. The studied structural motif of VEVE, when adopting an extended β-strand conformation, orients the hydrophilic and hydrophobic side chains on opposite sides of the peptide backbone. In aqueous conditions, the valine surfaces have a strong tendency to associate with each other in order to minimize their solvent exposure, resulting in dimerization of two peptides into a lipid-like molecule containing two hydrophobic alkyl tails. These dimeric lipid-like molecules favor a flat (bilayer) packing geometry over a cylindrical one when assembled. When we disrupted the alternating hydrophobic and hydrophilic amino acid sequence by exchanging the VEVE peptide segment with VVEE, the resulting nanostructures regain their interfacial curvature, forming traditional cylindrical nanofibers under exactly the same conditions. Another way of interpreting the change from curved to flat assemblies is to consider that, in some sequences, the forces of hydration between the alkylated β-sheets are dominant to the energetics of side chain contacts.
Figure 3.

Giant (ultralong and wide) nanobelts assembled from a peptide amphiphile containing four amino acids and an alkyl tail. (a) Chemical structure of the peptide amphiphile. (b–d) AFM images of peptide nanobelts at different scanning sizes. The assembled nanobelts are the dominant structures in the assembly system (almost artifact free). (e and f) AFM images of a single-layer and a double-layer nanobelt morphology. (g) AFM amplitude image of (f). (h) CD spectrum of the peptide nanobelt solution at a concentration of 0.05 wt % proves the existence of β-sheet secondary structure in the supramolecular assemblies15.
The second strategy to control PA nanostructures utilizes screening of the electrostatic repulsion among the charged groups. A variation of solution pH could induce a reversible transition between the unassembled PA state and the assembled PA state through the protonation or deprotonation of carboxylate or ammonium groups, respectively. Addition of inorganic salts, such as CaCl2, can also trigger self-assembly of PAs through charge screening. The process, however, is typically not reversible due to the difficulty of removing the added salts. Though charge screening has a great effect on the PA assembly kinetics, nanofibers are often observed to assume a cylindrical geometry with or without salts. On the other hand, the strategy of altering charge density on PA molecules is very effective not only in controlling nanostructure formation but also gel formation since it should promote bundling of the fibers. The concentration, electronic structure, and hydration of counterions were found to significantly influence PA self-assembly and the mechanical properties of gels41.
Co-assembly of multiple PAs offers a third method of producing novel nanostructures. The functional value of mixing different PA molecules is to form nanostructures that can multiplex bioactive signals in a synergistic way. Mixing of molecules bearing different epitopes offers the possibility to use this multiplexing of signals to regulate cellular activity. In this strategy, we have combined two oppositely charged PAs in aqueous solution at physiological pH, each bearing a different biological signal27. Electrostatic attractions produce mixed nanofibers that simultaneously present two biological signals on the nanofiber surface. The degree of mixing was further studied by CD and FRET42. The CD data suggest strong co-assembly of different PAs at various mixing ratios and indicates a possible appearance of microdomains along the length of the nanofibers.
We have also demonstrated the possibility of mixing PAs with opposite peptide polarities43. A new class of peptide amphiphiles was designed and synthesized in which the peptide sequence has a reverse polarity. Using solid-phase peptide synthesis, traditional PAs synthesized in our laboratory have either a free acid or a free amide group presented on the C-terminus, and the N-terminus is amidated with an alkyl chain. Considering the possibility that epitopes may require a free N-terminus for their bioactivity, we developed a new synthetic route by introducing the alkyl tail on an unnatural amino acid that can be coupled to the resin, allowing for the peptide synthesis to result in the N-terminal domain on the periphery of the molecule43. The secondary structure and thermal stability of co-assembled PAs with opposite peptide polarities were investigated using CD, NMR, and FTIR. These co-assemblies show unusual thermal stability when compared to assemblies composed of only one type of PA molecule.
It is also possible to control the nanofiber length by mixing PA molecules with a template molecule. We designed and synthesized a dumbbell-shaped template molecule with an oligo(p-phenylene ethynylene) core44. In this templating approach, the hydrophobic backbone of the molecule was designed to guide the self-assembly of PAs into nanofibers of a targeted length. AFM studies revealed the expected aggregate structures commensurate with the length of the template molecules when a PA molecule was mixed with the template molecule at a molar ratio of 200 to 1.
Another strategy that has been explored is to produce hybrid nanostructures by mixing PA molecules with polymers that carry opposite charges. In recent work, we reported that when a solution of high molecular weight hyaluronic acid (HA) was mixed with a solution of a PA bearing opposite charges, immediate formation of a solid membrane was observed at the interface (Figure 4)45. When the denser HA solution is placed on top of the PA solution, the HA fluid slowly sinks into the PA solution. Continuous growth of the membrane at the progressively renewed liquid-liquid interface eventually results in the formation of a liquid-filled sac. These sacs can be made instantly by injecting HA solution directly into the PA solution, and the resulting robust macroscopic structures offer opportunities in many research areas. This synergistic assembly also allows us to produce membranes of arbitrary shape and highly ordered strings. The strategy of utilizing the electrostatic attractions between a large charged molecule and oppositely charged self-assembling small molecules has great potential for the development of highly functional materials organized across multiple length scales.
Figure 4.
