

Abstract
Although congenic translocation of a segment from chromosome 10 from Lewis rat, containing an extracellular proteinase inhibitor gene, decreased blood pressure in Dahl-salt sensitive (DSS) rats, the relationship between the levels of matrix metalloproteinase (MMP), tissue inhibitor of metalloproteinase (TIMP), and cardiac function was unclear. In this study we investigated the cardiac effects of congenic translocation of a segment from chromosome 10 from Lewis rat, containing an extracellular proteinase inhibitor gene, in Dahl-salt sensitive rats. To test the hypothesis that left ventricular (LV) hypertrophy in DSS rats was due to high MMP and low TIMP levels and the decrease in blood pressure in congenic rats was associated with increase in proteinase inhibitor expression, cardiac function and levels of MMP and TIMP were determined in 16 weeks male DSS (D), Lewis (L) and congenic (CL-10) rats. Cardiac function was assessed by electrocardiography, echocardiography and a Millar catheter in LV cavity. LV MMP and TIMP levels were measured by Q-RT-PCR and Western blot analyses. In L, D and CL-10 rats, heart weight/body weight (g/g) were 3.73 ± 0.06, 4.45 ± 0.04 and 3.35 ± 0.05 × 10−3, respectively, suggesting significant (p < 0.05) LV hypertrophy (LVH) in D group. The ST duration was longer in D group compared with L group, suggesting coronary vasospasm, but normalized in CL-10 rats. The fractional shortening and ejection fraction were decreased in D group as compared with L group, but normalized in CL-10 groups. LV diameter was increased in D group as compared to L group, but normalized in CL-10 groups. The levels of MMP-9 were higher and TIMP were lower in D as compared to L groups, but normalized in CL-10 rats. Compared with control non-congenic Dahl rats, congenic rats exhibited lower blood pressure, amelioration of LV remodelling and dysfunction, as well as coronary abnormalities. In addition, congenic animals exhibited reduced myocardial expression of MMP-9, but increased expression of MMP-2 and TIMP-4 compared to non congenic animals. We concluded that the congenic transfer of TIMP ameliorated LV hypertrophy and cardiac dysfunction.
Keywords: MMP, TIMP, endothelial cardiomyocyte coupling, ECM remodelling, heart failure
Introduction
Despite the gigantic strides made towards the understanding of hypertensive heart disease, there is no link between the systemic hypertension, left ventricular hypertrophy (LVH) and extracellular matrix (ECM) remodelling. Remodelling by its very nature implies synthesis and degradation of ECM. Degradation of intricate connections between the cardiac muscle instigates the compensatory LVH (Mujumdar et al., 2001; Henderson et al., 2007). However, a critical balance between proteinase and anti-proteinase is necessary to maintain constitutive cardiac remodelling, LVH and regeneration of the connective tissue. Increase in proteinase/anti-proteinase ratio leads to chronic deterioration of muscle connections and systolic and diastolic heart failure. ECM remodelling interferes with integrin-mediated cell survival signalling (Hunt et al., 2002). This leads to formation of filamentous actin (F-actin), gaps between the cells, and disrupts endothelial-cardiomyocyte tight junctions (Lominadze et al., 2006), resulting in endothelial-cardiomyocyte (E-M) uncoupling. In the basement membrane of capillary endothelium of the heart, the MMPs reside in the latent ternary (MMP/NO/TIMP) complex (Tyagi & Hayden, 2003). During hypertensive heart disease TIMPs are oxidized and MMPs are activated. It is known that MMPs are good collagenases, but they are also excellent elastases. Therefore, because elastin turnover is relatively slower than collagen, it is replaced by oxidized collagen (Rucklidge et al., 1992). This leads to disruption of the cell-cell connection (Shastry & Tyagi, 2004) and alteration in the composition and concentration of inter cellular connective tissue proteins. This alters collagen/elastin ratio, disrupts connexin-43, and leads to accumulation of interstitial collagen (fibrosis) between the endothelium and cardiomyocyte. Fibrosis causes E-M disconnection (uncoupling) and attenuates NO diffusibility from endothelium to cardiomyocyte.
The homologous region on human chromosome 17 has been linked to chromosome 10 in DSS rats to blood pressure control (Julier et al., 1997). The rigorous analysis on the chromosome 10 in DSS rats and its relationship with blood pressure has demonstrated that QTL1 was responsible for blood pressure genes (Palijan et al., 2003). A congenic clone containing QTL1 (CL-10) rat was created by translocation of a segment of chromosome 10 from Lewis (normotensive control) rats to DSS rats. The blood pressure was normalized in CL-10 rats (Palijan et al., 2003). Interestingly, although both NOS2 and ACE genes were located in chromosome 10 in rats, they were found to be outside the blood pressure related genes (Deng, 1998; Garrett et al., 1998). The first gene in QTL1 in chromosome 10 was an extracellular proteinase inhibitor gene (Palijan et al., 2003). Therefore, we hypothesized that decrease in blood pressure in QTL1 congenic (CL-10) rats was associated with increase in proteinase inhibitor expression.
