

Abstract
Objective
Patent foramen ovale and pulmonary arteriovenous shunts are associated with serious complications such as cerebral emboli, stroke, and migraine with aura. The pathophysiological mechanisms that link these conditions are unknown. We aimed to establish a mechanism linking microembolization to migraine aura in an experimental animal model.
Methods
We introduced particulate or air microemboli into the carotid circulation in mice to determine whether transient microvascular occlusion, insufficient to cause infarcts, triggered cortical spreading depression (CSD), a propagating slow depolarization that underlies migraine aura.
Results
Air microemboli reliably triggered CSD without causing infarction. Polystyrene microspheres (10μm) or cholesterol crystals (<70μm) triggered CSD in 16 of 28 mice, with 60% of the mice (40% of those with CSD) showing no infarcts or inflammation on detailed histological analysis of serial brain sections. No evidence of injury was detected on magnetic resonance imaging examination (9.4T; T2 weighted) in 14 of 15 selected animals. The occurrence of CSD appeared to be related to the magnitude and duration of flow reduction, with a triggering mechanism that depended on decreased brain perfusion but not sustained tissue damage.
Interpretation
In a mouse model, microemboli triggered CSD, often without causing microinfarction. Paradoxical embolization then may link cardiac and extracardiac right-to-left shunts to migraine aura. If translatable to humans, a subset of migraine auras may belong to a spectrum of hypoperfusion disorders along with transient ischemic attacks and silent infarcts.
Migraine headaches are among the most common and debilitating conditions and occur in 10 to 12% of the general population.1 Migraine with aura accounts for 15% of cases, and the aura is characterized most commonly by visual or somatosensory symptoms that often anticipate the onset of headache by 20 to 40 minutes. Certain patients with migraine auras are at greater risk for stroke.2,3 Despite the multiplicity of potential mechanisms linking migraine aura and stroke,4 experimental evidence linking the triggering of migraine aura attacks to microvascular dysfunction is lacking. Cortical spreading depression (CSD) may be important to this link.
CSD is a slowly propagating intense neuronal and glial depolarization that spreads at a characteristic rate of 3 to 5mm per minute. It is a property of all mammalian cortices, but varies in susceptibility between the rodent and more resistant human brain.5 A number of high- and low-resolution brain imaging studies have led to the conclusion that CSD causes migraine aura.6–8 For example, during visual aura, a slowly propagating wave of cerebral blood oxygenation level-dependent (BOLD) signal change was recorded in calcarine cortex in a migraineur using near continuous high-resolution functional magnetic resonance imaging (MRI).6 The observed perturbations of the BOLD signal were retinotopically congruent with the patient’s visual percept, and displayed several CSD characteristics (eg, typical blood flow response and velocity of propagation). In experimental animals, CSD triggers specific gene programs to precondition the brain and render it more resistant to subsequent tissue injury.9 In patients, slowly propagating depolarizations closely resembling CSD have now been well documented following ischemic stroke,10 intracerebral hematoma, subarachnoid hemorrhage, and brain trauma,11,12 further suggesting that human brain can sustain slowly spreading depolarizations very much like CSD.
A pioneer of the vascular theory of migraine, Harold G. Wolff proposed that the neurological symptoms of migraine aura were caused by cerebral vasoconstriction and ischemia.13 Similarly, Van Harreveld and Stamm ascribed CSD to ischemia resulting from a spreading wave of vasoconstriction.14 Indeed, several vascular risk factors have been implicated in attacks of migraine aura.15 For example, migraine has been identified in several studies as an independent risk factor for ischemic stroke.3 Migraine with aura and stroke are also reportedly more common in patients with right-to-left cardiac shunts, usually in the setting of a patent foramen ovale (PFO),16 or less commonly, pulmonary arteriovenous malformations,17 although the strength of this relationship has been questioned recently.18 The mechanism for this association is not clear, but paradoxical embolization has been proposed as a possible explanation.
