

Abstract
Molecular modeling results indicate that the VDR contains two overlapping ligand binding pockets (LBP). Differential ligand stability and fractional occupancy of the two LBP has been physiochemically linked to the regulation of VDR-dependent genomic and non-genomic cellular responses. The purpose of this report is to develop an unbiased molecular modeling protocol that serves as a good starting point in simulating the dynamic interaction between 1α,25(OH)2-vitamin D3 (1,25D3) and the VDR LBP. To accomplish this goal, the flexible docking protocol developed allowed for flexibility in the VDR ligand and the VDR atoms that form the surfaces of the VDR LBP. This approach blindly replicated the 1,25D3 conformation and side-chain dynamics observed in the VDR x-ray structure. The results are also consistent with the previously published tenants of the vitamin D sterol (VDS)-VDR conformational ensemble model. Furthermore, we used flexible docking in combination with whole cell patch clamp electrophysiology and steroid competition assays to demonstrate that a) new non-vitamin D VDR ligands show a different pocket selectivity when compared to 1,25D3 that is qualitatively consistent with their ability to stimulate chloride channels and b) a new route of ligand binding provides a novel hypothesis describing the structural nuances that underlie hypercalceamia.
Keywords: VDR; non-genomic response; 1α,25(OH)2-vitamin D3; chloride channel; molecular modeling
Introduction
The vitamin D receptor (VDR) can act as both a nuclear localized classical ligand-dependent transcription factor and membrane receptor. As a transcription factor, the VDR acts as a primary regulator of genes that contain vitamin D response elements (i.e. sequence specific VDR binding sites). As a membrane receptor the VDR acts as a primary regulator of lipid and cytosolic second messengers that modulate the activity of kinases and phosphatases that in-turn control the opening of ion channels [14].
Vitamin D sterol ligands, the molecular tools commonly used to differentiate non-genomic (extra-nuclear) and genomic (nuclear) signaling differ in their chemistries [2]. Structure-function results obtained with these compounds suggest that the major physicochemical traits of a potent and efficient VDR genomic agonist are: 1) the ability to form hydrogen bonds (H-bonds) with the VDR S237 and R274 residues; 2) the ability to conform to a bowl-shaped molecular geometry (Fig. 1); and 3) a molecular volume that is ≤ to that calculated for 1α,25(OH)2-vitamin D3 (1,25D3, Fig. 2), the classic hormonal form of vitamin D3. Alternatively the major physicochemical traits of a potent and efficient non-genomic agonist are: 1) the ability to form H-bonds with S237 and R274; 2) the ability to conform to a planar-like molecular geometry (Fig. 1); and 3) a molecular volume that is ≤ to that calculated for 1,25D3 [14].
Figure 1. The Vitamin D Sterol (VDS)-VDR conformational ensemble model.

The VDS-VDR conformational ensemble model can be broken down into three interconnected parts: the VDS conformational ensemble (panel A); the VDR two pocket model (panel B); and the VDR conformational ensemble (panel C). A) The VDS conformational ensemble is dictated by the number of rotatable bonds in the molecules A-ring, seco-B-ring and side-chain regions. Two of the 6-s-trans molecular geometries of 1,25D3 that have been shown to form a stable complex with the VDR are depicted in the figure. B) The VDR two pocket model, posits that an additional steric space in the H2/β-sheet of the VDR molecule opens (blue transparent surface) when the hydrogen bond donor-acceptor relationship between H229 and Y295 switches. The switch is brought about by the rotation of the H229 imidazole R-group. The new pocket formed is termed the VDR alternative binding site (VDR-AP, blue transparent surface). The VDR-AP physically overlaps with the steric space of the VDR genomic pocket (VDR-GP, yellow transparent surface). The VDR-GP is defined by the 1,25D3-VDR x-ray data. The overlapping region of the VDR-GP and VDR-AP is termed the A-ring domain and the residues that form this region are labeled and their wireframes colored orange. Regions of the VDR that form the molecular surfaces of the VDR-GP and VDR-AP are highlighted in the VDR domain map and their locations indicated in the VDR tertiary structure by color coding the ribbon to the domain map. C) The implicit VDR conformational ensemble is depicted by using the x-ray coordinates of various apo- holo-NR structures (see the protein data bank). Comparison of the NR structures indicates that helix-12 (H12) is flexible and can sample a number of different conformational states. According to the tenants of the VDS/ VDR conformational ensemble model the population distribution of the H12 conformational states is dictated by a given ligands affinity for the VDR-AP and VDR-GP.
Figure 3. 1,25D3-VDR-GP and VDR-AP flexible docking results.