Methods
Reagents
Acetylcholine (ACH), nitroprusside (SNP), and endothelin-1 (ET-1) were obtained from Sigma Chemical Company. All reagents including the antibodies of MMPs and TIMPs were obtained from Sigma.
Animals
A congenic strain (CL-10) rat was created by replacing a segment of chromosome 10 in DSS rats and replacing with the segment from Lewis (normotensive control) rats. Two congenic strains male and female, designated CL-10, and were crossbred to produce F1 rats. Which then were intercrossed to produce F2 progeny. The F2 rats were genotyped for markers that resided in a region of interest. Male rats were weaned at 21 days of age, maintained on low salt diet (0.2% NaCl, Harlan Teklad 7034) for 2 weeks, and then fed a high-salt diet (2% NaCl) starting from 35 days (5 wks) and continue until 16 weeks. Sixteen-week-old male Dahl-salt sensitive (DSS) and Lewis controls rats were obtained from Harlan. All experiments were carried out in age-matched rats. A n = 6 were used in each group. Animals were maintained at 22 ± 2°C with 12:12 hour’s light dark cycle. Animals were handled in an accredited institute facility in accordance with the institutional animal care policies, and all of the research protocols conformed to the guiding principles for animal experimentation as enunciated by the Ethics Committee on Animal Research of the University of Louisville and the National Institutes of Health.
Electrocardiography
Rats were anaesthetized with intraperitoneal injection of Inactin (100 mg/kg body weight, Sigma Chemical Co), and were allowed to breath spontaneously during the procedures. A 3-leads ECG (Micro-Med Corp) was used to record animal electrocardiogram. The duration of S-T segments were recorded in milliseconds.
Echocardiography and hemodynamic parameters
Transthoracic echocardiographic studies were performed with a 12-MHz phased-array ultrasound system (Philips SONO-5500). M-Mode tracings of the left ventricle were recorded to measure inter-ventricular septal dimension, LV end-diastolic dimension, LV end-systolic dimension, and posterior wall dimension. The apical 4-chamber view was obtained, and, subsequently pulse-wave Doppler echocardiography was performed. To determine cardiac relaxation a pressure-tipped Millar catheter was inserted into the LV through the right common carotid artery. The aortic blood pressure (BP), heart rate (HR), and systolic and diastolic blood pressure (SBP & DBP) were measured. After arterial pressure measurements, the catheter was advanced to the LV and LVP, EDP and +dP/dt at maximum were measured. Pulsatile arterial pressure signal was analyzed by a computer using customized software (Micro-Med Corp) positioned at the level of heart. The duration of the in vivo experimental measurements was 2–3 hours. Because echocardiography and LV measurements were performed with inactin-anaesthetized rats, the issue of cardiodepression produced by anaesthesia must be acknowledged as a confounding factor in these experimental measurements.
Gravimetric parameters
At the end of in vivo measurements, body weight (BW), heart weight (HW), lung weight, liver weight, and kidney weight (KW) in grams were measured.
Endocardial endothelial-cardiomyocyte function
The “deli” shaped rings of the LV including the septum were cut transversely (i.e. O shaped), and were mounted in a tissue myobath. One of the two mounted wires was connected to a force transducer. The responses to endothelin-1 (ET-1), acetylcholine or nitroprusside were measured. Because capillary, including the endothelium, is embedded in the muscle (myocyte), the experiments in part measure contraction and/or relaxation of the tissue including the contribution of endothelium in cardiomyocyte relaxation. These experiments in part indicate cardiomyocyte endocardial endothelial coupling. The ET-1 is the primary cardiotonic agents in endothelium, therefore ET-1 was chosen. A dose–response curve of ET-1 was generated and maximum ET-1 dose was chosen. To prepare the stock solutions of concentrations of ET-1, acetylcholine and nitroprusside, the ET-1, ACH and SNP were based on weight measurements. A standard concentration was used across all tissues. In a typical experiment, the ring was stretched and brought to resting tension at which 1 μM ET-1 was added. At the maximum ET-1 contraction, acetylcholine (endothelial-dependent) or nitroprusside (endothelial independent) was added. To minimize the differences due to orientation of cardiac muscle, the rings were rotated 90° and contraction was measured again. The average of these two contractions was recorded. The percentage relaxation was calculated based on 100% contraction to 1 μM ET-1 and dose–response curves were generated. The ACH response tracings of CL-10 rats were similar to tracing of Lewis rats. The tension normalization is important for adequate comparison of responses of different preparations of cardiac rings to agonists. Therefore, we normalize the tension in grams with the weight of the ring in grams, prior to calculating the percentage relaxation to ACH. To avoid ischemia, oxygen at 20 psi was continuously bubbled through the myobath. This was sufficient to penetration of cardiotonic agent (Tyagi et al., 1999). The experiments were completed within 40 min during that time there was minimal injury due ischemia (Tyagi et al., 1999).