To establish a mechanism linking migraine aura to blood components, blood vessels and intracardiac or arteriovenous shunts, we examined whether small microemboli injected into the carotid artery could evoke CSD in otherwise normal mouse brain. We hypothesized that the injection of minute amounts of air, polystyrene microspheres, or cholesterol crystals into carotid artery could trigger CSD, and that CSD in this context could develop without requisite tissue damage. This work was reported previously in preliminary form.19
Materials and Methods
General Surgical Preparation
The care and handling of the animals and experimental protocols were in accordance with National Institutes of Health guidelines and approved by our Institutional Animal Care and Use Committee. Mice (C57BL/6J, 23–28g; Charles River Laboratories, Wilmington, MA) were anesthetized with 2% isoflurane in 70% N2O and 30% O2. Isoflurane (1%) was used for maintenance. Rectal temperature was maintained at 36.8 to 37.2°C. Blood pressure and arterial blood gases were measured. A PE-10 polyethylene catheter was inserted retrogradely into the right external carotid artery and advanced into the carotid bifurcation. The wound was then closed, and the animal was placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA).
Electrophysiological Recordings
A burr hole was opened over the right hemisphere, 1mm posterior and 1.5mm lateral to the bregma. The steady potential and electrocorticogram (ECoG) were recorded with a glass micropipette filled with 200mM NaCl, inserted 300μm below the dural surface (tip resistance 1–2MmΩ, Axoprobe-1A, Axon Instruments, Union City, CA; Fig 1). An Ag/AgCl reference electrode was placed subcutaneously in the neck.
FIGURE 1.
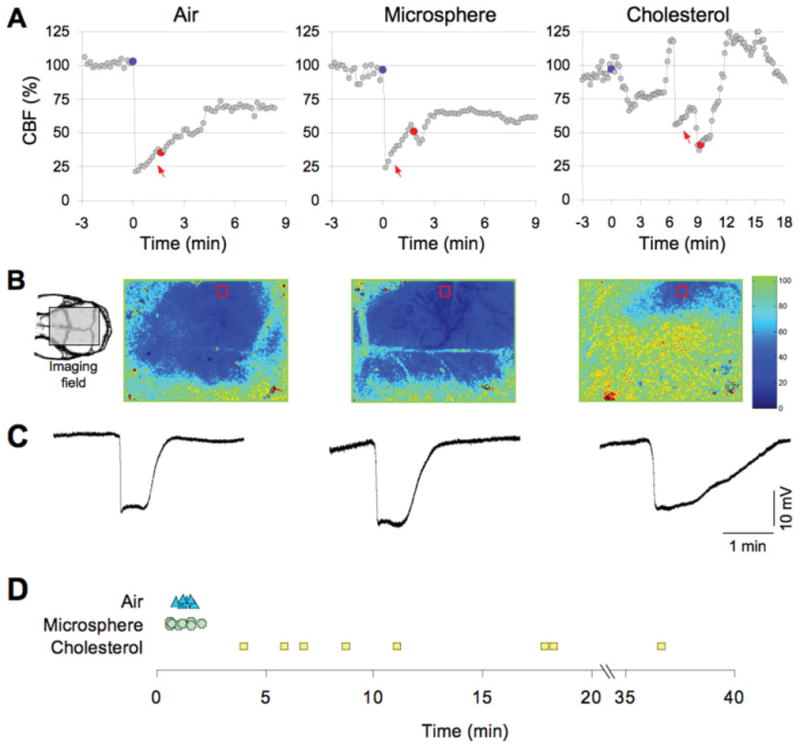
Microembolic hypoperfusion triggers cortical spreading depression (CSD). (A) Representative tracings show the time course of cerebral blood flow (CBF) reduction after intracarotid air, microsphere, or cholesterol injection (blue circles). Both air and microspheres abruptly but transiently decreased CBF to ≤25% of baseline, whereas the hypoperfusion after cholesterol microemboli was less predictable and fluctuated over time (note the compressed time scale for the cholesterol data). CSD (red circles) occurred within a few minutes after air or microsphere embolization, but was more delayed after cholesterol microemboli. CSDs were detected by the characteristic slow extracellular direct current (DC) shift shown in C and by a propagating wave of hypoperfusion. (B) Full-field images of CBF taken at the denoted time point between microembolization (red arrows in A) and CSD onset showing the spatial extent of hypoperfusion. Air and microspheres reduced CBF within the ipsilateral middle cerebral artery (MCA) and bilateral anterior cerebral artery territories, whereas cholesterol-induced hypoperfusion was more restricted to ipsilateral MCA branches. Red squares show the regions of interest within which the CBF time courses (shown in A) were quantified for each type of microemboli. The orientation of the imaging field over the mouse skull is shown in the inset (left). The inset shows the position of the rectangular imaging field. (C) Extracellular DC potential shifts characteristic of CSD were triggered by intracarotid microembolization with air (left), microsphere (middle), or cholesterol (right), and recorded by intracortical glass micropipettes within the ipsilateral MCA territory simultaneously with the laser speckle flowmetry. (D) The timing of CSD after air (triangles), microsphere (crosses), and cholesterol microemboli (squares) is shown for each animal. Both air and microspheres evoked a CSD within a few minutes after microembolization, whereas the onset of CSD was significantly delayed after cholesterol microemboli.
