

Abstract
A major challenge for neurological therapeutics is the development of small molecule drugs that can activate a panoply of downstream pathways without toxicity. Over the past decade our group has shown that a family of enzymes that regulate posttranscriptional and transcriptional adaptive responses to hypoxia are viable targets for neuronal protection and repair. The family is a group of iron, oxygen, and 2-oxoglutarate-dependent dioxygenases, known as the HIF prolyl 4-hydroxylases (HIF PHDs). We have previously shown that pluripotent protection offered by iron chelators is mediated, in part, via the ability of these agents to inhibit the HIF PHDs. Our group and others have implicated the transcriptional activator HIF-1 in some of the salutary effects of iron chelation-induced PHD inhibition. While some iron chelators are currently employed in humans for conditions such as hemochromatosis, the diverse utilization of iron in physiological processes in the brain makes the development of HIF activators that do not bind iron a high priority. Here we report the development of a high throughput screen to develop novel HIF activators and/or PHD inhibitors for therapeutic use in the central nervous system (CNS). We show that tilorone, a low-molecular weight, antiviral, immunomodulatory agent is the most effective activator of the HIF pathway in a neuronal line. We also show that tilorone enhances HIF protein levels and increases the expression of downstream target genes independent of iron chelation and HIF PHD inhibition in vitro. We further demonstrate that tilorone can activate an HIF-regulated reporter gene in the CNS. These studies confirm that tilorone can penetrate the blood–brain barrier to activate HIF in the CNS. As expected from these findings, we show that tilorone provides effective prophylaxis against permanent ischemic stroke and traumatic spinal cord injury in male rodents. Altogether these findings identify tilorone as a novel and potent modulator of HIF-mediated gene expression in neurons with neuroprotective properties.
Keywords: homeostasis, hypoxia, hypoxia inducible factor, iron, hypoxia response element, erythropoietin, vascular endothelial growth factor, tilorone, desferrioxamine, prolyl hydroxylase
Introduction
Therapeutic trials for acute stroke and traumatic spinal cord injury have been a source of disappointment to the clinical neuroscientific community.1 While the failures have come for many reasons, these experiences provide a firm foundation to reflect on how we should move forward to achieve success. One possible reason for failure is that the drugs we have historically developed to protect the brain and spinal cord affect only a small number of downstream targets.2 As stroke and spinal cord injury are heterogeneous disorders that influence many, if not scores, of pathophysiological pathways, a narrow approach is unlikely to be successful. A broader approach may increase the likelihood that we can prevent damage or sustain repair; however, such an attack holds the intrinsic disadvantage that when more targets are affected, more toxicity is likely to occur.
To develop a more comprehensive but safe therapeutic strategy, we have sought to develop a molecular understanding of how neurons and their associated cellular elements engage adaptive molecular machinery to compensate for oxidative or hypoxic stress. A canonical pathway for adapting to hypoxia or hypoxia ischemia involves the transcriptional activator hypoxia inducible factor-1 (HIF-1).3,4 HIF-1 is a heterodimeric, basic, helix-loop-helix, transcriptional activator that is stabilized in the hypoxic cell to trigger expression of 70–100 genes capable of compensating for a discrepancy in oxygen demand and supply.5,6 These genes include erythropoietin, vascular endothelial growth factor, and glycolytic enzymes; collectively and individually, these genes act to enhance the ability of cells to generate energy in the absence of oxygen and also to deliver oxygen more efficiently to cells.
How can we manipulate this pathway for therapeutic advantage? Over the past 10 years, elegant studies from the Semenza, Ratcliffe, McKnight, and Kaelin laboratories have demonstrated that HIF stability is regulated by a family of iron, oxygen, and 2-oxoglutarate-dependent dioxygenase enzymes called the HIF prolyl 4-hydroxylases.7,8 Three isoforms of HIF prolyl 4-hydroxylases exist in mammals: PHD-1, PHD-2, and PHD-3. Prior work from our group demonstrated that structurally diverse, low-molecular weight, peptide inhibitors of these enzymes prevent neuronal death induced by oxidative stress in vitro or by cerebral ischemia in vivo.9 As important, we demonstrated that neuroprotection by the canonical iron chelator, desferrioxamine mesylate (DFO), can be attributed, in part, to HIF prolyl 4-hydroxylase inhibition. These studies and others have stimulated a reexamination of DFO and its analogs as therapeutic for stroke and spinal cord injury, which is now in progress.10,11 However, since DFO does not penetrate the blood–brain barrier (BBB) well (its molecular weight is 657) and iron is a critical cofactor for many physiological processes, we decided to develop a screen to allow us to identify novel safeHIF activators that penetrate the BBB well and do not bind iron.
In this article, we describe the identification of tilorone12 as a novel and potent activator of the HIF pathway in the central nervous system (CNS). We show that tilorone is able to stabilize HIF in the nervous system. As expected, we also find that tilorone can establish tolerance to ischemic or traumatic injury to the CNS.
Materials and Methods
DFO and 3,4-dihydroxybenzoate (DHB) were purchased from Sigma chemicals; a library of 1040 compounds was purchased from MicroSource Discovery Systems, Inc. USA; tilorone or its analogs R10, 874-DA (below), R11, 567-DA, and R-9536-DA were purchased from Sigma or synthesized by Organix (Woburn, MA) (Fig. 1).