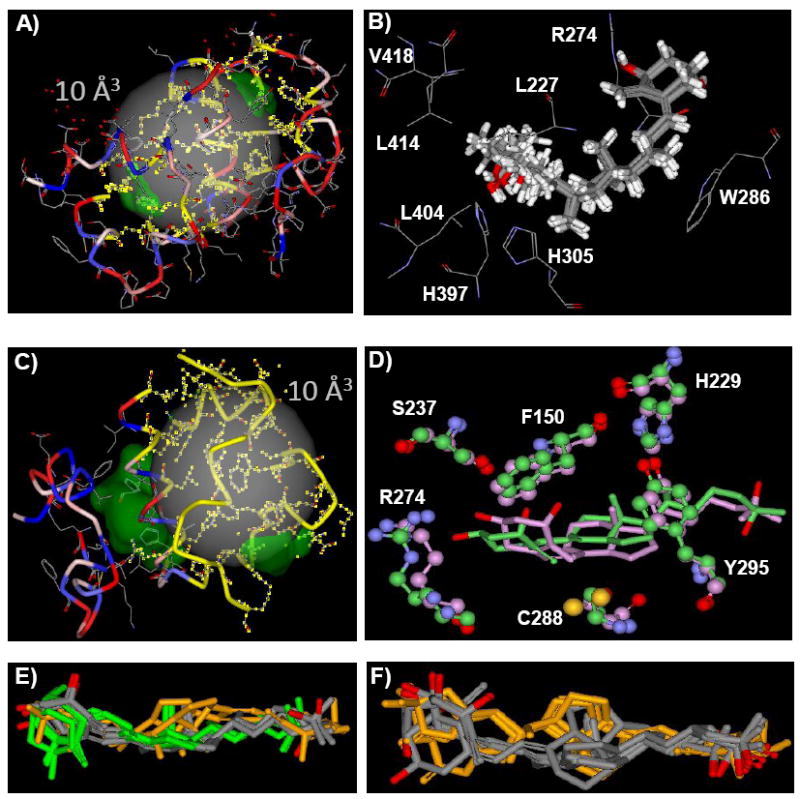
A) The VDR-GP 10 Å3 flexible docking site sphere is shown as a transparent gray sphere. The VDR-GP is rendered as a green solid surface. The VDR R-groups that form the VDR-GP are shown in wire-frame. The VDR backbone is rendered to show its secondary structure that is colored to indicate the relative hydrophobicity of each R-group (red polar to blue non-polar). The R-groups allowed to be re-drawn in the flexible docking simulation are highlighted in yellow. B) The top 10 1,25D3-VDR-GP flexible docking complexes are rendered to show the 1,25D3 molecules as thick gray tube structures and the VDR R-groups thin gray wire-frames. These R-groups are labeled with their one letter code. C) The VDR-AP 10 Å3 flexible docking site sphere is shown as a transparent gray sphere. The VDR-AP forms a continuous channel with the VDR-GP. This channel is rendered as a green solid surface. The VDR R-groups that form the VDR-AP are shown in wire-frame. The VDR backbone is rendered to show its secondary structure colored to indicate the relative hydrophobicity of each R-group (red polar to blue non-polar). The R-groups allowed to be re-drawn in the flexible docking simulation are highlighted yellow. D) The 1,25D3-VDR-AP complexes obtained using the computation methods outlined in Mizwicki et al. [13] (green) and the methods used herein (pink) are heavy atom (i.e. C, N, O, not H) overlaid for comparison. Key amino acids that line the VDR-AP are shown in ball and stick structure. Note the C288 residue forms a number of vdW contacts with the 1,25D3 triene (i.e. seco-B-ring) in both complexes. E) This presentation shows the top 10 1,25D3-VDR-AP flexible docking complexes. The A-ring first 6-s-trans poses are colored gray (n=5) (i.e. A-ring occupying the A-ring domain and side-chain oriented towards the H2/ β-sheet surface; see Fig. 1B). The A-ring first, 6-s-cis poses are colored green (n=3). The side-chain first, 1,25D3, 6-s-trans poses are colored orange (n=2). F) This presentation shows the top 10 BS-VDR-AP flexible docking complexes. The A-ring first, 6-s-trans poses are colored gray (n=6). The side-chain first, BS, 6-s-trans poses are colored orange (n=4).
Figure 2. Chemistry of the VDR ligands.
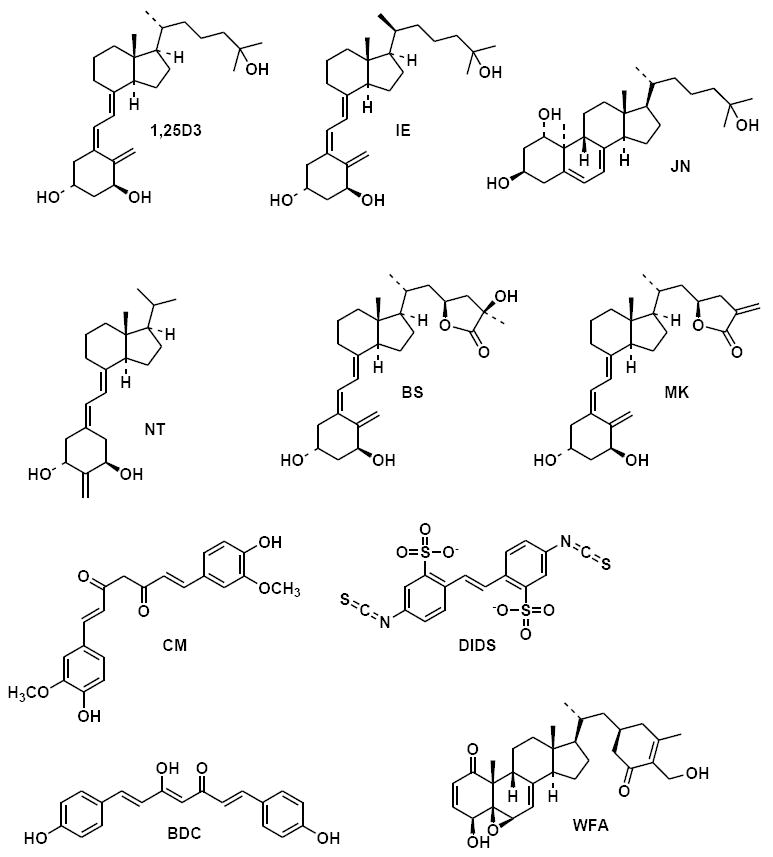
The chemical structures of 1α,25(OH)2-vitamin D3 (1,25D3), 20-epi-1α,25(OH)2-vitamin D3 (IE), 1α,25(OH)2-lumisterol D3 (JN), 2-Methylene-19-nor-1α-hydroxyhomopregnacalciferol (NT), 23S,25R-1α,25-(OH)2-D3-26,23-lactone (BS), (23S)-25-dehydro-1α-OH-D3-26,23-lactone (MK), curcumin (CM), 4,4’-Diisothiocyanostilbene-2,2’-disulfonic Acid (DIDS), bisdemethoxycurcumin (BDC) and withaferin A (WFA) are shown.
How the VDR molecule can serve as a receptor molecule for ligands with different shapes and physicochemical traits can be rationalized by the vitamin D sterol (VDS)-VDR conformational ensemble model (Fig. 1). This model posits that the VDR ligand binding domain (LBD) contains two overlapping ligand binding pockets (LBP). The VDR alternative pocket (VDR-AP, Fig. 1) was discovered by superimposing the non-genomic specific agonist 1α,25(OH)2-lumisterol D3 (JN, Fig. 2) on the 1,25D3 pose observed in the VDR x-ray crystal structure such that the side-chain of JN oriented towards the H2/ β-sheet surface of the VDR rather than the H3/H12 surface (Fig. 1). As shown in figure 1, if the VDR-AP is opened and H12 is closed, the VDR-AP and VDR-GP form a capped channel. According to the VDR ensemble principles this conformation represents one of many VDR local energy minima (i.e. conformational isomers, Fig. 1). From the perspective of the VDR ligand this conformation provides the largest internal surface area to sample. Alternatively, when the VDR-AP is closed or VDR-GP occluded by the C-terminal end of H11 (Fig. 1), the surface area of the channel is reduced. Thus different VDR ensembles are posited to be stabilized by different ligands and/or ligand pools (e.g. vitamin D metabolites), because the physicochemical interaction with the dynamic LBP surface will fundamentally differ [11, 13-15].