MMPs and TIMPs
Real time Q-RT-PCR will be performed for MMP-2, −9, and TIMP-4. The primers for MMP-2 are: 5′-gcactctggagcgaggatac-3′ and 5′-gccctcctaagc cagtctct-3′ (gene bank #NM-008610), 362 bp products. The primers for MMP-9 are: 5′-aagg caaaccctgtgtgttc-3′ and 5′-gtggttcagttgtggtggtg-3′, gene bank #NM-013599, 385 bp products. G3PDH (NM_001001303) sense 5′-TGAAGGTC GGTGTGAACGGATTTGGC-3′ and antisense 5′-CATGTAGGCCATGAGGTCCACCAC-3′; 650-bp amplification product. Western blot analysis of MMP-2, −9, TIMP-4 and actin was performed using respective antibodies (Sigma Chemical Company). The bands were scanned and normalized with actin and reported as arbitrary unit (AU).
Statistical analysis
Values are given as average+SD from n = 6 in each group. Differences between groups were evaluated by using a two-way ANOVA, followed by the Bonferroni post hoc test (Tarone, 1990), focusing on the respective effects of DSS-hypertension. The results of the two-way comparisons are reported as: ★, compared to Lewis; ★★, compared to DSS. A p < 0.05 was considered significant.
Results
Gravimetric data showed an increased heart weight/body weight ratio, indicating LVH in DSS rats. Interestingly, both LV and RV showed hypertrophy (Table I). There was increase in systemic blood pressure in DSS rats, suggesting both cardiac and vascular dysfunction in DSS rats. In CL- 10 rats, cardiac and vascular dysfunction was mitigated. There was decrease in liver but increase in kidney weights in DSS rats as compared to Lewis rats, indicating liver atrophy and renal hypertrophy, respectively, in DSS-hypertension. In CL-10 these parameters were improved (Table I).
Table I.
Hemodynamic, gravimetric and cardiac parameters of Lewis (L), DSS and CL-10 rats: Body weight (BW), heart weight (HW), lung weight, liver weight, and kidney weight (KW) in grams. Mean arterial pressure (MAP), systolic and diastolic (SBP and DBP, respectively) in mmHg and heart rate (HR) in beats/min were measured.
| L | DSS | CL-10 | |
|---|---|---|---|
| BW | 351 ± 8 | 364 ± 3 | 379 ± 36 |
| HW | 1.31 ± 0.06 | 1.62 ± 0.04★ | 1.27 ± 0.05★★ |
| HW/BW | 3.73 ± 0.06 | 4.45 ± 0.04★ | 3.35 ± 0.05★★ |
| LV | 1.06 ± 0.05 | 1.27 ± 0.06★ | 1.07 ± 0.06★★ |
| RV | 0.25 ± 0.04 | 0.35 ± 0.05★ | 0.20 ± 0.03★★ |
| KW | 1.46 ± 0.18 | 1.75 ± 0.22★ | 1.26 ± 0.11★★ |
| Lung | 1.48 ± 0.03 | 1.95 ± 0.10★ | 1.49 ± 0.03★★ |
| Liver | 18.5 ± 3.2 | 11.1 ± 0.6★ | 13.1 ± 1.5★★ |
| MAP | 96 ± 6 | 181 ± 6★ | 112 ± 5★★ |
| SBP | 106 ± 9 | 208 ± 7★ | 117 ± 6★★ |
| DBP | 85 ± 3 | 158 ± 7★ | 111 ± 4★★ |
| HR | 255 ± 5 | 311 ± 20 | 284 ± 13 |
p < 0.05 compared with L;
p < 0.05 compared with DSS.
Electrocardiography
The ECG data revealed increase in cardiac re-polarization (S-T) duration in DSS rats as compared to Lewis rats. In CL-10 rats, the S-T duration was normalized (Figure 1). It is known that during acute coronary occlusion in human and in rodents, there is increase in S-T duration. Although the ST segment elevation observed in DSS rats (Figure 1) could be due to many factors, we speculated that ST segment elevation may in part be due to coronary vasospasm.
Figure 1.

A typical 3-leads EKG of control (L), DSS and CL-10 rats: The bar graph represents S-T segment duration of cardiac re-polarization in milliseconds of L, DSS and CL-10 rats. Each bar is an average + SD from n = 6. ★p < 0.001 L vs DSS; ★★p < 0.002 DSS vs CL-10.
Echocardiography
The echocardiographic data revealed both systolic and diastolic dysfunction in DSS rats as compared to Lewis rats. There was increase in LV diameter in DSS rats as compared to Lewis rats. However, in CL-10 rats, both systolic and diastolic dysfunction was mitigated (Figure 2). The LV pressure was high in DSS rats as compared to Lewis rats. The time for cardiac relaxation (tau) was greater in DSS rats as compared to control rats. In CL-10 rats cardiac pressure and tau were normalized. The rate of rise (+dP/dt) and fall (−dP/dt) in cardiac pressure during cardiac cycle were decreased in DSS rats as compared to control rats. Interestingly, both +dP/dt and −dP/dt were normalized in CL-10 rats. Collectively, these data suggested that cardiac dysfunction in DSS rats was normalized by congene (Table II, Figure 3 and 4). Based on the representative pictures in Figure 3, there were differences in HR between the groups. Similar trend in HR was observed in average data shown in Table I. The max-P and EDP values for all three groups were different but similar to their respective groups, presented in Tables I and II. The bar graph in Figure 3 demonstrated the time for relaxation (Tau in ms). In Table II we presented the +dP/dt values. Because +dP/dt was preload dependent, in Figure 4 we plotted the ratio of +dP/dt/EDP. Because LV pressures in vivo were load-dependent, therefore, the apparent cardiac dysfunction in DSS rats was influenced by much higher afterload values.