Figure 1.

Scheme.
Cell Culture
Cell cultureswere obtained fromthe cerebral cortex of fetal Sprague-Dawley rats [embryonic day 17 (E17)] as described previously.13 HT22 murine hippocampal cells were cultured in Dulbecco modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, CA) with high glucose, L-glutamine, pyridoxine hydrochloride, and 10% fetal bovine serum (Invitrogen), supplemented with penicillin/streptomycin and 10% fetal bovine serum. For screening of novel HIF activators, HT22 cells were stably transfected with a luciferase promoter–reporter construct driven by a 68 bp region of a known HIF-1 regulated gene, enolase, containing a wild-type hypoxia response element (HRE, 5′-RCTGT-3′).
Luciferase Assay
HT22 cells, stably transfected with a luciferase reporter construct driven by an HRE promoter were plated in 96-well plates overnight prior to treatment with the compound library. All cell extracts were prepared and analyzed using the Luciferase assay system (Promega, Madison, WI), according to the manufacturer’s protocol. Luminescence was measured with a microplate luminometer, and results were expressed as relative light units per mg of total protein or relative light units as a function of number of viable cells.
Immunoblot Analysis
Following drug treatment, cells were scraped into cold phosphate-buffered saline (PBS) solution and centrifuged. After washing with cold PBS, the cell pellets were either used for whole cell lysate preparation using lysis buffer (Boston Bioproducts) added with a protease inhibitors cocktail (Sigma) or subjected to nuclear extraction procedure using NE-PER nuclear and cytoplasmic extraction reagents kit (Pierce Biotechnology). Samples were boiled in Laemmli buffer and electrophoresed under reducing conditions on 12% polyacrylamide gels. Proteins were then transferred to a polyvinylidene difluoride membrane (Bio-Rad, Hercules, CA). Nonspecific binding was inhibited by incubation in Odyssey blocking buffer (LI-COR Biosciences). Primary antibodies against HIF-1 (Upstate cell signaling solutions, Lake Placid, NY), was diluted 1:100 in odyssey blocking buffer and incubated overnight at 4 °C. Respective fluorophore-conjugated Odyssey IRDye-680 or IRD-800 secondary antibody (LI-COR Biosciences) was used at 1:10,000 dilution followed by incubation for 2 h at room temperature. Immunoreactive proteins were detected using an Odyssey infrared imaging system (LI-COR Biosciences).
Immunofluoroscence Staining
Indirect labeling methods were used to determine the levels of HIF-1 protein in HT22 and cortical neuron cultures. HT22 cells were cultured as described above, dissociated cells from cerebral cortex were seeded onto poly-D-lysine-coated eight-well culture slides (Becton Dickinson Labware, Bedford, MA) and treated with Tilorone (10 µM). Cells were washed with warm PBS and fixed at room temperature for 15 min with 4% paraformaldehyde. After washing, cells were incubated with blocking solution containing 0.3% Triton X-100 and 5% goat serum in PBS for one hour, followed by incubation with rabbit HIF-1 antibody (Upstate cell signaling solutions) (1:100 dilution) overnight. After three washes with PBS, cells were incubated with fluorescein isothiocyanate FITC-conjugated goat-anti-rabbit IgG antibody (Molecular Probes, Eugene, OR) (1:200 dilution) and 4′,6-diamidino-2-phenylindole. The slides were washed three times with PBS and mounted with fluorochrome mounting solution (Vector Laboratories, Burlingame, CA). Images were analyzed with a fluorescence microscope (Ziess, Germany). Control experiments were performed in the absence of the primary antibody.
Real-time Polymerase Chain Reaction
The levels of lactic acid dehydrogenase (LDH), erythropoietin (Epo), vascular endothelial growth factor (VEGF), and β-actin were analyzed using TaqMan gene expression assays, using a real-time polymerase chain reaction (PCR) (Applied Biosystems 7500).
Statistical Analysis
All data were entered into a computer database and statistical analysis was performed using the two-tailed paired Student’s t-test. Data are expressed as means ± standard error of the mean (SEM), where appropriate. Significance was accepted at P < 0.05.
In Vivo Experiments Bioluminescence Imaging
HRE–Luciferase Injection
HRE–luciferase adenovirus was delivered directly to the mouse brain (n = 8) via stereotaxic microinjection into the ventricles. Briefly, the right ventricle was targeted using a Benchmark™ digital stereotaxic instrument (Coretech Holdings Co., St. Louis, MO) and a small burr hole (<1-mm diameter) was drilled through the mouse skull to gain access to the ventricle with a 10 µL Hamilton syringe. HRE–luciferase adenovirus [1.0 × 1010 plaque-forming cell (PFU)] was injected into the ventricle using a motorized nanoinjector (KD Scientific Inc., Holliston, MA) at a rate of 0.25 µL/min for 20 min to achieve a final volume of 5μL.Themouse brainwas imaged 24 h post injection using 11.7T magnetic resonance imaging (MRI), allowing 100-µm resolution to assess damage caused by the injection. No discernable difference or injury was observed in comparing left and right ventricles following injection. Animals were kept for 72 h prior to tilorone injection.