Experimental Groups and Intervention
Mice were randomly assigned to undergo microembolization with saline (10μl; n = 8), air (0.5μl; n = 6), cholesterol crystals (3β-hydroxy-5-cholestene; <70μm-diameter, 4,000 particles; Sigma-Aldrich, St. Louis, MO; n = 12), or polystyrene microspheres (5 or 10μm diameter, up to 225,000 particles; Duke Scientific, Fremont, CA; n = 16 and 8, respectively), at a speed of 10μl/min into the carotid bifurcation (Table 1). Hemodynamic data, cerebral cortical blood flow, and ECoG were recorded continuously for 60 minutes (PowerLab; ADInstruments, Bella Vista, Australia). Animals were then decannulated and allowed to emerge from anesthesia. Systemic physiological parameters did not differ among experimental groups, and remained within normal range throughout the experiments.
TABLE 1.
Occurrence and Electrophysiological Properties of Microembolic CSDs
| Microemboli | Diameter (μm) | Volume (μl) | Particles | No. | Occurrence | Latency (min) | Duration (sec) | Amplitude (mV) |
|---|---|---|---|---|---|---|---|---|
| Saline | — | 10 | — | 8 | 0 | — | — | — |
| Air | — | 0.5 | — | 6 | 6 | 1.3 [1.2–1.6] | 56 [38–210] | 23 [21–34] |
| Microspheres | 20 | 5 | 7,000 | 10 | 10 | |||
| 10 | 0.5 | 10,000 | 5 | 0 | — | — | — | |
| 10 | 1 | 20,000 | 16 | 8 | 1.1 [0.6–1.6] | 62 [47–146] | 23 [18–25] | |
| 10 | 10 | 200,000 | 9 | 8 | 1.2 [0.8–1.5] | 77 [31–196] | 25 [22–27] | |
| 5 | 1.5 | 225,000 | 8 | 1 | 2 | 43 | 14 | |
| Cholesterol | <70 | 15 | 4,000 | 12 | 8 | 9.9 [6.4–18.1]a | 148 [122–470]a | 23 [18–28] |
CSD duration was measured from baseline to baseline. CSD latency is measured between injection of microemboli and onset of CSD. Data are presented as median [25–75% range].
p < 0.05 vs. air or microspheres.
CSD = cortical spreading depression; No. = number of animals.
Optical Imaging
Laser speckle flowmetry was used to study the spatiotemporal characteristics of cerebral blood flow (CBF) changes during the experiment.20 Relative CBF images were calculated from the ratio of baseline image of correlation time to subsequent images obtained approximately every 10 seconds.
MRI Study
Eleven mice with microsphere microembolization and 4 saline control animals were examined. MRI was performed on a 9.4T 21cm diameter horizontal bore animal magnet (Magnex Scientific, Yarnton, UK) with a AVANCE console; (Bruker BioSpin Corporation, Billerica, MA). The MRI protocols included: (1) rapid acquisition with relaxation enhancement (RARE) tripilot acquisition sequence for localization; and (2) multislice T2-weighted MRI (RARE sequence) in the rostralcaudal direction (20 contiguous slices with 0.5mm thickness), with repetition time = 7,000 milliseconds, echo time = 25 milliseconds, voxel size = 117×117 μm2, RARE factor = 8, and number of averages = 4.
Neurological Testing
Neurological function was assessed by a blinded neurologist in 5 animals after cholesterol embolization starting 1 hour after emergence from anesthesia. Exams were on days 1–3, 10, 14. Motor deficit, coordination, and fine movements were assessed quantitatively.21
Tissue Processing and Histological Evaluation
Brains from euthanized mice were fixed by immersion in 10% formalin. In preliminary studies, we found that the smallest lesion detected was approximately 100μm in diameter. Therefore, we obtained 5μm-thick paraffin sections at 100μm intervals throughout the forebrain, cerebellum, and brainstem, to detect a single 100μm infarct nearly 100% of the time.22 Sections were stained with hematoxylin/eosin and examined with optical microscopy by a pathologist blinded to the experimental groups.