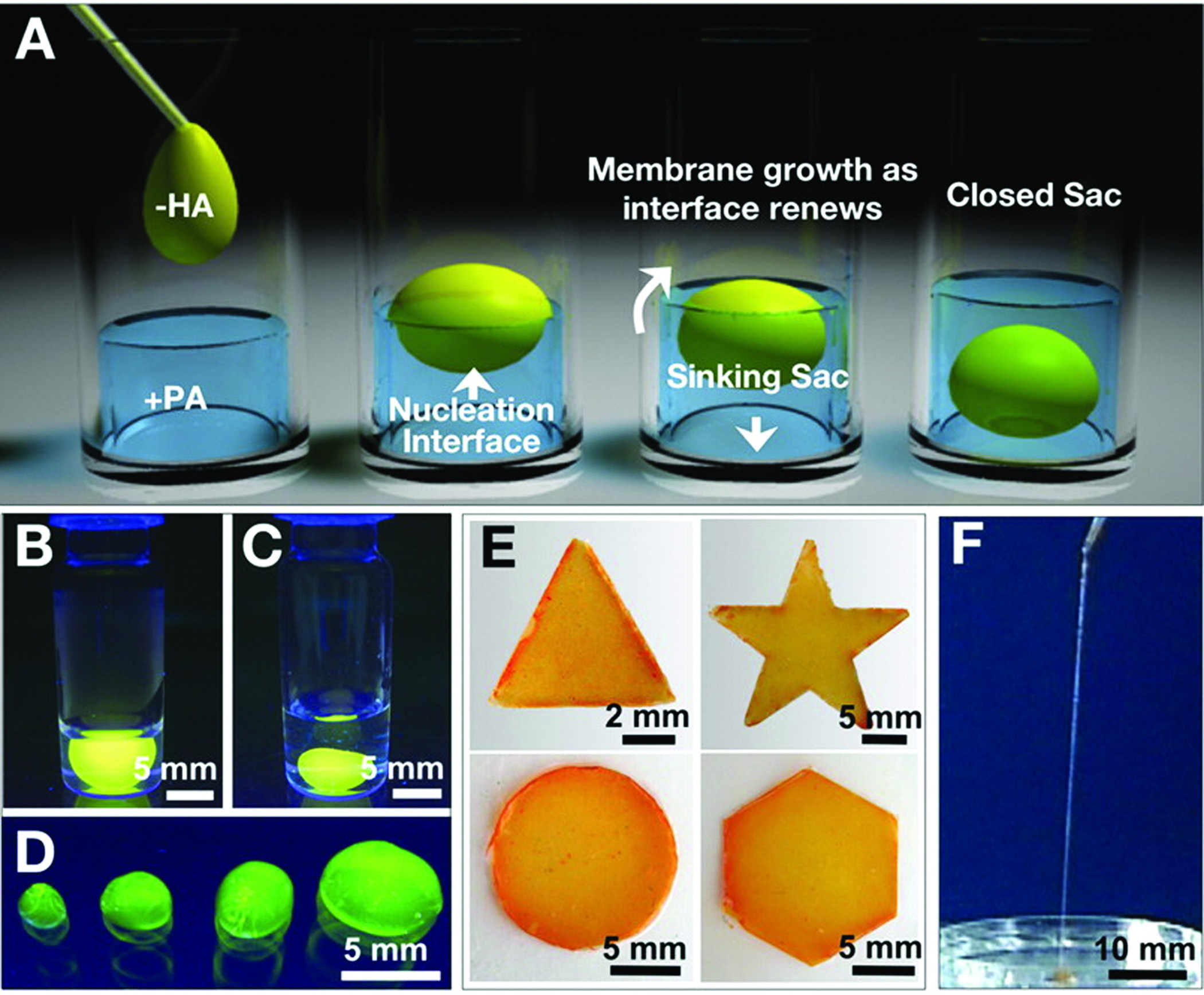
(A) Schematic representation of one method to form a self-sealing closed sac. A sample of the denser negatively charged biopolymer solution is dropped onto a positively charged peptide amphiphile solution. (B) Open and (C) closed sac formed by injection of a fluorescently tagged hyaluronic acid solution into a PA solution. (D) Self-assembled sacs of varying sizes. (E) PA-HA membranes of different shapes created by interfacing the large- and small-molecule solutions in a very shallow template (~1 mm thick). (F) Continuous strings pulled from the interface between the PA and HA solutions45.
3. Peptide Amphiphile Nanofibers
We are interested in the applications of individual assembled peptide amphiphile nanofibers as well as the nanofiber networks that they form. Given the strongly amphiphilic nature of the PA monomer, assembled nanofibers of PA molecules consist of a hydrophobic core and a hydrophilic shell in aqueous conditions. One obvious application is the encapsulation of small molecules, such as hydrophobic drugs or semiconducting organic molecules, or the surface decoration of other hydrophobic nanostructures such as carbon nanotubes. Another important application of PA nanofibers is the utilization of its charged surface to guide the assembly of inorganic nanoparticles and template inorganic mineralization.
3.1 Encapsulation of Small Molecules and Carbon Nanotubes
Pyrene was selected to establish the potential for encapsulation and delivery of hydrophobic small molecule drugs within the PA nanofiber46. The encapsulation and aggregation of pyrene within the core of nanofibers constructed from PA molecules with either cholesterol or palmitic acid hydrophobic domains was inferred by the formation of a pyrene excimer. This excimer formation is also observed using peptide amphiphiles in which the pyrene is covalently linked to the hydrophobic tail. In another study, 3,4-ethylenedioxythiophene (EDOT), a monomeric precursor to a conducting polymer, was also successfully encapsulated within the hydrophobic core of PA nanofibers47. Following oxidative polymerization, poly(3,4-ethylenedioxythiophene) (PEDOT) chains formed confined within the core of the insulating peptide amphiphile nanofiber. Such redox-active supramolecular nanostructures formed by self-assembly of PAs with epitopes could serve as both biologically and electronically active matrices for the transduction of biological signals.
Peptide amphiphiles have also been used for the dispersion of carbon nanotubes in water48. The encapsulation was confirmed using TEM and optical absorbance spectroscopy. This non-covalent functionalization process occurs without compromising the structural, electronic, or optical properties of carbon nanotubes. The assembly of peptide amphiphile molecules on the surfaces of carbon nanotubes has the potential for adding biofunctionality to a shell coating these one-dimensional conductors.
3.2 Cross-linking Chemistry for Enhanced Robustness
Various cross-linking chemistries have been incorporated into PA molecules in order to enhance the physical and chemical robustness of these supramolecular nanofibers. The first approach was to incorporate cysteine residues within the peptide sequence given their ability to form disulfide bonds under oxidizing conditions9,10. Peptide amphiphiles containing several cysteines were shown to be capable of intermolecular hydrogen bonding for nanofiber formation. After oxidation, the formation of intermolecular disulfide bonds effectively crosslinked the nanofiber into a high molecular weight polymer with fibrous morphology using the concept of covalent capture. Due to the nature of disulfide bonds, this cross-linking method is reversible using reducing agents. Another method, used by us and others, to polymerize PA molecules within their supramolecular assemblies is to incorporate diacetylene groups in the hydrophobic segment40,49,50. Using UV irradiation, we have successfully polymerized PA molecules within nanofibers through a polydiacetylene backbone while retaining their cylindrical shape. This polymerization strategy in filamentous PA nanostructures does not interfere with the use of β-sheets to control nanostructure shape, as the distance between flanking substituents is not changed through the diacetylene polymerization process. We also found that this topotactic polymerization yields more highly conjugated backbones when the peptidic segment of the PAs has a linear vs. a branched architecture. Most importantly, the efficient polymerization observed in assemblies of linear PA molecules offers strong evidence for the internal order that prevails in these supramolecular nanostructures.