Comparison of the apo-VDR-AP and the apo-VDR-GP structures identified only one major change in the VDR tertiary structure; this was the H-bonding network between the back and front of the VDR LBD [11] (Fig. 1). Breathing of the VDR LBP (i.e. dynamic fluctuation of LBP volume and surface profile) is controlled in-part by a molecular switch that exists between the Y295 (β-sheet)---H229 (H3)----R154 (loop)----K413/L414 (H12) residues, listed back to front respectively. When the VDR-GP is preferentially occupied by a ligand, these residues form H-bonds that are of paramount importance in holding together the H12 closed conformation (Fig. 1). When 1,25D3 occupies the VDR-AP, the communication between the back (Y295) and front (K413/L414) of the VDR LBD is fractured. The fracture is created by an H-bond donor-acceptor flip-flop between H229 and Y295 (Fig. 1). The H229 residue is therefore central to the communication between the β-sheet and H12 and the gating of the VDR-AP [9, 11].
The purpose of this study was to generate a more unbiased and rigorous in silico method for simulating the binding of 1,25D3 and other ligands to the VDR. In designing the protocol, three criteria were considered necessary: 1) flexibility in the VDR ligand must be allowed; 2) flexibility in the residues that contact the ligand must be allowed; and 3) superimposition of a VDR ligand on the 1,25D3 x-ray conformer must be avoided. The flexible docking method developed can replicate the 1,25D3 pose and atomic thermal factors observed in the holo-VDR x-ray structure [3]. The flexible docking results also support the concept that the VDR-AP exists and is kinetically favored by 1,25D3. Finally, the results also suggest a new mechanism for 1,25D3 binding that provides an explanation for how a genomic superagonist (IE) and a genomic antagonist (MK, Fig. 2) ligand are similar to 1,25D3 in their abilities to function as agonists that activate outwardly rectifying chloride channels.
Materials and Methods
Reagents
Withaferin A, Curcumin and Bisdemethoxycurcumin were purchased from ChromaDex, Inc. (Santa Ana, CA 92705) and stocks were made in ethanol. The vitamin D sterols were obtained from the following sources 1,25D3 (Milan Uskokavic, BioXell), MK and BS (Seiichi Ishizuka, Teijin Pharma, Japan) and NT (Abbot Inc., Chicago, IL) and stocks were made in ethanol. DIDS was purchased from Sigma-Aldrich and the stock made in DMSO.
Molecular Modeling
The protocol for generating vitamin D sterol conformational isomers and the flexible docking procedure is described in detail in Mizwicki et al. [12]. The only difference in the protocol used for flexibly docking ligands to the VDR-AP was the fact that 20 VDR R-groups were allowed to be redrawn during the docking process, whereas 15 VDR R-groups were allowed to be redrawn during the VDR-GP docking process. The number of R-groups allowed to be redrawn for each VDR LBP was determined by trouble shooting what value gave the best cDOCKER scores. The cDOCKER score ranks the complexes saved after a short molecular dynamics simulation is performed. The top 10 scored complexes were then energy optimized and the Gibbs free energy of ligand binding calculated. The top 5 Gibbs free energy of ligand binding values were averaged and the value presented in Table 2. Importantly, this protocol differs from the protocol described in Mizwicki et al. [13] in that solvation effects are considered in the GBMV algorithm used in calculating the ΔGbinding value. The procedure still lacks the assessment of the potential effect entropy changes have on the ΔGbinding value or calculated affinity.
Table 2. Flexible docking VDR ligand VDR-GP and VDR-AP Gibbs free energy of binding.
The table summarizes the region of chemical modification with respect to 1,25D3 and the Gibbs free energy of binding (ΔGbinding) values (see methods) calculated for the given VDR ligand (see Fig. 2 for structures) docked in the VDR-GP and VDR-AP. The average VDR-GP ΔGbinding value for each VDR ligand is calculated by averaging the values obtained from the top five of the 10 cDOCKER scored flexible docking complexes (see methods). The average A-ring first VDR-AP ΔGbinding value for each VDR ligand is calculated in the same manner. These average values are well above the theoretical ΔGbinding value that would be derived from the measured equilibrium constants for these sterols (e.g. 1,25D3 KD = 1.0 nM). The highest affinity side-chain first VDR-AP ΔGbinding value is provided for 1,25D3, BS and NT and is representative of a single VDR-AP complex. No average value is provided given the very low frequency of side-chain first complexes observed in the top 10 cDOCKER scored complexes. (*) Curcumin (CM) was flexibly docked to the VDR pockets using an ionized and unionized structure. The average CM-VDR-GP ΔGbinding shown in the table is generated from the unionized flexible docking results, if ionized the average VDR-GP ΔGbinding value is positive, reflective of an unfavored binding (e.g. BDC).
| VDR Ligand | Region of carbon skeleton modified | Average VDR-GP ΔGbinding (kcal/mole) |
Average A-ring first VDR-AP ΔGbinding (kcal/mole) |
Highest affinity side-chain first VDR-AP ΔGbinding (kcal/mole) |
|---|---|---|---|---|
| 1,25D3 | control | -50.7 | -40.4 | -34.7 |
| 25D3 | A-ring (C1) | -38.8 | -39.9 | --- |
| JN | Seco-B-ring | -40.5 | -36.2 | --- |
| BS | Side-chain | -59.2 | -39.9 | -43.3 |
| MK | Side-chain | -39.3 | -36.5 | --- |
| IE | Side-chain (C20) | -44.5 | -40.5 | --- |
| CM | --- | -21.5* | -26.0 | --- |
| BDC | --- | (+) | -45.7 | --- |
| WFA | A-ring/ seco-B-ring/ side-chain | -30.7 | -37.5 | --- |
| NT | A-ring/ seco-B-ring/ side-chain | -42.0 | -20.3 | -22.8 |
Ligand Binding
The hydroxyapatite protocol used has been published previously by Mizwicki et al. [12, 13].