Tilorone Injection
Tilorone analog R-10,874-DA (100 mg/kg body weight) was intraperitoneally injected 24 h prior to IVIS imaging (n = 3). Control animals (n = 3) received intraperitoneal (i.p.) saline injection at the same time.
IVIS Imaging
The 100 Series IVIS® Imaging System (Xenogen, Alameda, CA) incorporates a −90 °C-cooled CCD camera that is highly light sensitive. Prior to luciferin injection, a baseline light quantification was performed. Next, firefly d-luciferin potassium salt (Xenogen) was intraperitoneally injected at a concentration of 150 mg/kg body weight into each mouse. Luciferase-catalyzed oxidation of luciferin into oxyluciferin generates energy in the form of light (luminescence). Successful transfection of HRE–luciferase adenovirus and stabilization of HIF-1α by tilorone will produce light in the presence of luciferin substrate that can be measured with the IVIS imaging system. Integral luminescence from a defined region of interest around each animal’s brain was measured using Xenogen© Living Image® Software. Following luciferin injection, a luminescent signal was integrated every 5 min for 40 min, with the peak signal observed at 20min in most animals. The luminescent signal from control mice intraperitoneally injected with saline was similar to background noise.
Middle Cerebral Artery Occlusion
Animal Preparation and Monitoring
Adult male Sprague-Dawley rats weighing 250–280 g (n = 8) (Charles River Laboratories, Wilmington, MA) were operated on and analyzed for this study. Animals were allowed free access to food and water before and after surgery. Briefly, rats were anesthetized by an i.p. injection of chloral hydrate (400 mg/kg), followed 45 min later by a maintenance i.p infusion at a rate of 120 mg/kg/h, using a butterfly needle set. The animals were free breathing. Their body temperatures were kept stable at 36.5 ± 0.5 °Cusing a feedback-regulating heating pad and a rectal probe (Harvard Apparatus, MA). The right femoral artery was cannulated for measurement of arterial blood gases, glucose, and mean arterial blood pressure. These physiological parameters were monitored before and after middle cerebral artery occlusion (MCAO). In addition, laser Doppler Flowmetry (LDF) (Moor Instruments, Devon, UK) was used to monitor the regional cerebral blood flow (rCBF) through a burr hole 2 mm in diameter created in the right parietal bone (2mm posterior and 6 mm lateral to bregma).
Drug Administration
Tilorone (100 mg/kg body weight) was administered intraperitoneally to animals (n = 8) 24 h before permanent MCAO. The control animals received an equivalent volume of the vehicle (water) on a similar administration schedule.
Surgery
All rats were subjected to right MCAO. Under the operating microscope, the right common carotid artery was exposed through a midline incision in the neck. A 4–0 nylon suture with its tip rounded by heating over a flame and subsequently coated with poly-l-lysine (Sigma) was introduced into the external carotid artery and then advanced into the internal carotid artery for a length of 18–19 mm from the bifurcation. This method placed the tip of the suture at the origin of the anterior cerebral artery, thereby occluding the MCA. The placement of the suture tip was monitored by LDF measurements of rCBF. MCAO caused a sharp drop in rCBF to less than 30% of the preischemic baseline. The suture was left in place and the animals were allowed to awaken from the anesthesia following closure of the operation sites.
Infarct Measurement
The animals were anesthetized with ketamine (100 mg/kg, i.p.) and xylazine (50 mg/kg, i.p.) 24 h after MCAO and decapitated. The brain was rapidly removed, sliced into seven 2-mm coronal sections, using a rat matrix (RBM 4000C, ASI Instrument Inc., Warren, MI), and stained according to the standard 2, 3, 5-triphenyltetrazolium chloride (TTC) method.14,15 Each slice was drawn using a computerized image analyzer (Scion Corporation). The calculated infarction areas were then compiled to obtain the infarct volumes per brain (in mm3). Infarct volumes were expressed as a percentage of the contralateral hemisphere volume to compensate for edema formation in the ipsilateral hemisphere.16,17
Rodent Spinal Cord Injury Contusion Model
To examine whether tilorone could have tissue protection in spinal cord contusion injury, we treated Long–Evans hooded rats (n = 8 male, n = 8 female) with 100 mg/kg of tilorone i.p. 40 min before a 25-mm weight drop injury. Five spinal cord tissue pieces at and surrounding the impact site were collected and assayed for lesion volume 24 h after injury.18,19
Results
A screen was developed to identify compounds that activate the HIF pathway as well as or superior to the canonical HIF activator, DFO. We used a promoter–reporter construct that contained 68 bp of a known hypoxia and HIF-1 regulated gene, enolase.20 This 68 bp sequence, containing a canonical HRE (5′-RCTGT-3′), mediated a nearly twofold increase in luciferase reporter expression following transient transfection into an immortalized hippocampal neuroblast cell line (HT22) and treatment with 10 µM DFO (not shown) for 12–16 h. Mutation of the HRE sequence, but not other sequences outside the HRE, abolished the response. Similarly, addition of the vehicle showed no increase in luciferase. To reduce biological variability that results from the use of transfection reagents and to increase the signal-to-noise ratio of the primary screen, immortalized mouse hippocampal neuroblastoma cells were stably transfected with the HRE and HRE-mutant reporter. We verified that the canonical activators of the HIF pathway, such as 1% hypoxia, DFO (10 µM), and dihydroxy-benzoic acid (DHB, 10 µM), increased the expression of the HRE (approximately twofold) but not mutant HRE–luciferase reporter in this cell type (not shown). As expected, classical antioxidants, such as butylated hydroxyanisole or N-acetylcysteine, did not activate either reporter.