Figure 2.
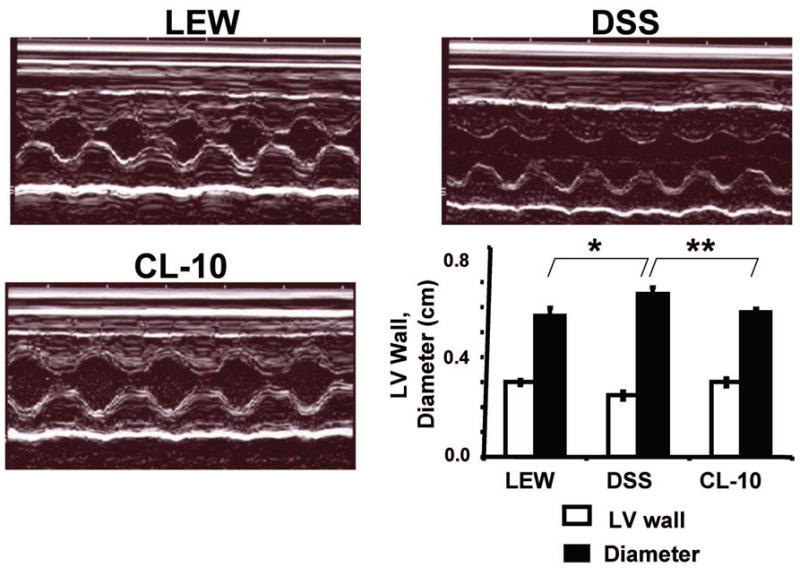
M-mode echocardiogram of L, DSS, and CL-10 rats: Bare graph represents LV wall and diameter in centimeters. Each bar is an average + SD from n = 6. LV diameter, ★p < 0.005 L vs DSS; ★★p < 0.02 DSS vs CL-10.
Table II.
Ejection fraction (EF) and fractional shortening (FS), left ventricle (LV) end-diastolic pressure (EDP), and maximum LV pressure (LVP) in mmHg. The first derivative in fall of LV pressure (−dP/dt) and rise in LV pressure (+dP/dt) in mmHg/sec were measured.
| L | DSS | CL-10 | |
|---|---|---|---|
| FS (%) | 54 ± 3 | 45 ± 3★ | 58 ± 1 |
| EF | 0.89 ± 0.02 | 0.80 ± 0.10★ | 0.91 ± 0.01★★ |
| EDP | 10 ± 3 | 35 ± 4★ | 15 ± 2★★ |
| LVP | 135 ± 4 | 186 ± 7★ | 140 ± 6★★ |
| −dP/dt | 6024 ± 510 | 2505 ± 298★ | 5000 ± 350★★ |
| +dP/dt | 6274 ± 260 | 2705 ± 287★ | 5048 ± 210★★ |
Figure 3.

LV pressure wave of L, DSS and CL-10 rats: Millar catheter inserted into LV of anesthetized rats and LV pressure was recorded. The bar graph represents time of LV relaxation (tau) in milliseconds. Each bar is an average + SD from n = 6. ★p < 0.002 L vs DSS; ★★p < 0.005 DSS vs CL-10.
Figure 4.
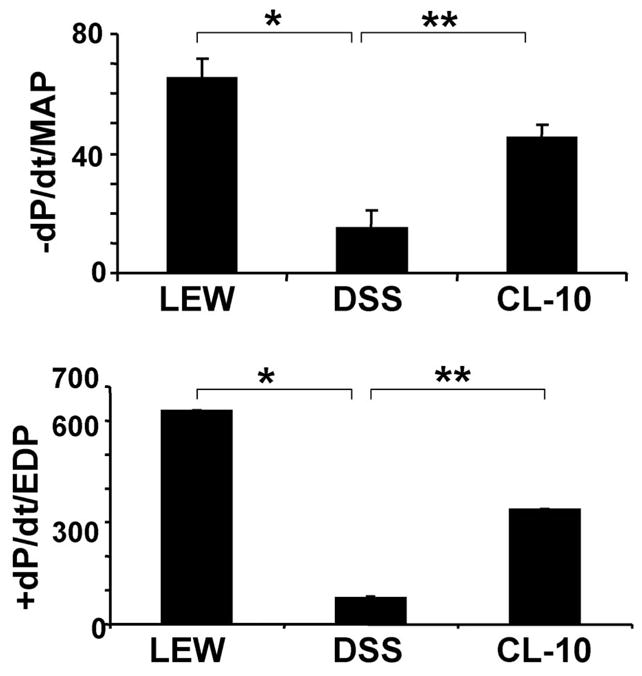
Rates of cardiac diastolic relaxation (−dP/dt) and systolic contraction (+dP/dt) of L, DSS and CL-10 rats: Because cardiac diastolic function depends on after load, therefore, −dP/dt was normalized with MAP, and because cardiac contractile function depends on preload, +dP/dt was normalized with EDP. Each bar is an average + SD from n = 6. ★p < 0.001 L vs DSS; ★★p < 0.002 DSS vs CL-10.