Electrophysiology
TM4 cells (American Type Culture Collection, ATCC, Manasas, VA, USA) were cultured in a 1:1 mixture of Dulbecco’s modified Eagle’s media (DMEM) and Ham’s F12 with the addition of 1 mM L-glutamine, 15 mM HEPES, 1.2 g/L sodium bicarbonate, 100 U/ml penicillin, 100 μg/ml streptomycin, 5% (v/v) horse serum and 2.5% (v/v) fetal calf serum, in a 5% CO2 humidified atmosphere. For patch-clamp experiments the cells were plated at ~10% density and used within 24-48 hrs of plating. Cells were washed 3x prior to obtaining a giga seal. Chloride currents were studied in the whole-cell patch-clamp configuration with a Heka EPC-9 amplifier (ALA Scientific Instruments Inc., West bury, NY) as described by (Hamill et al., 1991). Recording pipettes with resistances ranging between 3-5 MΩ were fabricated with a DMZ Universal micropipette puller from Drummond capillaries (Drummond Scientific Co., Broomall, PA), coated with Sylgard elastomer (Dow Corning Corp., Midland, MI) to reduce capacitative transients, and fire-polished. Seal resistances ranged from 3 to 15 GΩ. Experiments were carried out at room temperature. Currents were low-pass-filtered at 1 kHz and digitized every 100 μs. Cell membrane capacitance and series resistances were electronically compensated prior to the recording of currents. For the recording of chloride currents, it was used an external solution consisting of (mM): 150 NaCl, 10 BaCl2, 2 MgCl2, 10 glucose, 10 HEPES, pH 7.3. The corresponding pipette solution consisted of (mM): 160 CsCl, 10 MgCl2, 10 HEPES/TEAOH buffer, pH 7.2. Cs+ and TEA+ were used to block K+ channel activity. Currents were activated with 100 ms-pulses between − 60 and 80 mV, from a holding potential of − 30 mV.
Results/ Discussion
The Vitamin D Sterol (VDS) Ensemble
Vitamin D3 (D3) is a seco-steroid and contains an aliphatic cholesterol side-chain; therefore, it differs from most other NR ligands in that it is highly flexible (Fig. 1). To simulate changes in the flexibility or shape of the vitamin D sterol PC_Model GMMX conformational search calculations were performed using the whole molecule (see methods). The relative population of 1,25D3 A-ring α/β-chair conformations produced by this method compared well with the equilibrium observed by solution state nuclear magnetic resonance (Table 1) [24]. The 1,25D3 side-chain shows the same population distribution when compared to that obtained in previously published side-chain dot map calculations [13], where the A and seco-B-rings are not present (i.e. only the CD-ring fragment is used, Fig. 1). Lactonization of the 1,25D3 side-chain (e.g. MK and BS, Fig. 2) reduced the disorder of the side-chain, consistent with the reduction in the number of degrees of rotational freedom (Fig. 2) [12].
Table 1. The 1,25D3 Conformational Ensemble Used in Flexible Docking Experiments.
1,25D3 conformational isomers were produced using the GMMX conformational search calculation provided by PC_Model v8.0. Using the search criterion (see methods) over 1,000 1,25D3 conformations were saved and the top 500 were used in the flexible docking calculation. The breakdown of the A-ring, seco-B-ring and side-chain configurations observed for the top 500 structures are provided in the table. The A-ring α and β-chair population distributions include α and β-twist boats, no boat configurations were observed. The side-chain populations (Pop. A, Pop. B and Pop. C) are defined by the C16-C17-C20-C22 dihedral angles or anti, gauche(+) and gauche(-) geometric isomers (see [11, 13] for more details).
| A-ring | Seco-B-ring | Side chain | |||||
|---|---|---|---|---|---|---|---|
| VDR Ligand | α-chair | β-chair | 6-s-cis | 6-s-trans | Pop. A | Pop. B | Pop. C |
| 1,25D3 (500) | 52% | 48% | 26% | 74% | 68% | 14% | 18% |
The VDR LBP Ensemble
Current computational methodologies do not allow for an assessment of global conformational changes in the VDR molecule such as those depicted in the VDR ensemble in figure 1. However, R-group (i.e. amino acid side-chain) molecular dynamics or local conformational changes can be simulated. To address how R-group flexibility could alter the dynamics of vitamin D sterol binding, fifty different VDR starting conformations were generated using both the 1,25D3 bound VDR-GP and VDR-AP energy optimized complexes from Mizwicki et al. [13] as the VDR starting structures (see methods). In addition, 15 VDR-GP and 20 VDR-AP R-groups respectively were allowed to be redrawn for each of the 5,000 theoretical complexes generated by the flexible docking calculation.
1,25D3-VDR-GP Flexible Docking Results
The 1,25D3 molecular ensemble (Table 1) was docked to the VDR-GP by generating a 10 Å3 site sphere that encapsulates the VDR-GP LBP (Fig. 3A). After flexible docking, the top ten scored complexes (Fig. 3B) were energy optimized and the Gibbs free energy of binding (ΔGbinding) calculated (see methods). The 1,25D3 molecule in the highest affinity 1,25D3-VDR-GP complex superimposes well with the 1,25D3 conformation observed in the x-ray structure (RMSD = 0.85Å). Therefore the bowl-shaped geometry of 1,25D3 observed in the VDR x-ray study is produced in the generation of the VDS ensemble and selected by the VDR-GP during the flexible docking simulation. The only difference was observed in the position of the terminal side-chain atoms (Fig. 3B). Importantly, the x-ray B-value for these 1,25D3 atoms are significantly higher when compared to the average B-value of all of the 1,25D3 heavy atoms (see pdb:1DB1). Thus the flexible docking results are consistent with the dynamics observed for the 1,25D3 atoms in the x-ray.
The average ΔGbinding value for the 1,25D3-VDR-GP flexible docking complex is in good agreement with the value obtained when the 1,25D3 x-ray pose is used as the only ligand conformation in the flexible docking simulation (Table 2). The flexible docking results indicate that a β-chair A-ring with an equatorial 1α-OH group and a 6-s-trans seco-B-ring are selected by the VDR-GP (Table 2), consistent with the x-ray data [21] and structure-function results obtained prior to the solving of the holo-VDR crystal structure [16, 17, 20]. Replication of the 1,25D3 x-ray pose and ΔGbinding value was not achieved when the 1,25D3 ensemble was generated using the Discover Studio 2.0 (Accelrys, Inc., San Diego) BEST and CAESER algorithms (Table 2).
1,25D3-VDR-AP Flexible Docking Results
The 1,25D3 conformational ensemble (Table 1) was docked to the VDR-AP by generating a 10 Å3 site sphere that encapsulates the VDR-AP LBP (Fig. 3C). The highest affinity 1,25D3-VDR-AP complex superimposes well with the best 1,25D3-VDR-AP complex predicted by calculating the potential interaction energy between 1,25D3 and the VDR-AP. Both methods indicated that the preferred conformation of the 1,25D3 molecule accepted by the VDR-AP is the α-chair, 6-s-trans conformer. The 1,25D3 side-chain shows more disorder, but remains in a low energy, population A orientation (Fig. 3D), giving the molecule an overall planar-like geometry (Fig. 1).