3.3 Mineralization Templated on Peptide Amphiphile Nanofiber Surfaces
The chemistry on the surface of the PA nanofibers can be customized to create templates for mineralization. The first PAs designed by our laboratory templated biomimetic hydroxyapatite crystals similar to those found in bone and dentin9. In this initial work, PAs containing phosphoserine residues were used to template mineralization of hydroxyapatite, the inorganic component of mammalian bone and teeth51. Phosphorylated serine is known to be abundant in proteins that template calcium phosphate in mineralized tissues51–53. This templating process produced hydroxyapatite mineral in two-dimensional systems with the crystallographic c-axis aligned with the long axis of PA nanofibers, mimicking a similar crystallographic orientation of hydroxyapatite crystals in bone with respect to the long axis of collagen fibers. The significance of the enzyme alkaline phosphatase was later established for the biomineralization of PA networks in three dimensions54. The temporal control from enzymatically mediated harvesting of phosphate ions enables PA nanofibers presenting phosphorylated serine to nucleate hydroxyapatite on their surface, allowing for spatially controlled biomimetic mineralization of a three-dimensional PA scaffold.
The robustness and pH-stability of cross-linked PA fibers allow for mineralization under different conditions. PAs have been utilized as effective substrates to template the nucleation and growth of cadmium sulfide (CdS) nanocrystals (Figure 5)55. In this study, gels of cross-linked nanofibers were first broken up through a combination of mechanical agitation and sonication to form a dilute aqueous suspension. Cadmium nitrate tetrahydrate (Cd(NO3)2·4H2O) was then added as a source of Cd2+. The mixture was subsequently exposed to hydrogen sulfide (H2S) gas, initiating the templated growth of cadmium sulfide nanocrystals. Nanostructures with different morphologies were formed when H2S was diffused through suspensions containing various molar ratios of Cd2+/PA. In systems containing modest amounts of cadmium (Cd2+/PA = 2.4:1), individual CdS nanocrystals of 3–5 nm in diameter can be clearly seen decorating the PA fibers.
Figure 5.

Bright field TEM micrographs of CdS mineralized suspensions of PA fibers at various Cd2+:PA molar ratio. (a) Cd2+:PA = 2.4:1; (b) Cd2+:PA = 24:1. Left inset shows an electron diffraction pattern corresponding to the CdS zinc blende structure. Right inset shows an enlargement of a portion of an encapsulated fiber highlighting the strip of lower electron density through the middle corresponding to the hydrophobic core. (c) Cd2+:PA = 240:1. (d) High-resolution TEM micrograph showing the lattice structure of CdS nanocrystals grown on a PA fiber. Fiber axis is parallel to the long axis of the micrograph55.
3.4 Peptide Amphiphile Nanofibers for Magnetic Resonance Imaging (MRI)
Magnetic resonance imaging has advanced to be one of the most powerful diagnostic techniques in clinical radiology, providing three-dimensional structures of living tissue at near cellular resolution. A current difficulty in developing new generations of MR agents is to obtain significant contrast over long periods of time for in vivo studies. We have covalently attached a derivative of 1,4,7,10-tetraazacyclododecane-1,4,7,10 tetraacetic acid (DOTA), a molecule that chelates gadolinium, to PA molecules in order to increase the relaxivity of the MR agent21,22. These MR agent-conjugated PA molecules can assemble into either cylindrical nanofibers or spheres in aqueous solution. As a result of self-assembly, the rotational correlation time of the MR agent was increased, suggesting an enhanced relaxivity. It was also found that the molecule’s relaxivity and imaging utility can be influenced by the tethered position of the DOTA derivative on the PA molecules. The relaxivity was enhanced when the DOTA/ Gd(III) complex was closer to the hydrophobic region. We have also shown that the scaffolds containing these MR agent-conjugated PAs can be imaged by MRI techniques22.
3.5 Light-Triggered Self-Assembling Peptide Amphiphiles
Light provides a non-invasive stimulus to trigger the self-assembly of PAs into nanofibers. To enable the self-assembly of the pH-responsive PAs inside liposomes, a photoacid generator (PAG) was introduced to lower the pH upon exposure to light. PA self-assembly triggered by light activation was achieved within this spatially confined environment using various model PAs designed to self-assemble under acidic conditions56. The PAs were first encapsulated within the aqueous interior of liposomes simply by using a PA solution to hydrate a phospholipid film. The encapsulation and light-triggered PA assembly were supported through the use of confocal fluorescent microscopy, SEM, FTIR and CD. This method could enable the preparation of nanofiber bundles displaying bioactive epitopes to be delivered as densely packed nanofibers within the interior of liposomes for use in targeting specific tissues or tumors.
We have also designed PA molecules incorporating photo-cleavable groups between the hydrophobic and peptide domains57. The designed PA contains a palmitoyl tail, a 2-nitrobenzyl group, and an oligopeptide segment GV3A3E3, in which the 2-nitrobenzyl group is covalently linked to the N-terminal amide of the glycine residue and can be cleaved by irradiation at 350 nm. TEM of these derivatives revealed interesting hierarchical assemblies of PA molecules consisting of quadruple helices of PA assemblies with a uniform helical pitch of ~92 nm. The quadruple helical structures were found to be comprised of four individual nanofibers of 11 nm diameter. Interestingly, after a 5-min irradiation at 350 nm, which cleaves the nitrobenzyl group from PA molecules, the helical structures transform into the typical cylindrical nanofibers.