Endothelial-cardiomyocyte function
There was impairment in endothelial-dependent acetylcholine response in cardiac rings from DSS rats. The congenic translocation of chromosome-10 ameliorated the endothelial-dependent cardiac relaxation. There was no change on the response to nitroprusside in DSS, Lewis and CL-10 rats. These data suggested attenuation in endothelial-dependent cardiac function in DSS rats. The translocation of a part of chromodome-10 from Lewis to DSS rats mitigated the endothelial-dependent cardiac dysfunction (Figure 5).
Figure 5.
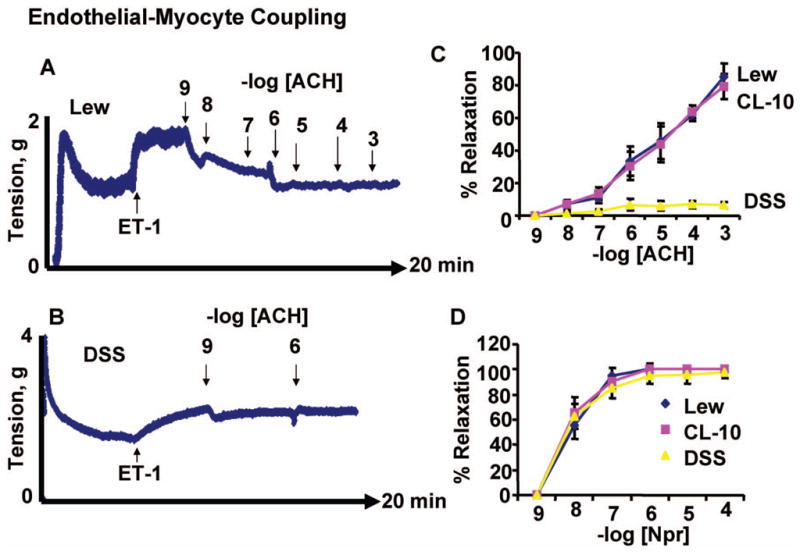
Endothelial-dependent cardiac relaxation: LV rings from L (A), DSS (B) and CL-10 rats were contracted with ET-1. Different doses of endothelial-dependent (acetylcholine, ACH, C) and endothelial-independent (nitroprusside, SNP, D) were added to the rings in a tissue myobath. The % cardiac relaxation was estimated. C and D, each data point represents average + SD from n = 6. Representative pictures of L and DSS groups are shown in panel A and B. In all experimental protocol rings were allowed fully to contract to ET-1 prior to adding ACH and measuring the responses to ACH. The concentration-response curve presented is the average of six independent experimental data points.
Expression of MMP and TIMP
Although we did not measure the MMP activity by zymography, we measured MMP levels by both PCR (gene expression) and by Western blot analysis (protein levels). The data suggested that MMP-2 and TIMP-4 were decreased; MMP-9 was increased in DSS rats as compared to controls. There was normalization of the expression and the levels of MMPs and TIMPs in CL-10 rats (Figure 6). Although the MMP and TIMP experiments only investigated gene expression and protein levels, and did not assess the activities of MMP and TIMP and therefore our conclusions may be speculative.
Figure 6.
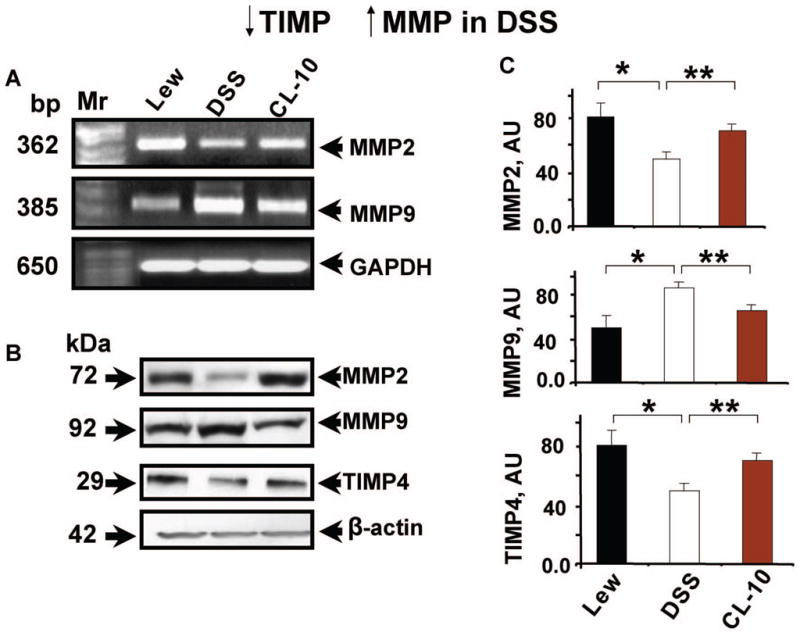
Q-RT-PCR analysis of MMP-2, −9 and GAPDH (A); and Western blot (B) analysis of MMP-2, −9, TIMP-4 and beta-actin in LV of L, DSS and CL-10 rats. Molecular weight markers (base pair, bp) and kDa are shown to the left (C): bar graph represents the levels of MMP-2, −9 and TIMP-4 in the hearts of L, DSS and CL-10 rats. Each bar is an average ± SD from n = 6. ★p < 0.05 L vs DSS; ★★p < 0.05 DSS vs CL-10.