The average1,25D3-VDR-AP ΔGbinding value is calculated to be -40.4 Kcal/mole (Table 2); therefore, the 1,25D3-VDR-GP/AP ΔGbinding values are in good agreement with the previously reported, more rudimentary potential interaction energies that suggested 1,25D3 thermodynamically favored the VDR-GP and kinetically favored the VDR-AP. The 1,25D3-VDR-AP flexible docking results also confirm and expand upon the evidence that the VDR-AP can accept more 1,25D3 conformational isomers, including 1,25D3 6-s-cis rotomers, as compared to the VDR-GP (Fig. 3D) [13-15].
In two of the top 10 scored 1,25D3-VDR-AP complexes, the ligand orientation is reversed within the LBP (Fig. 3E). This places the hydrophobic, highly flexible side-chain atoms in the relatively hydrophilic A-ring domain (Fig. 1); therefore, it is possible that 1,25D3 could access the more hydrophobic VDR-GP through the VDR-AP portal (Fig. 1). This process would be more favored for a ligand with flexibility in its seco-B-ring and side-chain, because it must wiggle around H3 to enter the VDR-GP (Fig. 1). The shape of 1,25D3 required to move around H3 is bowl-like (refer to shape of the two pockets together, Fig. 1); therefore, the geometry of the channel may induce the bowl-like shape of 1,25D3 observed in the thermodynamically favored VDR-GP.
The VDR-AP side-chain first VDR-GP binding mechanism differs from the current induced-fit hypothesis that suggests 1,25D3 binds to a somewhat opened H12 conformational state and upon binding induces H12 closure. If a ligand does enter the VDR-GP first, it would require that R274 become exposed by the movement of the C-terminal end of H11 (Fig. 1). Alternatively, the enthalpy barrier is null for VDR-AP opening/ closing (i.e. gating) (Fig. 1) [9, 11]. Another physicochemical feature that is consistent with the hypothesis that ligands first sample the VDR-AP is the observation that the residues forming the surface and internal walls of the VDR-AP are flexible when compared to the VDR-GP (Fig. 1). Thus the intrinsically disordered 1,25D3 ligand is proposed to be sensed by an intrinsically disordered region of the VDR. Accordingly the VDR-AP could function as a selectivity filter for the VDR-GP and thereby control the fractional occupancy of the VDR-GP and subsequently the transregulation of genes that contain a VDRE [14].
VDS-VDR-AP vs. VDR-GP Ligand A-ring Selectivity
We have previously proposed that 25D3 has the same stability in the VDR-AP as 1,25D3 [13]. This was based on the observation that the 3β-OH group of 25D3 and 1,25D3 form H-bonds with S237 and R274 in the VDR-AP and that no H-bonds were formed by the 1α-OH group [10, 13, 14] (see Fig. 3D). Comparison of the average 1,25D3 and 25D3-VDR-AP flexible docking ΔGbinding values (-40.4 and -39.9 kcal/mole respectively, Table 2) further support this hypothesis. Comparison of the average VDR-GP flexible docking ΔGbinding values for 25D3 and 1,25D3 also substantiate the fact that in order for a ligand to bind to the VDR-GP and therefore be capable of driving gene transcription, it must possess a 1α-OH group and be capable of sampling a bowl-like molecular geometry [15]. Furthermore, the VDR-GP exclusively requires the A-ring be in a β-chair conformation, while in contrast the VDR-AP accepts both A-ring chair forms and their intermediate rotomers (i.e. half-chairs, data not shown).
VDS-VDR-AP vs. VDR-GP Ligand Seco-B-ring Selectivity
This laboratory has previously been proposed that 1α,25(OH)2-lumisterol D3 (JN, Fig. 2) can form a complex with the VDR-AP that is of equivalent stability when compared to 1,25D3 [13]. The average JN-VDR-AP ΔGbinding value of -36.2 kcal/mole are consistent with this hypothesis (Table 2). In addition, the JN-VDR-AP and 6-s-cis 1,25D3-VDR-AP highest affinity complexes superimpose well with one another (data not shown). Unlike, 25D3, JN contains a 1-OH group and therefore, like 1,25D3 is predicted to thermodynamically favor the VDR-GP (Table 2); however, given its fused B-ring JN would be anticipated to have a much slower VDR-GP association rate if access to the VDR-GP was through the VDR-AP side-chain first complex, because this is anticipated to require flexibility in the A/B-ring region of a VDS ligand (Fig. 1).
VDS-VDR-AP vs. VDR-GP Ligand Side-chain Selectivity
It has been previously proposed that side-chain oxidation of 1,25D3 increases the VDR-AP and VDR-GP stability when compared to 1,25D3 [11]. This hypothesis may explain why downstream metabolites of 1,25D3 can have similar genomic function, but reduced VDR affinity when compared to 1,25D3. The most stable VDR-AP side-chain metabolite complex was formed by BS (Fig. 2) [11]. The protocol used in our previous work was lacking in that solvation and entropic variables were not considered in the computation. The algorithm used in this study to determine the ΔGbinding values considers the effect of solvation.
The average BS-VDR-AP ΔGbinding values indicated that BS was only as stable as 1,25D3, rather than more stable in the VDR-AP, as originally proposed [11]. It was observed that BS formed a more stable side-chain first complex with the VDR-AP when compared with 1,25D3 -43.3 kcal/mole versus -34.7 kcal/mole respectively (Table 2 and Fig. 3F). In addition, the reduced flexibility of the BS side-chain (see Fig. 2) could also hinder the ability of BS to wiggle into the VDR-GP when compared to 1,25D3. Thus the original hypothesis that BS has a higher VDR-AP fractional occupancy when compared to 1,25D3 is supported by the flexible docking results.
The 1,25D3 side-chain lactone genomic antagonist [12] (MK, Fig. 2) is somewhat similar in chemistry when compared to 1,25D3. MK formed a complex of similar affinity when compared to 25D3, JN, 1,25D3 and BS (Table 2). Like BS, MK formed a more stable side-chain first VDR-AP complex when compared to 1,25D3 (Table 2). Given that the VDR-AP has been physiochemically associated with regulating non-genomic signaling [13, 14], MK may be a capable VDR-dependent non-genomic agonist.