We then examined the ability of 1040 compounds in a Microsource library collection to activate the HRE reporter system at levels equivalent to or superior to DFO.21 The drugs were diluted from 10 mM stocks to a final concentration of 10 µM. For each plate, DFO was used as a positive control and 0.1% dimethyl-sulfoxide as a negative control. A number of new putative activators of the HIF pathway were identified. Of note, a number of established HIF activators contained in the library were confirmed by the screen, including DFO (Table 1). Interestingly, one compound at 10 µM activated the HRE–luciferase reporter significantly more effectively than DFO at 10 µM (Table 1). The compound was identified as tilorone, an oral immunomodulatory agent with antiviral and antitumor effects.22,23 Tilorone inhibits the infection of mice with a host of viruses. One putative mode of action of tilorone is mediated via its DNA intercalating properties. As an intercalator, tilorone is believed to facilitate the transcriptional induction of interferons.
TABLE 1.
Hypoxia Response Element–Luciferase Reporter Activation by Desferrioxamine Mesylate and Tilorone (Background Signal Taken as 1.0)
| Compound | Fold activation | Chemical structure |
|---|---|---|
| Deferoxamine mesylate | 1.89 | 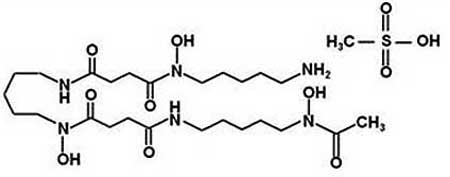 |
| Tilorone | 9.67 | 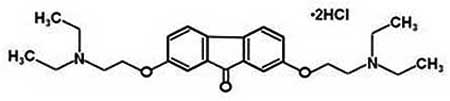 |
To begin to define whether a structure–activity relationship exists among known analogs of tilorone, we analyzed the effects of tilorone and a few of its commercially available analogs on stabilization of the HIF protein. Under normoxia, HIF protein levels are generally low owing to constitutive degradation. Treatment of immortalized hippocampal HT22 cells or primary cortical neurons with tilorone (or tilorone analogs) or hypoxia resulted in an increase in HRE-driven luciferase expression (Table 1). As expected from these findings, tilorone and its analogs also induced HIF-1α protein levels as measured by immunoblot (Fig. 2A) or immunofluorescence (Fig. 2B). Unlike DFO, the stabilization of HIF-1α by tilorone is unlikely related to inhibition of the HIF PHDs for several reasons. First, tilorone or its analogs are unable to stabilize an oxygen-dependent domain–luciferase fusion protein in SH-SY5Y human neuroblastoma cells, while the canonical PHD inhibitors do stabilize it (not shown). Second, PHD inhibitors induce HRE-dependent luciferase reporter activity in a host of cell types, including Hep3b cells. Tilorone does not induce significant increases of an HRE reporter in Hep3b cells (not shown). Third, at concentrations delivered to cells (10 µM), tilorone does not significantly inhibit recombinant PHD-1, PHD-2, or PHD-3 in a test tube [PHD-1, 6.7% inhibition at 200 µM tilorone; PHD-2, 11.2% inhibition at 200 µM tilorone (1 µM iron added to the test tube), 8.2% inhibition at 200 µM (40 µM iron added to the test tube); PHD-3, 1.5% inhibition at 200 µM tilorone]. Together these results suggest that neither tilorone nor one of its cellular metabolites are able to inhibit HIF PHD function with the efficacy of known PHD inhibitors (the IC50 for DFO or DHB in inhibiting recombinant PHD-1, PHD-2, or PHD-3 is 0.1–1 µM).
Figure 2.

Tilorone induces hypoxia inducible factor (HIF) protein levels in the nucleus of mouse hippocampal neuroblasts (HT22) or rat primary cortical neurons. (A) Immunoblot analysis with an antibody to HIF-1α in lysates from nontreated (control) mouse HT22 cells, canonical HIF activators [1% 02, cobalt chloride (CoCl2), or desferrioxamine mesylate (DFO)], or tilorone and its analogs (see Materials and Methods). (B) HT22 cells or primary cortical neurons stained with 4′,6-diamidino-2-phenylindole to label nuclei blue (A, control; D, tilorone) or stained for HIF-1α (green; B, control; E, tilorone). Overlay (C, control; F, tilorone) indicates that tilorone increases nuclear and extranuclear HIF-1α (Abbreviations: Epo, erythropoietin; LDH, lactic acid dehydrogenase; VEGF, vascular endothelial growth factor).