3.6 Surface Patterning of Peptide Amphiphile Nanofibers
Surfaces formed from peptide-based patterns could be useful in molecular recognition events for biosensing or in control of the growth and orientation of nanocrystals on aligned nanofiber arrays. Recently, we demonstrated a novel technique, termed sonication-assisted solution embossing (SASE), that allows the simultaneous self-assembly, alignment, and patterning of PA nanofibers over large areas26,58. In this soft lithographic technique, the PA solution was first placed within the topographical features of an elastomeric stamp. The self-assembly is driven by solvent evaporation while subjecting the system to ultrasonic agitation. After removing the stamp, bundles of aligned nanofibers were observed to orient parallel to the channels. This alignment is the result of a synergistic interaction between a liquid crystal transition that occurs during solvent evaporation and the spatial confinement imposed by the substrate topology. With a properly designed mold, it is even possible to guide nanofibers around turns using this SASE technique. These PA nanofibers can also be deposited using a dip-pen direct patterning technique59.
In addition, we have used microfabrication techniques to develop well-defined topographical patterns of bioactive PA gels with microscale and nanoscale resolution (Figure 6)60. Self-assembly of PAs containing both the polymerizable diacetylene groups and the RGDS cell adhesion epitope were designed for use within the molds, yielding networks of photo-polymerized nanofibers that were shaped into topographical patterns containing holes, posts, or channels. Various topographical patterns were fabricated from both networks of randomly dispersed PA nanofibers or pre-aligned PA nanofibers. In both cases, silica slabs were chosen as rigid substrates due to their transparency to UV light of 254 nm that was used to polymerize PA nanofibers. For the fabrication of topographical patterns consisting of random PA nanofibers, a drop of fresh PA solution was placed on top of the silica substrates that had been modified to be hydrophilic by exposure to oxygen plasma. PDMS molds were then placed over the PA solutions. The PA was self-assembled inside the molds by placing the whole silica/PA/PDMS setup into a sealed chamber filled with ammonium hydroxide (NH4OH) vapor. Following the self-assembly, PA nanofibers were polymerized by UV irradiation. Finally, the PDMS mold was removed to yield topographically patterned PA substrates. In the case of fabricating microtextures of aligned PA nanofibers, PA solutions were aged for a sufficiently long time to allow the formation of PA nanofibers. Then, the large-area alignment of PA nanofibers was achieved by directly pipetting the aged PA solutions while dragging the pipette tip onto the silica substrate. The molds were then stamped and polymerized as in the unaligned samples. These prepared surfaces were then used as cell substrates, examining the effect of the topographical and nanoscale features on the migration, alignment, and differentiation of mesenchymal stem cells (MSCs). It was found that the topographical patterns containing aligned PA nanofibers are able to promote the alignment of MSCs. Osteoblastic differentiation of these cells was enhanced when cultured on a substrate of randomly oriented nanofibers patterned into microtextures of holes.
Figure 6.

Fabrication techniques and resulting PA structures. (a–e) The fabrication process starts by either (a) dropping freshly dissolved PA for microtextures with randomly oriented nanofibers, or (b) dragging an aged PA solution for microtextures with aligned nanofibers on a silica substrate. (c) Then, a PDMS mold was used to cover the PA solution while allowing it to conform to the mold, self-assemble into nanofibers, and gel by exposure to ammonium hydroxide (NH4OH). The PA gel was then polymerized under UV irradiation and released from the mold to realize the PA microtextures. The process in (a,c) was used to achieve well-defined three-dimensional PA structures with (d) randomly oriented nanofibers including (f) removable layers with microtextures or (g) pores and surface microtextures such as (g) channels, (i) holes, (j) posts, and (k) two-level topographies with features down to 5 µm in size. On the other hand, following the process in (b,c), microtextures with (l,m) channels and holes were also achieved but with aligned nanofibers (inset in m)60.
4. Peptide Amphiphile Nanofibers for Regenerative Medicine
PA nanofibers can bundle and entangle to form self-supporting three-dimensional networks under physiological conditions at concentrations on the order of 1% by weight. This transition typically requires charge screening by multivalent ions within physiologic media, and enables the encapsulation of cells within this nanofiber matrix20. Cells entrapped within PA nanofiber matrices have been found to be viable for weeks and can continue to proliferate, indicating no cell cycle arrest. Embedded TEM images have revealed internalization of the PA by these encapsulated cells, and biochemical assays demonstrated evidence of PA metabolism (Figure 7). This suggested that networks of PA nanofibers could serve as components of an artificial extracellular matrix to signal cells. The architectural resemblance of PA nanofibers to filamentous structures in natural mammalian extracellular matrices allows them to function as a highly biomimetic artificial matrix. PA nanofibers not only display signals to cell receptors, but also provide structural support to the encapsulated cells and could be eventually metabolized into nutrients. The gelation kinetics of peptide amphiphile nanofiber networks were also found to be tunable by changing the internal peptide sequence while keeping the bioactive domain constant, producing nanofiber gels with a range of gelation times.14 When the region promoting β-sheet formation was changed to include more bulky and hydrophilic residues, SLSLGGG instead of AAAAGGG, the time for gelation was found to increase significantly. This design gives flexibility to these systems as an injectable biomaterial, allowing for the gelation time to be tuned depending on the desired application. The bioactivity of the terminal IKVAV epitope was unaffected by modifying the β-sheet domain, with no significant change observed for the viability or neurogenesis of encapsulated neural progenitor cells.
Figure 7.

(A) TEM micrograph of a cell entrapped in the nanofibrillar matrix internalizing the PA nanofibers. (B) Intermediate magnification of the region marked b in (A) showing the formation of intracellular membrane delineated compartments filled with nanofibers. (C) High magnification micrograph of the nanofibers in the area marked c in (B)20.
In order to enhance matrix-cell adhesion, we have incorporated the RGDS epitope into the PA molecular design. The covalent architecture to present this epitope on the nanofiber surface can be varied through orthogonal protecting group chemistry23. We have synthesized PAs that can present RGDS in linear, branched, double-branched or cyclic geometries. PAs designed with a branched architecture show improved cell attachment and migration, owing to the decreased molecular packing density of epitopes on the nanofiber surfaces31. Importantly, co-assembly of bioactive and non-bioactive PA molecules can generally enable control over the epitope densities presented to cells. This characteristic of PA systems has been specifically studied by our laboratory using the RGDS epitope31. A PA system presenting RGDS was also developed and optimized for biological adhesion as a matrix for the therapeutic delivery bone marrow mononuclear cells61. In addition, branched RGDS-presenting PAs can be used to coat a traditional tissue engineering scaffold, poly(glycolic acid). The PA-modified scaffold enhanced the adhesion of primary human bladder cells62. This demonstrated the potential to use PAs to functionalize polymeric materials traditionally used for tissue engineering in order to enhance their bioactivity.