The VDR-AP and Superagonist Function
The concept that VDR ligands access the VDR-GP through the VDR-AP provides a new mechanistic route to understanding VDR superagonist function. 20-epi-1,25D3 (IE, Fig. 1) is a well studied VDR genomic superagonist. IE differs from 1,25D3 only in the stereochemistry about carbon-20, but this change converts IE into a 10-100-fold more potent genomic agonist when compared to 1,25D3 as determined by the measured EC50 of IE and 1,25D3 in inducing expression of reporter genes and differentiation of HL60 cells. Epimerization of C20 also makes 1,25D3 more hypercalcaemic, a therapeutic drawback that remains a mechanistic mystery [1]. Comparison of the 1,25D3 and IE VDR x-ray structures indicates only a minor difference in contacts made with the VDR-GP [23]. Thus no definitive structural evidence correlates with IE superagonism.
According to side-chain dot map calculations IE alters the side-chain conformation distribution to favor more so a northerly orientation with respect to the CD-ring (Fig. 1, Pop. B) [25]. It is this orientation that the VDR two pocket model suggests is required to enter the VDR-GP by wrapping around H3 (see Fig. 1). Therefore it is proposed that IE is a superagonist ligand because a) it is capable of more quickly occupying the VDR-GP when compared to 1,25D3 and b) does not undergo C23(S) side-chain oxidation [22], which produces VDS metabolites that have been shown to bind more stably to the VDR-AP when compared to their C24(R) metabolic counterparts [11]. Thus globally IE and its downstream metabolites are proposed to shift the VDS ligand pool to increase the VDR-GP fractional occupancy when compared with 1,25D3 and its pool (i.e. metabolites). In addition, the IE-VDR-AP average ΔGbinding values indicate that IE can form an A-ring first complex with the VDR-AP that has similar affinity to that predicted for 1,25D3 (Table 2).
Novel VDR Ligands Favor Binding the VDR-AP
Recent evidence demonstrates that curcuminoids (CM and BDC, Fig. 2) bind specifically to the VDR with a low micromolar affinity and form a stable VDR-AP complex [8]. The CM/ BDC-VDR-GP and VDR-AP flexible docking results suggest both energetically preferred the VDR-AP (Table 2). This is consistent with CM and BDC possessing a planar molecular shape and identical carbon spacing between the two phenolic –OH groups when compared to the spacing between the C3-OH and the 25-OH group of 1,25D3. Thus CM and BDC contain the physicochemical attributes that have been linked to the VDR-AP [14].
In searching for other potential VDR ligands present in plant extracts we identified withaferin A (WFA). WFA was selected because of its similar structure to JN and BS (Fig. 2) and because it is known to be an anti-inflammatory [7], an attractive function associated with 1,25D3. WFA bound specifically to the VDR with a measured affinity of ~ 2 μM (Fig. 4A). The flexible docking results demonstrated that WFA, like CM and BDC energetically preferred the VDR-AP according to the methodology used (Table 2).
Figure 4. DIDS and WFA bind specifically to the VDR LBD.
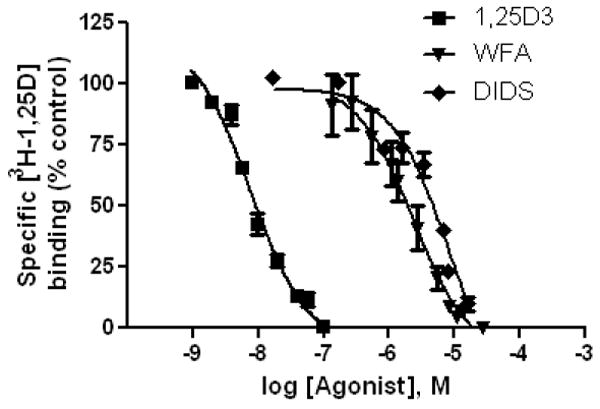
Radiolabeled steroid competition was used to generate VDR IC50 curves for 1,25D3, WFA and DIDS (see methods). The curves represent data pooled from three separate experiments where the concentration of protein was held constant. The IC50 values for 1,25D3, WFA and DIDS were measured to be ~8 nM, 2 μM and 8 μM respectively.
1,25D3, CM, IE and MK Regulation of Outwardly Rectifying Chloride Currents in TM4 Cells
Given CM, IE and MK form similar complexes with the VDR-AP when compared to 1,25D3, they were screened for their ability to stimulate outward rectifying chloride currents (ORCC) in TM4 Sertoli cells. Like 1,25D3 and CM, MK and IE stimulated the opening of ORCC channels (Fig. 5A). Furthermore, the currents were blocked by co-incubation with equal molar HL, the non-genomic specific antagonist (Fig. 5A). It is observed that IE is a non-agonist when added at 1 nM, but has equal activity when compared to 1,25D at 100 nM. This suggests that IE has the same ability to stimulate the non-genomic response, but that it is not as efficient as 1,25D3. This may indicate that the side-chain population A conformational isomer (see Fig. 1 and [13]) is better at accommodating the planar shape requirement suggested to be the optimal geometry for the binding to the VDR-AP A-ring first.
Figure 5. 1,25D3, MK and IE opening of outwardly rectifying chloride current (ORCC) channels in TM4 Sertoli cells.
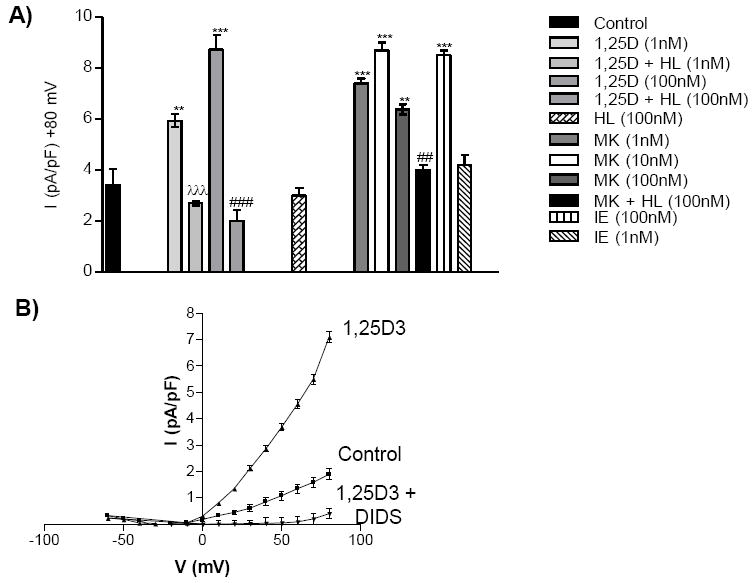
A) Depicts the ORCC measured by whole cell patch clamp of TM4 cells in the presence or absence of the given concentrations of 1,25D3 and MK (± equal molar HL) or IE. The average mean chloride current measured at +80 mV is depicted in the figure (n ≥ 3); **p< 0.01 compared with control; ***p< 0.001 compared with control; ##p< 0.001 compared with VDR ligand group; ###p< 0.001 compared with VDR ligand group. B) Whole cell patch clamp recordings obtained from TM4 cells. The graph plots the applied voltage (mV) against the intensity (I) of the ORCC (pA/pF) observed when 1,25D3 is applied to the bath solution in the presence or absence of DIDS.