We next examined whether tilorone or its analogs could induce robust expression of known HIF-dependent targets, such as VEGF, Epo, and glycolytic enzymes in vitro. Indeed, we found that 10 µM tilorone was significantly more effective in inducing the expression of canonical HIF targets than 100 µM DFO (Fig. 3), consistent with our findings from the reporter studies. Interestingly, the panoply of gene targets induced by tilorone included putative prosurvival (VEGF, Epo) and prodeath (BNIP3) genes. The increase in prodeath gene expression occurred in the absence of any change in basal viability in the cultures (not shown).
Figure 3.
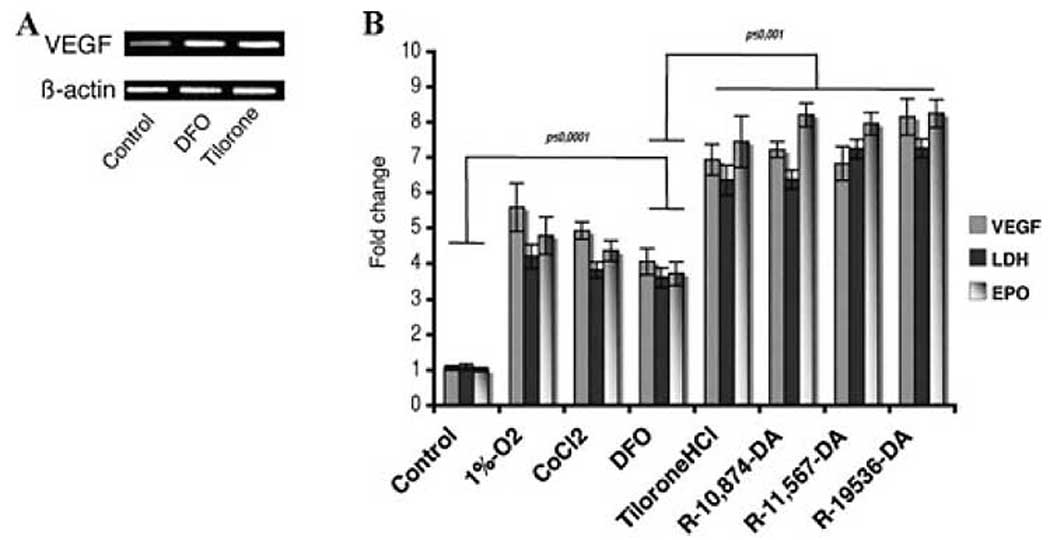
Tilorone induces the expression of canonical HIF-activated genes in HT22 cells (A) or primary neurons (B). (A) Total RNA was collected following treatment DFO (100 µM) or Tilorone (10 µM). Real-time polymerase chain reaction (RT-PCR) using specific primers determined the level of mRNA. (B) Primary neurons were exposed to normoxia (control), canonical HIF activators (1% hypoxia, CoCl2, or DFO), or tilorone or its analogs. Total RNA was collected. Quantitative PCR using specific primers for VEGF, Epo, and LDH (known HIF targets) was performed. Results shown are mean ± SEM; P < 0.05.
Together these studies argue that tilorone induces HIF activation more potently than DFO or other putative PHD inhibitors. The lack of shift of the high IC50 for inhibiting PHD-2 by tilorone in the presence of low or high iron concentrations suggests that, even if tilorone has weak PHD-2 inhibitory properties, this inhibition occurs independently of iron chelation to mediate its HIF activating effects.
To begin to assess the in vivo effects of tilorone, we examined the effects of various concentrations of tilorone delivered intraperitoneally on HIF stabilization in the CNS. Consistent with our in vitro observations, we found that above 50 mg/kg increased HIF-1α protein could be observed in lysates from brain tissue (Fig. 4A). As expected, this increase in HIF protein was associated with an increased expression of a HIF reporter gene as measured by in vivo bioluminescence imaging (Fig. 4B and C). Thus, tilorone crosses the BBB, in contrast to DFO, which penetrates the BBB poorly and fails to activate the reporter (Sen et al., unpublished observations).
Figure 4.

Tilorone stabilizes HIF-1 protein levels in brains of animals in a dose-dependent manner (A) and increases hypoxia response element (HRE)–luciferase expression in rat brains (B). (A) Immunoblot analysis with an antibody to HIF-1α in lysates from brains of nontreated (control) animals or following treatment with indicated doses of tilorone or 1% oxygen. (B) Bioluminescence imaging of animals treated with tilorone analog R-10,874-DA (100 mg/kg body weight) was injected intraperitoneally 24 h prior to IVIS imaging (n = 3). Control animals (n = 3) received i.p. saline injection at the same time. (C) Quantification of emitted light (see Materials and Methods for details). Results shown are mean ± SEM.
To examine whether tilorone could prevent neuronal injury, we treated male Sprague-Dawley Rats (n=8) with 100 mg/kg of tilorone intraperitoneally and then exposed these animals to permanent focal ischemia (MCAO) 24 h later, as previously described.9 Tilorone was associated with a significant reduction in infarct volume as measured histologically by TTC staining [74% reduction in infarct volume (P < 0.005), Fig. 5A and B]. Protection could not be attributed to tilorone-induced changes in body temperature, plasma glucose levels, plasma pH, or blood flow (not shown). Together these studies demonstrate that tilorone is a potent HIF activator in vivo and also confers prophylaxis against cerebral ischemia.