4.1 Neural Regeneration
The laminin-derived IKVAV sequence has been incorporated into PAs in order to enhance neural attachment, migration, and neurite outgrowth. Our previous work showed that neural progenitor cells cultured within our IKVAV PA quickly undergo selective and rapid differentiation into neurons while the formation of astrocytes was largely suppressed13. This observed selective differentiation was even greater within our PA networks relative to that observed when cells are cultured with laminin, the natural protein bearing the IKVAV sequence on which the synthetic peptide is based. The markers used to establish cell lineage in these experiments were the proteins β-tubulin III and glial fibrillary acidic protein (GFAP), markers for neurons and astrocytes, respectively (Figure 8). This observed response is presumably due to the high epitope density presented on the nanofiber surface. Control experiments using a mixture of soluble IKVAV peptide and nanofibers of a PA without the IKVAV epitope did not reveal the same effect.
Figure 8.

Neural Progenitor Cells (NPCs) cultured under different experimental conditions. (A and B) The same field of view in two different planes of focus showing immunocytochemistry of NPCs encapsulated in IKVAV-PA gels at 1 day. Differentiated neurons were labeled for β-tubulin (in green) and differentiated astrocytes (glial cells) were labelled for GFAP (in orange). All cells were Hoechst stained (in blue). (C) Immunocytochemistry of an NPC neurosphere encapsulated in an IKVAV-PA nanofiber network at 7 days. The large extent of neurite outgrowth was typical of the cells examined. (D) Percentage of total cells that differentiated into neurons (β-tubulin+). The IKVAV-PA gels had significantly more neurons compared to both laminin and poly-D-lysine (PDL) controls at both 1 and 7 days (*P<0.05, **P<0.01). (E) Percentage of total cells that differentiated into astrocytes (GFAP+). The IKVAV-PA gels had significantly fewer astrocytes compared to both laminin and PDL controls by 7 days (*P<0.05)13.
These findings in vitro encouraged us to use this material to treat spinal cord injury, where the formation of a glial scar by astrocytes and inhibitory factors prevents axonal regeneration after injury63. When applied in a mouse spinal cord injury model, the IKVAV nanofiber showed very promising results34. The PA was found to reduce cell death at the injury site and decrease astrogliosis involving a hyperplastic state of astroctyes. Additionally, the injected nanofiber gel increased the number of oligodendroglia, the cells responsible for the formation of the myelin sheath around neurons in the central nervous system. Histologically we also observed regeneration of descending motor neurons and ascending sensory neurons in animals treated with the self-assembling IKVAV PA networks at the site of injury. This observed histological change was also accompanied by observations of behavioral improvement, demonstrating enhanced hind limb functionality in treated rodent models34.
4.2 Hard Tissue Replacement and Regeneration
Because of their mechanical properties and reasonable biocompatibility, metals such as titanium and its alloys are frequently used to restore function in the human skeleton, for example to stabilize fractures or reconstruct joints. PA nanofibers have been used to functionalize metal implants for bone replacement. A nickel-titanium (NiTi) shape-memory alloy, frequently used for stents, bone plates, and artificial joints, was modified through covalent attachment of peptide amphiphile nanofibers30,64. This modification with RGDS-presenting PAs significantly increased the number of adhered pre-osteoblastic cells, while most cells detached from the non-functionalized NiTi. In another method, RGDS-presenting PAs were gelled within porous titanium scaffolds28,29. The PA nanofibers were used to fill the pores of the titanium foam, providing support for pre-osteoblastic cells seeded within the foams. Cells within these PA-titanium foam hybrids were viable, proliferative, and exhibited signs of osteogenic differentiation.
Enamel is the outermost coating of vertebrate teeth and the hardest tissue in the body. Regeneration of this tissue remains a very challenging task in regenerative medicine, in part because ameloblasts that are responsible for the production of enamel during development subsequently apoptose, preventing enamel regeneration during adulthood. Branched RGDS PAs were used as a scaffold in vitro for ameloblast-like cells and primary enamel organ epithelial (EOE) cells which initiate the process of enamel formation26. When incubated in the presence of branched RGDS PA nanofibers, these cells demonstrated enhanced proliferation and increased expression of amelogenin and ameloblastin, both proteins secreted by ameloblasts during enamel formation. In the organ culture model, PA was injected into embryonic mouse incisors. EOE cell proliferation and differentiation into ameloblasts was observed, as evidenced by their expression of enamel specific proteins such as integrin 6, amelogenin, and ameloblastin (Figure 9).
Figure 9.

Branched RGD-peptide amphiphiles (BRGD-PA) influencing ameloblast cell behaviour after injection into the enamel organ epithelia (solid arrow) of incisor primordial maintained for 2 days in organ culture. The complete incisor is shown at low power in A, B, and C with the injection site marked by a dashed line box. (A–G) Higher-magnification images of representative tissue sections from the site of injection (solid arrow). Od, odontoblasts; Am, ameloblasts; SI, stratum intermedium. A–D show cell proliferation using BrdU incorporation into cellular DNA. The stem cell compartment at the caudal end of the incisor is marked with an open arrow and corresponds to sites of high cell division. (A) Sham treatment consisting of incisor primordia injected with PBS showed little BrdU incorporation into the DNA of enamel organ epithelial cells at the injection site. (B) Incisor primordia injected with control PA showed but a few BrdU-labelled cells at the injection site. (C) Incisor primordia injected with BRGD-PA showed cells have incorporated BrdU as a consequence of increased cell division. (D) Bar graph of the total number of cells incorporating BrdU from the injections sites shown in A–C. (E–G) Immunodetection of integrin 6 protein expression from representative sections taken from incisor primordia at the site of PA injection (solid arrow). (E) Relatively low levels of integrin 6 protein expression localized to enamel organ cells and part of dental papillae in sham treated incisor primordia. (F) Integrin 6 protein expression at the injection site of the control-PA. (G) Increased integrin 6 protein expression among the cells at the injection site of the BRGD-PA. The cells at the injected site are shown to be proliferating in C26.