Finally, the stilbene derivative DIDS (Fig. 2) blocked the activation of ORCC channels by 1,25D3 (Fig. 5B) and was shown to bind specifically to the VDR (Fig. 4). The DIDS VDR IC50 (~8 μM) is far lower than the concentrations traditionally used to block ORCC channels (50-500 μM).
The VDR-AP and Hypercalcemia
According to the VDS/VDR conformational ensemble model (Fig. 1), VDS exert their cellular effects via regulating non-genomic and genomic cellular signaling cascades [14]. Of the VDR ligands addressed in this study only 1,25D3 and IE trigger hypercalcemia when administered at therapeutic doses. Meanwhile, the VDR ligands MK and BS have both been shown to reduce the calcemic effect of 1,25D3 in vivo [4, 5]. All of these ligands shared similar VDR-AP affinity and nearly all stimulated ORCC channels (Fig. 4B); however, they differed in their VDR-GP affinity (Table 2) and the ability to stimulate gene expression [11]. Thus, increasing the concentration (genomic effect) and maintaining the function (non-genomic effect) of ion channels may be the underlying cause of hypercalcemia.
If hypercalcemia results from a shift in the balance of VDR non-genomic and genomic signaling, then according to the ensemble model increasing the concentration of free 1,25D3 or IE would facilitate an increase in VDR-GP fractional occupancy (the thermodynamically favored 1,25D3-VDR complex). In turn genomic signaling (e.g. expression of calcium channels and VDR [6, 18]) is enhanced. MK blocks 1,25D3-VDR gene expression [12]; therefore, it could augment the calcemic effect of 1,25D3 by reducing the expression of calcium channels and/or the VDR itself. Alternatively, BS formed a more stable side-chain first VDR-AP complex and VDR-GP complex when compared to MK and 1,25D3. Therefore the BS-VDR-AP models suggest that BS could act to allosterically temper the 1,25D3 calcemic effect by sterically (i.e. physically) blocking 1,25D3 access to the VDR-GP.
Finally, it was recently shown that analog NT (Fig. 2) can suppress PTH and enhance CYP24-OHase without raising serum calcium levels in vivo [19]. The later is a trait not observed for 1,25D3 when administered at pharmacologically relevant doses [1]. NT differs from other VDR ligands in this study in that it lacks a side-chain and has a 2-methylene group (Fig. 2). According to the flexible docking results NT differed from 1,25D3, BS, MK and IE in that it lacked the ability to form a viable complex with the VDR-AP. Thus the inability of NT to stimulate an increase in serum calcium and its ability to activate VDR mediated gene transcription may be a functional consequence of its reduced fractional occupancy of the VDR-AP.
Conclusions
The unbiased flexible docking protocol and the results generated from its application support the original tenants of the VDS-VDR conformational ensemble model [11, 13-15]. The flexible docking technique represents a start point to build upon in future theoretical VDS-VDR ligand binding simulations. The ΔGbinding values presented do not quantitatively correlate to real-life ligand-receptor affinities; however, they qualitatively provide a rationale physicochemical explanation for many VDS-VDR structure-function conundrums. In order to substantiate the claims made, each VDR ligand must be further assayed for both non-genomic and genomic function and additional biochemical characterization of the VDR-GP and VDR-AP complexes obtained. In closing, studying the details of how VDR ligands differ with respect to 1,25D3 in the way they interact with the VDR may provide novel structural/ molecular insights into structure-function conundrums within the vitamin D and perhaps other NR-fields.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bouillon R, Okamura WH, Norman AW. Structure-function relationships in the vitamin D endocrine system. Endocr Rev. 1995;16(2):200–257. doi: 10.1210/edrv-16-2-200. [DOI] [PubMed] [Google Scholar]
- 2.Dormanen MC, Bishop JE, Hammond MW, Okamura WH, Nemere I, Norman AW. Nonnuclear effects of the steroid hormone 1α,25(OH)2-vitamin D3: Analogs are able to functionally differentiate between nuclear and membrane receptors. Biochem Biophys Res Commun. 1994;201:394–401. doi: 10.1006/bbrc.1994.1714. [DOI] [PubMed] [Google Scholar]
- 3.Hmama Z, Nandan D, Sly L, Knutson KL, Herrera-Velit P, Reiner NE. 1α,25-Dihydroxyvitamin D3-induced myeloid cell differentiation is regulated by a vitamin D receptor-phosphatidylinositol 3-kinase signaling complex. J Exp Med. 1999;190(11):1583–1594. doi: 10.1084/jem.190.11.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishizuka S, Ishimoto S, Norman AW. Biological activity assessment of 1α,25-dihydroxyvitamin D3-26, 23-lactone in the rat. J Steroid Biochem. 1984;20:611–615. doi: 10.1016/0022-4731(84)90131-6. [DOI] [PubMed] [Google Scholar]
- 5.Ishizuka S, Miura D, Ozono K, Chokki M, Mimura H, Norman AW. Antagonistic actions in vivo of (23S)-25-dehydro-1α-hydroxyvitamin D3-26,23-lactone on calcium metabolism induced by 1α,25-dihydroxyvitamin D3. Endocrinology. 2001;142(1):59–67. doi: 10.1210/endo.142.1.7925. [DOI] [PubMed] [Google Scholar]
- 6.Karnauskas AJ, Van Leeuwen JP, Van den Bemd GJ, Kathpalia PP, DeLuca HF, Bushinsky DA, Favus MJ. Mechanism and function of high vitamin D receptor levels in genetic hypercalciuric stone-forming rats. J Bone Miner Res. 2005;20(3):447–454. doi: 10.1359/JBMR.041120. [DOI] [PubMed] [Google Scholar]