Figure 5.

Tilorone administration reduces infarct volume in a permanent middle cerebral artery occlusion model. (A) Male Sprague-Dawley rats (n = 8) nontreated controls or treated intraperitoneally (n = 8) with 100 mg/kg of tilorone 24 h prior to permanent middle cerebral artery occlusion infarct volume as measured histologically by 2, 3, 5-triphenyltetrazolium chloride staining. (B) Quantification of infarct volume. Results shown are mean ± SEM (P < 0.005).
To determine whether tissue protection by tilorone could be extended to other CNS injury paradigms, such as spinal cord injury, we examined the effects of tilorone in a rodent spinal cord contusion model. Tilorone (100 mg/kg) was injected into the peritoneum and increased HIF-dependent gene expression in the spinal cord similar to supratentorial sites (not shown). In Long–Evans hooded rats tested for whether tilorone could have tissue protection in spinal cord contusion injury, there was a 33% lesion volume reduction in the male tilorone-treated group (Fig. 6).
Figure 6.
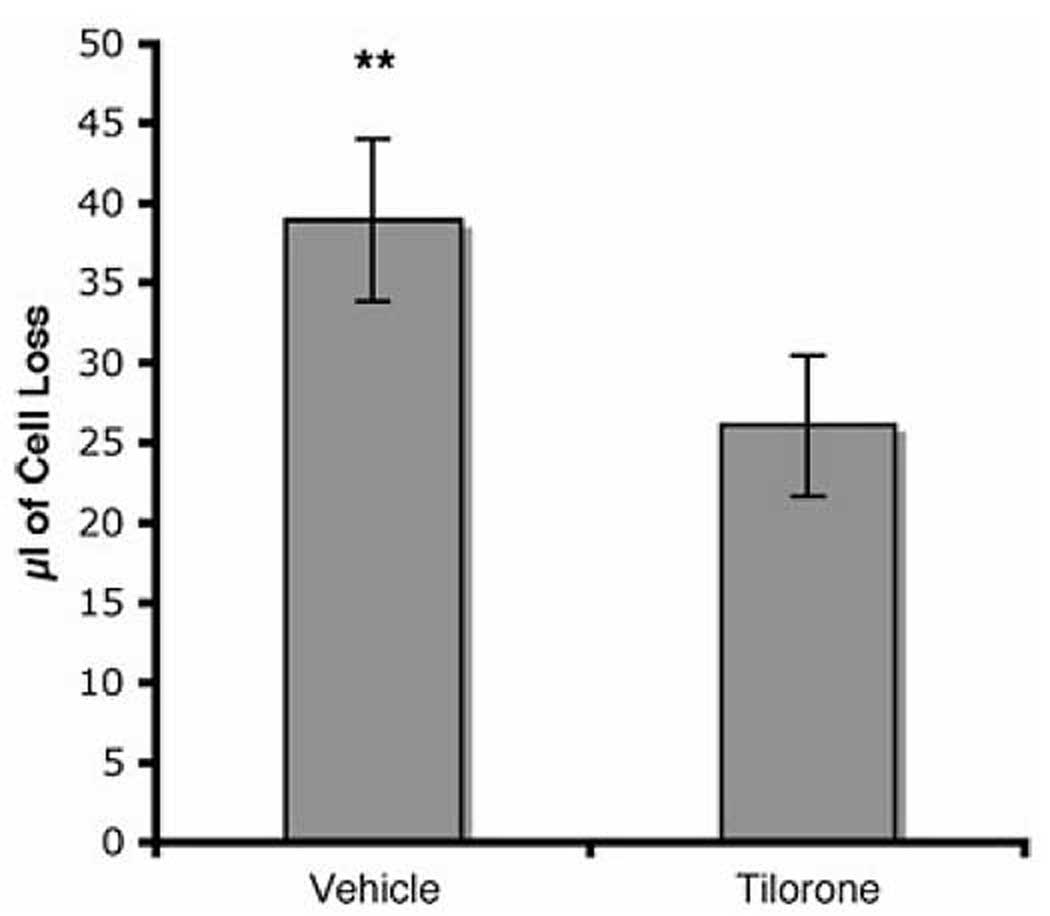
Tilorone administration reduces lesion volume in a spinal cord contusion injury model. Long–Evans hooded rats (n = 8) treated intraperitoneally with 100 mg/kg of tilorone 40 min prior to a 25-mm weight drop injury. Lesion volume was assayed 24 h after injury. Results shown are mean ± SEM.