Discussion
LV hypertrophy, alterations in LV morphology, structure and function assessed in the current study were related to LV remodelling. LV remodelling represented a more evolved phase of hypertensive heart disease, and in this patho-physiological scenario, the unbalance between MMPs and their tissue inhibitors has been proposed as a major mechanism of cardiac alterations (Cox et al., 2004). Due to more severe interstitial fibrosis (Matsui et al., 2006) there was E-M uncoupling in DSS rats. Studies (Matsui et al., 2006) reported interstitial collagen deposition, collagen content or collagen metabolites in heart of DSS rats. Here, we reported that the interstitial fibrosis in DSS rats was due in part to increased MMP-9 and decreased TIMP. Because DSS-hypertension and LV hypertrophy are associated with marked interstitial fibrosis (Matsui et al., 2006), a decrease in MMP associated with reduced LV hypertrophy may not justify the hypothesis that LV hypertrophy was due to increase in MMPs. Also it was possible that increased MMP was a compensatory response to increased collagen production, rather than a cause of LV hypertrophy. However, it was a paradox that MMP activation and cardiac fibrosis track together. This paradox can be explained by considering turnover of the degraded matrix proteins. It is well known that MMPs are collagenases, as well as excellent elastases. Because turnover of elastin is slower than collagen, therefore, degraded elastin is replaced by oxidatively modified collagen, leading to fibrosis (Rucklidge et al., 1992).
We are the first to suggest a role of ST segment duration as a measure of coronary vasospasm (collapse) independent of vessel reactivity. However, in clinical practice, QT duration is used as measure of both depolarization and re-polarization. The ST duration is well defined as re-polarization, indicative of coronary reserve. The elevation in T-wave is associated with coronary collapse during myocardial ischemia. There are numerous studies demonstrating the use of both 12-leads and 3-leads ECG to measure the ST duration. In addition, we routinely observed elevation of T wave in acute coronary ligation in rodent. We observed similar ST duration in all DSS rats. Previously, we suggested a link between cardiac fibrosis/scar and impaired Q-wave post myocardial infarction (Tyagi, 2000). Here we demonstrated that LVH and changes in cardiac remodelling were associated with increase in ST segment duration, indicating coronary vasospasm, and were ameliorated in congenic rats.
Since there is more contractile protein due to hypertension and hypertrophy in DSS, the response to ET is greater in DSS rats (Figure 5B, please note the scale on y-axis). However, the response duration was slower than Lewis or in the CL-10 rats. In DSS rats, abnormalities in electrical properties of the heart, impaired LV relaxation, blunted −dP/dt and increase in tau were accompanied by E-M uncoupling and the augmentation of MMP/TIMP ratio. In congenic rats, increase in TIMP-4 levels were associated with mitigation of abnormal electrical properties, impaired LV relaxation, E-M uncoupling, blunted −dP/dt and increase in tau. Interestingly, our results showed for the first time E-M dysfunction in salt-induced hypertension. The response to acetylcholine and not to nitroprusside was attenuated in LV rings from DSS rats. There was no change in Lewis rats. However, in congenic rats response to acetylcholine was normalized. These results suggested E-M uncoupling in DSS rats, and in congenic rats E-M uncoupling was mitigated.
Previous studies have shown E-M uncoupling in chronic pressure overload (Mujumdar & Tyagi, 1999). In DSS rats during salt overload, LV systolic and diastolic dysfunction was observed (Matsui et al., 2006). The LV diameter was increased in DSS rats (Matsui et al., 2006). Our results suggested LV dilatation in DSS rats, and in congenic rats, there was normalization in LV dysfunction and induction of TIMP-4. Previous studies have shown decrease in MMP-2 activity and TIMP-1 expression in hypertension and associated cardiac fibrosis, stiffness and impaired cardiac diastolic relaxation (Lindsey et al., 2002). The MMP-9 activation and decrease in TIMP-4 expression was linked to cardiac dilatation and heart failure (Mujumdar & Tyagi, 1999). Here we suggested that although MMP-2 was decreased, the levels of MMP-9 were increased in DSS rats. This led to robust increase in MMP-9/TIMP-4 ratio causing cardiac systolic cardiomyocyte slippage and failure. However, in congenic rats over expression of TIMP-4 was associated with mitigation of cardiac systolic failure.