4.3 Angiogenesis
Enhancing angiogenesis, the process by which existing blood vessels remodel into new vasculature, is of great interest to treat ischemic diseases of the heart, peripheral vasculature, and chronic wounds65,66. The heparin-binding peptide amphiphile (HBPA) was designed with a Cardin-Weintraub heparin-binding domain to specifically bind heparan sulfate-like gylcosaminoglycans (HSGAG). This polysaccharide screens charges on the PA molecules and triggers PA self-assembly into nanofibers, thus displaying the polysaccharide on the nanofiber surface42,67. This gives these nanofibers the capability to capture the many proteins known to have heparin-binding domains such as fibroblast growth factor 2 (FGF-2), bone morphogenetic protein 2 (BMP-2) and vascular endothelial growth factor (VEGF). The heparin-binding character of this HBPA was verified using isothermal titration calorimetry (ITC) measurements, with an association constant of 1.1×107. Due to charge screening by the negatively charged heparin, the binding effect leads the positively charged PA molecules to self-assemble into a nanofiber gel containing both heparin and HBPA. FGF-2 and VEGF are well known to participate in angiogenesis signaling and thus HBPA nanofibers were hypothesized by our laboratory as promoters of blood vessel formation67,68. When nanogram quantities of FGF-2 was added, the heparin-presenting HBPA prolonged release of growth factor relative to a PA network prepared using divalent phosphate ions to screen charges and promote self-assembly. Moreover, the material, when combined with VEGF and FGF-2, induced significant angiogenesis in a rat corneal assay (Figure 10). Scrambling of the heparin-binding sequence presented on HBPA was found to diminish the observed angiogenic effect in a tube formation assay using endothelial cells67. Since the scrambled molecule had a similar association constant with heparin as the HBPA, this difference in bioactivity was attributed to a slower off-rate for the interaction between heparin and HBPA, leading to more efficient growth factor signaling and also stabilizing the protein from enzymatic degradation.
Figure 10.

In vivo angiogenesis assay. Rat cornea photographs 10 days after the placement of various materials at the site indicated by the black arrow. Heparin-nucleated PA nanofiber networks with growth factors show extensive neovascularization. Controls of collagen, heparin, and growth factors (B) and collagen with growth factors (C) show some neovascularization. Heparin with growth factors (D), and collagen with heparin. The bar graph (F) contains values for the average and maximum length of new blood vessels and the area of corneal neovascularization. A 100% value in the area measurement indicates that the cornea is completely covered, and a 100% value in the length parameters indicates that the new vessels are as long as the diameter of the cornea (bars are 95% confidence levels, * p < 0.05 when PA-heparin gel was compared to collagen gel with growth factors, ** p < 0.005 when PA-heparin gel with growth factors was compared to all of the other controls). PA nanofibers with heparin, PA solution with growth factors, and growth factors alone did not result in measurable neovascularization (values not shown in graph)68.
Type I diabetes mellitus is a condition resulting from autoimmune destruction of the body’s insulin-producing beta cells. One therapeutic strategy involves transplantation of donor islets, pancreatic cell aggregates of which beta cells are a part, to treat this condition. However, the efficacy of this strategy is limited, at least in part, by poor islet viability and engraftment following transplant.69 Given these limitations with engraftment, the angiogenic potential of HBPA was explored to potentially enhance vascularization at the transplant site and promote islet engraftment.70 At the present time, islets from donors have to be transplanted into the liver because of its high degree of vasculature. HBPA, combined with FGF-2 and VEGF, significantly enhanced vasculature in the omentum, an intraperitoneal fat mass that in principle would be a more desirable, less invasive site for transplantation of islets. When islets were transplanted with HBPA-heparin nanofiber networks and angiogenic growth factors into the omentum, the cure percentage of diabetic mice restored to a normoglycemic state was significantly enhanced relative to untreated diabetic mice. This was not the case for the administration of islets with HBPA-heparin alone or with growth factors alone.
5. Summary and Future prospects
This paper has reviewed most of our published work on peptide amphiphiles as the molecular building block to generate bioactive nanofibers of high aspect ratio. We highlighted our strategies to control the self-assembly of these molecules through rational design of the molecular structures as well as manipulation of their self-assembly environment. We also discussed recent progress made in the application of these materials as cell scaffolds, drug carriers, and bioactive networks for central nervous system regeneration, angiogenesis, as well as regeneration of bone and enamel. Using in vivo models, we are currently testing these materials to regenerate bone, cartilage and heart tissue.
There are many interesting challenges ahead with these systems, particularly a supramolecular level understanding of why they are so effective at cell signaling, so they can be further improved to tackle dynamically much more complex signaling pathways in stem cells and cancer cells. With advances in systems biology and proteomics, much more sophisticated signaling devices will become accessible through this supramolecular platform. Another interesting direction is the formulation of hybrid systems with macromolecules and/or inorganic components. In many of these future materials, we expect the crafting of highly dynamic systems far from equilibrium that exhibit adaptable behavior in their environment.
Acknowledgments
Funding for our work on peptide amphiphiles has been supported by the U.S. Department of Energy-Basic Energy Sciences (DE-FG02-00ER45810), by the National Science Foundation (DMR-0605427, MRSEC at Northwestern University DMR-0520513, and NSEC at Northwestern University NSEC EEC-0647560), and by National Institute of Health (NIH/NIBIB 5 R01 EB003806, NIH/NIDCR 5 R01 DE015920-3, NIH/NCI 5 U54 CA119341-04, NIH/NINDS 5 P50 NS054287-04). M.J. Webber is supported by the Northwestern Regenerative Medicine Training Program (RMTP) NIH award 5T90-DA022881. The authors are grateful to the Biological Imaging Facility (BIF), Integrated Molecular Structure Education and Research Center (IMSERC), NUANCE Center (EPIC, NIFTI, Keck-II) and Keck Biophysics Facility at Northwestern University.