Discussion
We screened a library of Food and Drug Administration-approved compounds to identify novel compounds that activate the HIF pathway for potential use as neuroprotectants in humans.21 The most potent and effective activator of the HIF pathway in our screen was identified as tilorone, a potent oral immunomodulatory and interferon-inducing agent. Tilorone (10 µM) induced a nearly 10-fold increase in HRE-dependent luciferase activity, while DFO (10 µM) induced less than twofold (Table 1).These observations in the primary screen were supported by several studies that demonstrated that tilorone can increase HIF-1α protein levels in the nucleus of neurons (Fig. 2) and stimulate the expression of known HIF-dependent gene expression (Fig. 2–Fig 4). Of note, tilorone also appears to traverse the BBB to stabilize HIF-1α protein and increase the activity of a reporter driven by the HRE in vivo (Fig. 4). These studies establish tilorone as a novel activator of the HIF pathway in the brain.
The ability of tilorone to activate HIF in the CNS in vivo was correlated with its ability to confer significant resistance to stroke and spinal cord injury (Fig. 5 and Fig. 6). Although it is unclear whether there is a causal relationship between the neuroprotective properties of tilorone and its ability to induce HIF, the ability of tilorone to penetrate the BBB represents a significant advance over other known HIF activators, such as DFO or cobalt chloride. Indeed, tilorone was able to stabilize HIF and induce the expression of a HIF-dependent reporter gene in the CNS in the absence of injury, suggesting that it can penetrate a normal BBB (Fig. 4). An interesting but unexpected result was our inability to detect the upregulation of known HIF-dependent genes in the region where we observed clear induction of an HIF reporter gene in vivo (not shown). The findings suggest that other factors besides HIF stabilization may govern the expression of HIF-dependent genes in the CNS. A search is underway to define those factors.
Tilorone was marketed in the early 1970s as the first low-molecular weight interferon-inducing agent.22 This property of tilorone appears to derive from its ability to intercalate into DNA and thereby enhance the expression of certain cytokines. It was removed from the market because of in vitro studies that indicated that it could induce storage of sulphated glycosaminoglycans (GAGs). These side effects were attributed to symmetrical basic side chains in the 2 and 7 positions. The model is that these side chains interface with GAGs and prevent them from lysosomal attack. Monobasic, asymmetrically substituted, tilorone derivatives appear to have no effect on GAG synthesis while maintaining their intercalating and immunomodulatory properties.24 Studies are currently underway to determine whether the monobasic tilorone derivatives still are capable of activating the pathway.
The above discussion raises the important question of how tilorone is stabilizing HIF and activating gene expression. The dibasic symmetrically substituted compounds would be expected to target the lysosome. As the lysosome is now known to be the site of degradation of many cytosolic proteins, it is possible that tilorone influences HIF stability by altering lysosomal function rather than by canonical pathways involving HIF hydroxylation and proteasomal degradation.25 An alternative and equally attractive possibility is that tilorone is inducing increases in HIF levels not by regulating degradation but rather by influencing synthesis. 26,27 One possible mechanism by which tilorone might induce HIF synthesis is via activation of cell surface nicotinic receptors leading to increased activity of Erk or Akt.28 Akt has been shown to increase HIF levels via mTOR activation and increased translation. Future studies will distinguish among these and other possibilities.
In closing, we have used a simple promoter-based screen to identify tilorone or some of its analogs as novel activators of the HIF pathway in the CNS. While the precise mechanism by which tilorone protects neurons in vivo remains unclear, the correlation between the protective effects of tilorone and its ability to activate the HIF pathway is striking. Studies are underway to determine the optimal mode of delivery to determine whether tilorone can be delivered post injury and mediate protective or reparative effects expected from HIF activation in the CNS.
Acknowledgments
We would like to gratefully acknowledge the technical and intellectual input of Dr. Juan Chavez who contributed several figures to this manuscript and Dr. Joe LaManna. The work was funded by New York State Department of Health Contract (CO19772), the Center for Research Excellence in Spinal Cord Injury, and the Adelson Foundation for Neurorehabilitation and Repair.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Savitz SI, Fisher M. Future of neuroprotection for acute stroke: in the aftermath of the SAINT trials. Ann Neurol. 2007;61:396–402. doi: 10.1002/ana.21127. [DOI] [PubMed] [Google Scholar]
- 2.Ratan RR, et al. Translation of ischemic preconditioning to the patient: prolyl hydroxylase inhibition and hypoxia inducible factor-1 as novel targets for stroke therapy. Strok. 2004;35(11 Suppl 1):2687–2689. doi: 10.1161/01.STR.0000143216.85349.9e. [DOI] [PubMed] [Google Scholar]