Co-ordination between angiogenesis and cardiac hypertrophy (LVH) is the hallmark of compensatory response to heart failure; however, a disruption in coordination between cardiac hypertrophy and angiogenesis contributes to the transition to heart failure (Shiojima et al., 2005). Angiogenesis, LVH and fibrosis track together; therefore, in the absence of blood supply to the myocardium at risk, the lack of angiogenesis by increased angiostatic factors leads to fibrotic and LVH conditions. It is a paradox that activation of MMP is needed to disrupt the matrix during angiogenesis and to create new blood vessels, or opening the collaterals, however, the peptides generated by MMP activation are angiostatic. Early studies by O’Reilly (1997) and
O’Reilly et al. (1997) suggested endostatin and angiostatin generated by MMP action on collagen-18 and plasminogen, respectively, inhibit tumor angiogenesis. In addition, the vasostatin is generated by degradation of calreticulin (Pike et al., 1998), tumstatin (Maeshima et al., 2002), arresten (Colorado et al., 2000) and canstatin (Kamphaus et al., 2000) by degradation of collagen-4 and restin (Ramchandran et al., 1999) is generated by degradation of collagen-15. The decrease in MMP-2 and increase in MMP-9 (Figure 6) in hypertension is consistent with the notion that MMP-2 is constitutive and MMP-9 is inducible and causes dis-co-ordinated matrix degradation leading to decompensatory hypertrophy and heart failure by generating angiostatic factors. This demonstrates, however, that MMP, TIMP and LVH may co-exist.
Novelty, significance and limitations
Although this provocative study expands the current view on the role of extracellular matrix degradation in the structural and functional cardiac alterations that are present in hypertension, it is difficult to exclude the possibility that reduced hypertrophy in CL-10 rats is related to decreased blood pressure.
However, the paradox of differential elastin versus collagen remodelling by MMP can be very detrimental in vessel wall where 50% of the protein is elastin, leading to systemic vascular stiffness and hypertension. Therefore, increased levels of TIMP decreases proteolytic and oxidative stresses and may decrease LVH and blood pressure. Although we show mitigation of cardiac dysfunction and endothelial cardiomyocyte uncoupling in congenic rats, there is no data demonstrating a direct link between TIMP-4 expression and decrease in LVH and hypertension. The normalization of the cardiac function in CL-10 rat hearts may be due entirely to the normalization of hypertension, and that the changes in MMPs and TIMP-4 may be the secondary mechanism in the DSS hearts. The study of administration of GM6001 (a specific MMP-9 inhibitor) in DSS rats is warranted to demonstrate a cause and effect relationship of inhibitor expression and decrease in hypertension. These studies are in progress.
Acknowledgments
This study was supported by NIH grants HL-71010, HL-74185 and HL-88012. The work was presented at American Heart Association-Council on high blood pressure research, Annual Meeting, San Antonio, TX, Oct 4-7, 2006.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Colorado PC, Torre A, Kamphaus G, Maeshima Y, Hopfer H, Takahashi K, Volk R, Zamborsky ED, Herman S, Sarkar PK, et al. Anti-angiogenic clues from vascular basement membrane collagen. Cancer Res. 2000;60(9):2520–26. [PubMed] [Google Scholar]
- Cox MJ, Hawkins UA, Hoit BD, Tyagi SC. Attenuation of oxidative stress and remodelling by cardiac inhibitor of metalloproteinase protein transfer. Circulation. 2004;109(17):2123–28. doi: 10.1161/01.CIR.0000127429.53391.78. [DOI] [PubMed] [Google Scholar]
- Deng AY. Is the NOS system involved in genetic hypertension in Dahl rats? Kidney Intern. 1998;53:1501–11. doi: 10.1046/j.1523-1755.1998.00933.x. [DOI] [PubMed] [Google Scholar]
- Garrett MR, Dene H, Walder R, Zhang Q, Cicila GT, Assadnia S, Deng AY, Rapp JP. Genomic scan and congenic strains for blood pressure quantitative trait loci using Dahl salt-sentive rats. Genome Res. 1998;8:711–23. doi: 10.1101/gr.8.7.711. [DOI] [PubMed] [Google Scholar]
- Henderson BC, Tyagi N, Ovechkin A, Kartha GK, Moshal KS, Tyagi SC. Oxidative remodelling in pressure overload induced chronic heart failure. Euro J Heart Failure. 2007;9(5):450–7. doi: 10.1016/j.ejheart.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Hunt MJ, Aru GM, Hayden MR, Moore CK, Hoit BD, Tyagi SC. Induction of oxidative stress and disintegrin metalloproteinase in human heart end-stage failure. Am J Physiol. 2002;283(2):L239–L245. doi: 10.1152/ajplung.00001.2002. 2002. An editorial on this paper, L237–L238. [DOI] [PubMed] [Google Scholar]