- 7.Maitra R, Porter MA, Huang S, Gilmour BP. Inhibition of NFkappaB by the natural product Withaferin A in cellular models of Cystic Fibrosis inflammation. J Inflamm (Lond) 2009;6:15. doi: 10.1186/1476-9255-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masoumi A, Goldenson B, Ghirmai S, Avagyan H, Zaghi J, Abel K, Zheng X, Espinosa-Jeffery A, Mahanian M, Liu P, Hewison M, Hewison M, Mizwicki M, Cashman J, Fiala M. 1a,25-dihydroxyvitamin D3 interacts with curcuminoids to stimulate amyloid-b clearance by macrophages of Alzheimer’s disease patients. Journal of Alzheimer’s Disease. 2009 doi: 10.3233/JAD-2009-1080. [DOI] [PubMed] [Google Scholar]
- 9.Mizwicki MT, Bishop JE, Norman AW. Applications of the vitamin D sterol-Vitmain D receptor (VDR) conformational ensemble model. Steroids. 2005 doi: 10.1016/j.steroids.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 10.Mizwicki MT, Bula CM, Bishop JE, Norman AW. A perspective on how the vitamin D sterol/vitamin D receptor (VDR) conformational ensemble model can potentially be used to understand the structure-function results of A-ring modified vitamin D sterols. J Steroid Biochem Mol Biol. 2005 Oct 1; doi: 10.1016/j.jsbmb.2005.06.025. Published On-line. [DOI] [PubMed] [Google Scholar]
- 11.Mizwicki MT, Bula CM, Bishop JE, Norman AW. New insights into Vitamin D sterol-VDR proteolysis, allostery, structure-function from the perspective of a conformational ensemble model. J Steroid Biochem Mol Biol. 2007;103(3-5):243–262. doi: 10.1016/j.jsbmb.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mizwicki MT, Bula CM, Mahinthichaichan P, Henry HL, Ishizuka S, Norman AW. On the mechanism underlying (23S)-25-dehydro-1{alpha}(OH)-vitamin D3-26,23-lactone antagonism of hVDRwt gene activation and its switch to a superagonist. J Biol Chem. 2009 doi: 10.1074/jbc.M109.042069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizwicki MT, Keidel D, Bula CM, Bishop JE, Zanello LP, Wurtz JM, Moras D, Norman AW. Identification of an alternative ligand-binding pocket in the nuclear vitamin D receptor and its functional importance in 1α,25(OH)2-vitamin D3 signaling. Proc Natl Acad Sci USA. 2004;101(35):12876–12881. doi: 10.1073/pnas.0403606101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mizwicki MT, Norman AW. The vitamin D sterol-vitamin D receptor ensemble model offers unique insights into both genomic and rapid-response signaling. Sci Signal. 2009;2(75):re4. doi: 10.1126/scisignal.275re4. [DOI] [PubMed] [Google Scholar]
- 15.Norman AW, Mizwicki MT, Norman DPG. Steroid hormone rapid actions, membrane receptors and a conformational ensemble model. Nature Reviews Drug Discovery. 2004;3:27–41. doi: 10.1038/nrd1283. [DOI] [PubMed] [Google Scholar]
- 16.Okamura WH, Mitra MN, Procsal DA, Norman AW. Studies on vitamin D and its analogs. VIII. 3-Deoxy-1,25-dihydroxy-vitamin D3, a potent new analog of 1,25-(OH)2-D3. Biochem Biophys Res Commun. 1975;65:24–30. doi: 10.1016/s0006-291x(75)80056-8. [DOI] [PubMed] [Google Scholar]
- 17.Okamura WH, Mitra MN, Wing RM, Norman AW. Chemical synthesis and biological activity of 3-deoxy-1α-hydroxyvitamin D3 an analog of 1α,25-(OH)2-D3, the active form of vitamin D3. Biochem Biophys Res Commun. 1974;60:179–185. doi: 10.1016/0006-291x(74)90189-2. [DOI] [PubMed] [Google Scholar]
- 18.Pike JW, Meyer MB, Watanuki M, Kim S, Zella LA, Fretz JA, Yamazaki M, Shevde NK. Perspectives on mechanisms of gene regulation by 1,25-dihydroxyvitamin D3 and its receptor. J Steroid Biochem Mol Biol. 2007;103(3-5):389–395. doi: 10.1016/j.jsbmb.2006.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plum LA, Prahl JM, Ma X, Sicinski RR, Gowlugari S, Clagett-Dame M, DeLuca HF. Biologically active noncalcemic analogs of 1alpha,25-dihydroxyvitamin D with an abbreviated side chain containing no hydroxyl. Proc Natl Acad Sci USA. 2004;101(18):6900–6904. doi: 10.1073/pnas.0401656101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Procsal DA, Okamura WH, Norman AW. Studies on the mode of action of calciferol IX: Structural requirements for the interaction of 1α,25-(OH)2-vitamin D3 with its chick intestinal receptor system. J Biol Chem. 1975;250:8382–8388. [PubMed] [Google Scholar]
- 21.Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell. 2000;5(1):173–179. doi: 10.1016/s1097-2765(00)80413-x. [DOI] [PubMed] [Google Scholar]
- 22.Siu-Caldera M-L, Sekimoto H, Peleg S, Nguyen C, Kissmeyer A-M, Binderup L, Weiskopf A, Vouros P, Uskokovic M, Reddy GS. Enhanced biological activity of 1α,25-dihydroxy-20-epi-vitamin D3, the C-20 epimer of 1α,25-dihydroxyvitamin D3, is in part due to its metabolism into stable intermediary metabolites with significant biological activity. J Steroid Biochem Mol Biol. 1999;71:111–121. doi: 10.1016/s0960-0760(99)00130-2. [DOI] [PubMed] [Google Scholar]
- 23.Tocchini-Valentini G, Rochel N, Wurtz JM, Mitschler A, Moras D. Crystal structures of the vitamin D receptor complexed to superagonist 20-epi ligands. Proc Natl Acad Sci USA. 2001;98(10):5491–5496. doi: 10.1073/pnas.091018698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wing RM, Okamura WH, Rego A, Pirio MR, Norman AW. Studies on vitamin D and its analogs VII: Solution conformations of vitamin D3 and 1,25-dihydroxyvitamin D3 by high resolution proton magnetic resonance spectroscopy. J Am Chem Soc. 1975;97:4980–4985. doi: 10.1021/ja00850a036. [DOI] [PubMed] [Google Scholar]
- 25.Yamada S, Yamamoto K, Masuno H, Ohta M. Conformation-function relationship of vitamin D: Conformational analysis predicts potential side-chain structure. Journal of Medicinal Chemistry. 1998;41(9):1467–1475. doi: 10.1021/jm970761l. [DOI] [PubMed] [Google Scholar]