- 3.Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci. STKE. 2007 doi: 10.1126/stke.4072007cm8. cm8. [DOI] [PubMed] [Google Scholar]
- 4.Ratan RR, et al. Harnessing hypoxic adaptation to prevent, treat, and repair stroke. J. Mol. Med. 2007;85:1331–1338. doi: 10.1007/s00109-007-0283-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freeman RS, Barone MC. Targeting hypoxia-inducible factor (HIF) as a therapeutic strategy for CNS disorders. Curr. Drug Targets CNS Neurol. Disord. 2005;4:85–92. doi: 10.2174/1568007053005154. [DOI] [PubMed] [Google Scholar]
- 6.Semenza GL. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem. J. 2007;405:1–9. doi: 10.1042/BJ20070389. [DOI] [PubMed] [Google Scholar]
- 7.Stolze IP, Mole DR, Ratcliffe PJ. Regulation of HIF: prolyl hydroxylases. Novartis Found. Symp. 2006;272:15–25. discussion 25–36. [PubMed] [Google Scholar]
- 8.Siddiq A, Aminova LR, Ratan RR. Hypoxia inducible factor prolyl 4-hydroxylase enzymes: center stage in the battle against hypoxia, metabolic compromise and oxidative stress. Neurochem. Res. 2007;32:931–946. doi: 10.1007/s11064-006-9268-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siddiq A, et al. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J. Biol. Chem. 2005;280:41732–41743. doi: 10.1074/jbc.M504963200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freret T, et al. Delayed administration of deferoxamine reduces brain damage and promotes functional recovery after transient focal cerebral ischemia in the rat. Eur. J. Neurosci. 2006;23:1757–1765. doi: 10.1111/j.1460-9568.2006.04699.x. [DOI] [PubMed] [Google Scholar]
- 11.Woo KJ, et al. Desferrioxamine, an iron chelator, enhances HIF- 1alpha accumulation via cyclooxygenase-2 signaling pathway. Biochem. Biophys. Res. Commun. 2006;343:8–14. doi: 10.1016/j.bbrc.2006.02.116. [DOI] [PubMed] [Google Scholar]
- 12.Krueger RE, Mayer GD. Tilorone hydrochloride: an orally active antiviral agent. Science. 1970;169:1213–1214. doi: 10.1126/science.169.3951.1213. [DOI] [PubMed] [Google Scholar]
- 13.Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. 1990;4:1624–1633. [PubMed] [Google Scholar]
- 14.Bederson JB, et al. Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke. 1986;17:1304–1308. doi: 10.1161/01.str.17.6.1304. [DOI] [PubMed] [Google Scholar]
- 15.Belayev L, et al. Middle cerebral artery occlusion in the rat by intraluminal suture. Neurological and pathological evaluation of an improved model. Stroke. 1996;27:1616–1622. doi: 10.1161/01.str.27.9.1616. discussion 1623. [DOI] [PubMed] [Google Scholar]
- 16.Swanson RA, et al. A semiautomated method for measuring brain infarct volume. J. Cereb. Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- 17.Ayoub IA, et al. Nicotinamide reduces infarction up to two hours after the onset of permanent focal cerebral ischemia in Wistar rats. Neurosci. Lett. 1999;259:21–24. doi: 10.1016/s0304-3940(98)00881-7. [DOI] [PubMed] [Google Scholar]
- 18.Hashimoto M, et al. Osteopontin-deficient mice exhibit less inflammation, greater tissue damage, and impaired locomotor recovery from spinal cord injury compared with wild-type controls. J. Neurosci. 2007;27:3603–3611. doi: 10.1523/JNEUROSCI.4805-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Constantini S, Young W. The effects of methylprednisolone and the ganglioside GM1 on acute spinal cord injury in rats. J. Neurosurg. 1994;80:97–111. doi: 10.3171/jns.1994.80.1.0097. [DOI] [PubMed] [Google Scholar]
- 20.Semenza GL, et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996;271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- 21.Heemskerk J, Tobin AJ, Bain LJ. Teaching old drugs new tricks. Meeting of the Neurodegeneration Drug Screening Consortium, 7–8 April 2002, Washington, DC, USA. Trends Neurosci. 2002;25:494–496. doi: 10.1016/s0166-2236(02)02236-1. [DOI] [PubMed] [Google Scholar]
- 22.Chandra P, Wright GJ. Tilorone hydrochloride: the drug profile. Top Curr. Chem. 1977;72:125–148. doi: 10.1007/BFb0048451. [DOI] [PubMed] [Google Scholar]
- 23.Pollard RB. Usages of interferon and interferon inducers in man. Med. Biol. 1980;58:57–62. [PubMed] [Google Scholar]
- 24.Alcaro S, Ortuso F, Coleman RS. Molecular modeling of DNA cross-linking analogues based on the azinomycin scaffold. J. Chem. Inf. Model. 2005;45:602–609. doi: 10.1021/ci0496595. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh P, Kornfeld S. The GGA proteins: key players in protein sorting at the trans-Golgi network. Eur. J. Cell Biol. 2004;83:257–262. doi: 10.1078/0171-9335-00374. [DOI] [PubMed] [Google Scholar]
- 26.Treins C, et al. Insulin stimulates hypoxiainducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J. Biol. Chem. 2002;277:27975–27981. doi: 10.1074/jbc.M204152200. [DOI] [PubMed] [Google Scholar]
- 27.Nishi K, et al. LPS induces hypoxia-inducible factor 1 activation in macrophage-differentiated cells in a reactive oxygen species-dependent manner. Antioxid. Redox Signal. 2008;10:983–996. doi: 10.1089/ars.2007.1825. [DOI] [PubMed] [Google Scholar]
- 28.Briggs CA, et al. alpha7 nicotinic acetylcholine receptor agonist properties of tilorone and related tricyclic analogues. Br. J. Pharmacol. 2008;153:1054–1061. doi: 10.1038/sj.bjp.0707649. [DOI] [PMC free article] [PubMed] [Google Scholar]