References
- 1.Palmer LC, Stupp SI. Accounts of Chemical Research. 2008;41:1674–1684. doi: 10.1021/ar8000926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palmer LC, Velichko YS, de la Cruz MO, Stupp SI. Philosophical Transactions of the Royal Society a-Mathematical Physical and Engineering Sciences. 2007;365:1417–1433. doi: 10.1098/rsta.2007.2024. [DOI] [PubMed] [Google Scholar]
- 3.Stupp SI, Pralle MU, Tew GN, Li LM, Sayar M, Zubarev ER. Mrs Bulletin. 2000;25:42–48. [Google Scholar]
- 4.Service RF. Science. 2005;309:95–95. doi: 10.1126/science.309.5731.95. [DOI] [PubMed] [Google Scholar]
- 5.Hartgerink JD, Zubarev ER, Stupp SI. Current Opinion in Solid State & Materials Science. 2001;5:355–361. [Google Scholar]
- 6.Hwang JJ, Iyer SN, Li LS, Claussen R, Harrington DA, Stupp SI. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:9662–9667. doi: 10.1073/pnas.152667399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klok HA, Hwang JJ, Hartgerink JD, Stupp SI. Macromolecules. 2002;35:6101–6111. [Google Scholar]
- 8.Klok HA, Hwang JJ, Iyer SN, Stupp SI. Macromolecules. 2002;35:746–759. [Google Scholar]
- 9.Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684–1688. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 10.Hartgerink JD, Beniash E, Stupp SI. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:5133–5138. doi: 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lowik D, van Hest JCM. Chemical Society Reviews. 2004;33:234–245. doi: 10.1039/b212638a. [DOI] [PubMed] [Google Scholar]
- 12.Kokkoli E, Mardilovich A, Wedekind A, Rexeisen EL, Garg A, Craig JA. Soft Matter. 2006;2:1015–1024. doi: 10.1039/b608929a. [DOI] [PubMed] [Google Scholar]
- 13.Silva GA, Czeisler C, Niece KL, Beniash E, Harrington DA, Kessler JA, Stupp SI. Science. 2004;303:1352–1355. doi: 10.1126/science.1093783. [DOI] [PubMed] [Google Scholar]
- 14.Niece KL, Czeisler C, Sahni V, Tysseling-Mattiace V, Pashuck ET, Kessler JA, Stupp SI. Biomaterials. 2008;29:4501–4509. doi: 10.1016/j.biomaterials.2008.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cui H, Muraoka T, Cheetham AG, Stupp SI. Nano Letters. 2009;9:945–951. doi: 10.1021/nl802813f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giancotti FG, Ruoslahti E. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 17.Zaidel-Bar R, Cohen M, Addadi L, Geiger B. Biochem Soc Trans. 2004;32:416–420. doi: 10.1042/BST0320416. [DOI] [PubMed] [Google Scholar]
- 18.Pierschbacher MD, Ruoslahti E. Proc Natl Acad Sci U S A. 1984;81:5985–5988. doi: 10.1073/pnas.81.19.5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pierschbacher MD, Ruoslahti E. Nature. 1984;309:30–33. doi: 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- 20.Beniash E, Hartgerink JD, Storrie H, Stendahl JC, Stupp SI. Acta Biomaterialia. 2005;1:387–397. doi: 10.1016/j.actbio.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 21.Bull SR, Guler MO, Bras RE, Meade TJ, Stupp SI. Nano Letters. 2005;5:1–4. doi: 10.1021/nl0484898. [DOI] [PubMed] [Google Scholar]
- 22.Bull SR, Guler MO, Bras RE, Venkatasubramanian PN, Stupp SI, Meade TJ. Bioconjugate Chemistry. 2005;16:1343–1348. doi: 10.1021/bc050153h. [DOI] [PubMed] [Google Scholar]
- 23.Guler MO, Hsu L, Soukasene S, Harrington DA, Hulvat JF, Stupp SI. Biomacromolecules. 2006;7:1855–1863. doi: 10.1021/bm060161g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guler MO, Pokorski JK, Appella DH, Stupp SI. Bioconjugate Chemistry. 2005;16:501–503. doi: 10.1021/bc050053b. [DOI] [PubMed] [Google Scholar]
- 25.Guler MO, Soukasene S, Hulvat JF, Stupp SI. Nano Letters. 2005;5:249–252. doi: 10.1021/nl048238z. [DOI] [PubMed] [Google Scholar]
- 26.Huang Z, Sargeant TD, Hulvat JF, Mata A, Bringas P, Koh CY, Stupp SI, Snead ML. Journal of Bone and Mineral Research. 2008;23:1995–2006. doi: 10.1359/JBMR.080705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niece KL, Hartgerink JD, Donners J, Stupp SI. Journal of the American Chemical Society. 2003;125:7146–7147. doi: 10.1021/ja028215r. [DOI] [PubMed] [Google Scholar]
- 28.Sargeant TD, Guler MO, Oppenheimer SM, Mata A, Satcher RL, Dunand DC, Stupp SI. Biomaterials. 2008;29:161–171. doi: 10.1016/j.biomaterials.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 29.Sargeant TD, Oppenheimer SM, Dunand DC, Stupp SI. Journal of Tissue Engineering and Regenerative Medicine. 2008;2:455–462. doi: 10.1002/term.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sargeant TD, Rao MS, Koh CY, Stupp SI. Biomaterials. 2008;29:1085–1098. doi: 10.1016/j.biomaterials.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Storrie H, Guler MO, Abu-Amara SN, Volberg T, Rao M, Geiger B, Stupp SI. Biomaterials. 2007;28:4608–4618. doi: 10.1016/j.biomaterials.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 32.Sephel GC, Tashiro KI, Sasaki M, Greatorex D, Martin GR, Yamada Y, Kleinman HK. Biochem Biophys Res Commun. 1989;162:821–829. doi: 10.1016/0006-291x(89)92384-x. [DOI] [PubMed] [Google Scholar]
- 33.Tashiro K, Sephel GC, Weeks B, Sasaki M, Martin GR, Kleinman HK, Yamada Y. J Biol Chem. 1989;264:16174–16182. [PubMed] [Google Scholar]
- 34.Tysseling-Mattiace VM, Sahni V, Niece KL, Birch D, Czeisler C, Fehlings MG, Stupp SI, Kessler JA. Journal of Neuroscience. 2008;28:3814–3823. doi: 10.1523/JNEUROSCI.0143-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsonchev S, Schatz GC, Ratner MA. Nano Letters. 2003;3:623–626. [Google Scholar]