- Julier C, Delepine M, Keavney B, Terwilliger J, Davis S, Weeks DE, Bui T, Jeunemaitre X, Velho G, Froguel P. Genetic susceptibility for human familial essential hypertension in a region of homology with blood pressure linkage on rat chromosome 10. Hum Mol Genet. 1997;6:2077–86. doi: 10.1093/hmg/6.12.2077. [DOI] [PubMed] [Google Scholar]
- Kamphaus GD, Colorado PC, Panka DJ, Hopfer H, Ramchandran R, Torre A, Maeshima Y, Mier JW, Sukhatme VP, Kalluri R. Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J Biol Chem. 2000;275(2):1209–15. doi: 10.1074/jbc.275.2.1209. [DOI] [PubMed] [Google Scholar]
- Lindsey MM, Maxwell P, Dunn FG. TIMP-1: a marker of LV diastolic dysfunction and fibrosis in hypertension. Hypertension. 2002;40:136–41. doi: 10.1161/01.hyp.0000024573.17293.23. [DOI] [PubMed] [Google Scholar]
- Lominadze D, Roberts AM, Tyagi N, Moshal KS, Tyagi SC. Homocysteine causes cerebrovascular leakage in mice. Am J Physiol Heart & Circulatory Physiol. 2006;290:H1206–H1213. doi: 10.1152/ajpheart.00376.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima Y, Sudhakar A, Lively JC, Ueki K, Kharbanda S, Kahn CR, Sonenberg N, Hynes RO, Kalluri R. Tumstatin, an endothelial cell specific inhibitor of protein synthesis. Science. 2002;295(5552):140–3. doi: 10.1126/science.1065298. [DOI] [PubMed] [Google Scholar]
- Matsui I, Shimosawa T, Uetake Y, Wang H, Ogura S, Kaneko T, Liu J, Ando K, Fujita T. Protective effect of potassium agonist the hypertensive cardiac dysfunction. Hypertension. 2006;48:225–31. doi: 10.1161/01.HYP.0000232617.48372.cb. [DOI] [PubMed] [Google Scholar]
- Mujumdar VS, Smiley LM, Tyagi SC. Activation of matrix metalloproteinase dilates and decreases cardiac tensile strength. Intern J Cardiol. 2001;79(2–3):277–86. doi: 10.1016/s0167-5273(01)00449-1. [DOI] [PubMed] [Google Scholar]
- Mujumdar VS, Tyagi SC. Temporal Regulation of ECM Components in transition from compensatory hypertrophy to decompensatory heart failure. J Hypertension. 1999;17:261–70. doi: 10.1097/00004872-199917020-00011. [DOI] [PubMed] [Google Scholar]
- O’Reilly MS. Angiostatin: an endogenous inhibitor of angiogenesis and tumor growth. Exs. 1997;79:273–94. [PubMed] [Google Scholar]
- O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88(2):277–85. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- Palijan A, Lambert R, Dutil J, Sivo Z, Deng AY. Comprehensive congenic coverage revealing multiple blood pressure quantitative trait loci on Dahl rat chromosome 10. Hypertension. 2003;42:515–22. doi: 10.1161/01.HYP.0000090096.88509.15. [DOI] [PubMed] [Google Scholar]
- Pike SE, Yao L, Jones KD, Cherney B, Appella E, Sakaguchi K, Nakhasi H, Teruya-Feldstein J, Wirth P, Gupta G, et al. Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J Exp Med. 1998;188(12):2349–56. doi: 10.1084/jem.188.12.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramchandran R, Dhanabal M, Volk R, Waterman MJ, Segal M, Lu H, Knebelmann B, Sukhatme VP. Antiangiogenic activity of restin, NC10 domain of human collagen XV: comparison to endostatin. Biochem Biophys Res Commun. 1999;255(3):735–9. doi: 10.1006/bbrc.1999.0248. [DOI] [PubMed] [Google Scholar]
- Rucklidge GJ, Milne G, McGaw BA, Milne E, Robins SP. Turnover rates of different collagen types measured by isotope ratio mass spectrometery. Biochim Biophys Acta. 1992;11:1156–7. doi: 10.1016/0304-4165(92)90095-c. [DOI] [PubMed] [Google Scholar]
- Shastry S, Tyagi SC. Homocysteine induces metalloproteinase and shedding of beta-1 integrin in microvessel endothelial cells. J Cell Biochem. 2004;93:207–13. doi: 10.1002/jcb.20137. [DOI] [PubMed] [Google Scholar]
- Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115(8):2059–64. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarone RE. A modified Bonferroni method for discrete data. Biometrics. 1990;46:515. [PubMed] [Google Scholar]
- Tyagi SC. Proteinases and myocardial extracellular matrix turnover. Mol Cell Biochem. 1997;168:1–12. doi: 10.1023/a:1006850903242. [DOI] [PubMed] [Google Scholar]
- Tyagi SC. Physiology of extracellular matrix: Cardiovascular adapation and remodelling. Pathophys. 2000;7:177–82. doi: 10.1016/s0928-4680(00)00046-8. [DOI] [PubMed] [Google Scholar]
- Tyagi SC, Hayden MR. Role of nitric oxide in matrix remodelling in diabetes and heart failure. Heart Failure Rev. 2003;8:23–8. doi: 10.1023/a:1022138803293. [DOI] [PubMed] [Google Scholar]
- Tyagi SC, Smiley LM, Mujumdar VS. Homocyst(e)ine impairs endocardial endothelial function. Canad J Physiol & Pharmacol. 1999;77:950–7. [PubMed] [Google Scholar]