- 36.Velichko YS, Stupp SI, de la Cruz MO. Journal of Physical Chemistry B. 2008;112:2326–2334. doi: 10.1021/jp074420n. [DOI] [PubMed] [Google Scholar]
- 37.Paramonov SE, Jun HW, Hartgerink JD. Journal of the American Chemical Society. 2006;128:7291–7298. doi: 10.1021/ja060573x. [DOI] [PubMed] [Google Scholar]
- 38.Jiang HZ, Guler MO, Stupp SI. Soft Matter. 2007;3:454–462. doi: 10.1039/b614426h. [DOI] [PubMed] [Google Scholar]
- 39.Tovar JD, Claussen RC, Stupp SI. Journal of the American Chemical Society. 2005;127:7337–7345. doi: 10.1021/ja043764d. [DOI] [PubMed] [Google Scholar]
- 40.Hsu L, Cvetanovich GL, Stupp SI. Journal of the American Chemical Society. 2008;130:3892–3899. doi: 10.1021/ja076553s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stendahl JC, Rao MS, Guler MO, Stupp SI. Advanced Functional Materials. 2006;16:499–508. [Google Scholar]
- 42.Behanna HA, Rajangam K, Stupp SI. Journal of the American Chemical Society. 2007;129:321–327. doi: 10.1021/ja062415b. [DOI] [PubMed] [Google Scholar]
- 43.Behanna HA, Donners J, Gordon AC, Stupp SI. Journal of the American Chemical Society. 2005;127:1193–1200. doi: 10.1021/ja044863u. [DOI] [PubMed] [Google Scholar]
- 44.Bull SR, Palmer LC, Fry NJ, Greenfield MA, Messmore BW, Meade TJ, Stupp SI. Journal of the American Chemical Society. 2008;130 doi: 10.1021/ja710749q. 2742-+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Capito RM, Azevedo HS, Velichko YS, Mata A, Stupp SI. Science. 2008;319:1812–1816. doi: 10.1126/science.1154586. [DOI] [PubMed] [Google Scholar]
- 46.Guler MO, Claussen RC, Stupp SI. Journal of Materials Chemistry. 2005;15:4507–4512. [Google Scholar]
- 47.Tovar JD, Rabatic BM, Stupp SI. Small. 2007;3:2024–2028. doi: 10.1002/smll.200600645. [DOI] [PubMed] [Google Scholar]
- 48.Arnold MS, Guler MO, Hersam MC, Stupp SI. Langmuir. 2005;21:4705–4709. doi: 10.1021/la0469452. [DOI] [PubMed] [Google Scholar]
- 49.Biesalski M, Tu R, Tirrell MV. Langmuir. 2005;21:5663–5666. doi: 10.1021/la0504558. [DOI] [PubMed] [Google Scholar]
- 50.Lowik D, Shklyarevskiy IO, Ruizendaal L, Christianen PCM, Maan JC, van Hest JCM. Advanced Materials. 2007;19 1191-+ [Google Scholar]
- 51.Palmer LC, Newcomb CJ, Kaltz SR, Spoerke ED, Stupp SI. Chemical Reviews. 2008;108:4754–4783. doi: 10.1021/cr8004422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Addadi L, Weiner S. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:4110–4114. doi: 10.1073/pnas.82.12.4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.George A, Bannon L, Sabsay B, Dillon JW, Malone J, Veis A, Jenkins NA, Gilbert DJ, Copeland NG. Journal of Biological Chemistry. 1996;271:32869–32873. doi: 10.1074/jbc.271.51.32869. [DOI] [PubMed] [Google Scholar]
- 54.Spoerke ED, Anthony SG, Stupp SI. Advanced Materials. 2009;21 doi: 10.1002/adma.200802242. 425-+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sone ED, Stupp SI. Journal of the American Chemical Society. 2004;126:12756–12757. doi: 10.1021/ja0499344. [DOI] [PubMed] [Google Scholar]
- 56.Lee HK, Soukasene S, Jiang HZ, Zhang SM, Feng WC, Stupp SI. Soft Matter. 2008;4:962–964. doi: 10.1039/b719486b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Muraoka T, Cui H, Stupp SI. Journal of the American Chemical Society. 2008;130:2946–2947. doi: 10.1021/ja711213s. [DOI] [PubMed] [Google Scholar]
- 58.Hung AM, Stupp SI. Langmuir. 2009;25:7084–7089. doi: 10.1021/la900149v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang HZ, Stupp SI. Langmuir. 2005;21:5242–5246. doi: 10.1021/la0501785. [DOI] [PubMed] [Google Scholar]
- 60.Mata A, Hsu L, Capito R, Aparicio C, Henrikson K, Stupp SI. Soft Matter. 2009;5:1228–1236. doi: 10.1039/b819002j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Webber MJ, Tongers J, Renault MA, Roncalli JG, Losordo DW, Stupp SI. Acta Biomaterialia. 2009 doi: 10.1016/j.actbio.2009.07.031. doi:10.1016/j.actbio.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harrington DA, Cheng EY, Guler MO, Lee LK, Donovan JL, Claussen RC, Stupp SI. Journal of Biomedical Materials Research Part A. 2006;78A:157–167. doi: 10.1002/jbm.a.30718. [DOI] [PubMed] [Google Scholar]
- 63.Silver J, Miller JH. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 64.Bansiddhi A, Sargeant TD, Stupp SI, Dunand DC. Acta Biomaterialia. 2008;4:773–782. doi: 10.1016/j.actbio.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Folkman J. Curr Mol Med. 2003;3:643–651. doi: 10.2174/1566524033479465. [DOI] [PubMed] [Google Scholar]
- 66.Folkman J, Klagsbrun M. Science. 1987;235:442–447. doi: 10.1126/science.2432664. [DOI] [PubMed] [Google Scholar]
- 67.Rajangam K, Arnold MS, Rocco MA, Stupp SI. Biomaterials. 2008;29:3298–3305. doi: 10.1016/j.biomaterials.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rajangam K, Behanna HA, Hui MJ, Han XQ, Hulvat JF, Lomasney JW, Stupp SI. Nano Letters. 2006;6:2086–2090. doi: 10.1021/nl0613555. [DOI] [PubMed] [Google Scholar]
- 69.Robertson RP. N Engl J Med. 2004;350:694–705. doi: 10.1056/NEJMra032425. [DOI] [PubMed] [Google Scholar]
- 70.Stendahl JC, Wang LJ, Chow LW, Kaufman DB, Stupp SI. Transplantation. 2008;86:478–481. doi: 10.1097/TP.0b013e3181806d9d. [DOI] [PMC free article] [PubMed] [Google Scholar]