Statistical Analysis
Data are expressed as mean ± standard deviation or median (25–75% range). Hemodynamic and blood gas parameters were compared between the groups using factorial analysis of variance. Mann-Whitney rank sum test was used to test group differences in CSD parameters, as well as the impact of the CBF deficit (area under the curve) on the probability of CSD occurrence or brain infarction. Fisher exact test was used to assess differences in CSD occurrence between groups or the presence of microinfarcts. Bonferroni correction was used to preserve the overall type I error rate at 0.05; p < 0.05 was considered statistically significant.
Results
CSD Is Readily Triggered by Intravascular Cerebral Microemboli
Injections of air, cholesterol crystals, and microspheres all triggered CSD, whereas intracarotid infusion of vehicle (normal saline, n = 8) did not. Air microemboli caused CSD in all 6 mice (see Table 1), and was detected by characteristic slow direct current (DC) potential shifts (see Fig 1C) and a concomitant spreading wave of oligemia typical for a mouse brain. Cholesterol crystals and polystyrene microspheres caused CSD in 8 of 12 and 8 of 16 mice, respectively. Injecting a higher number of microspheres (200,000 particles) increased CSD probability (8 of 9 mice). Larger microspheres (20μm diameter; n = 10) caused severe and sustained CBF reduction with anoxic depolarization and tissue infarction, with early death after embolization. Conversely, when fewer (10μm diameter; 10,000 particles) or smaller-diameter microspheres (5μm diameter; 225,000 particles) were administered, CSDs were less frequent (0 of 5 and 1 of 8 mice, respectively) suggesting dose-response and size-response relationships. Air microemboli always triggered a single CSD. Recurrent CSDs were observed 10 to 30 minutes after injecting microspheres (2 of 16 mice; 20,000 particles) or cholesterol crystals (1 of 12), and in 4 of 9 mice when testing a larger number of microspheres (200,000 particles). A mild and transient contralateral weakness was detected immediately after surgery in animals exhibiting a CSD; deficits were not detected at 24 hours.
The duration of CSDs triggered by air and microspheres did not differ significantly from those triggered by topical KCl application to normal cortex (55 [49–61] seconds, tested in a separate group of mice; median [25–75% range]) (see Table 1, Fig 1C). By contrast, CSDs following cholesterol crystal microembolization were significantly prolonged, possibly indicating prolonged vascular blockage. The latency from injection to CSD onset also differed among microemboli. Air microemboli and microspheres triggered CSD within 1 to 2 minutes after injection, whereas the onset was significantly delayed (9.9 minutes) after cholesterol microemboli (see Fig 1D, Table 1).
CSD Is Triggered by Microembolic Hypoperfusion
Hypoperfusion and CSD followed the injection of all 3 microemboli, and its timing and point of origin corresponded to the onset, duration, magnitude, and location of microembolic hypoperfusion. Although not all hypoperfusion episodes were followed by a CSD, all microembolic CSDs were preceded by global or regional hypoperfusion. Air abruptly decreased CBF to 19% (25–75% range, 13–26%) of baseline, often within the entire ipsilateral hemisphere and contralateral anterior cerebral artery (ACA) territory (see Fig 1A, B). The hypoperfusion was followed by a CSD within 2 minutes (see Table 1), originating from the arterial territories that showed the most severe CBF reduction. The spatial extent, magnitude, and time course of hypoperfusion after microspheres (10μm; 20,000 particles) were similar to air microembolism (23% [25–75% range, 16–30%] of baseline; see Fig 1A, B). Regarding the clinical relevance of this volume of air, 0.5μl was roughly equivalent to 1–2ml in the human, which is larger than the clinical experience in the echocardiography laboratory using 5–10ml of agitated saline to diagnose PFO. However, injections of < 0.5μl air were not feasible in the mouse.
Contrasting with the blood flow response to air or microspheres, cholesterol microemboli caused milder hypoperfusion (56% [25–75% range, 38–66%]), affecting a smaller area of cortex usually limited to the ipsilateral hemisphere (see Fig 1B). This mild hypoperfusion, however, was longer-lasting, and showed transient, abrupt, and apparently spontaneous fluctuations (see Fig 1A). These CBF fluctuations, when severe and prolonged, evoked a CSD from the cortical focus of hypoperfusion (Fig 2). As a result, cholesterol microemboli frequently triggered CSDs, albeit in a delayed fashion (see Table 1; Fig 1).
FIGURE 2.
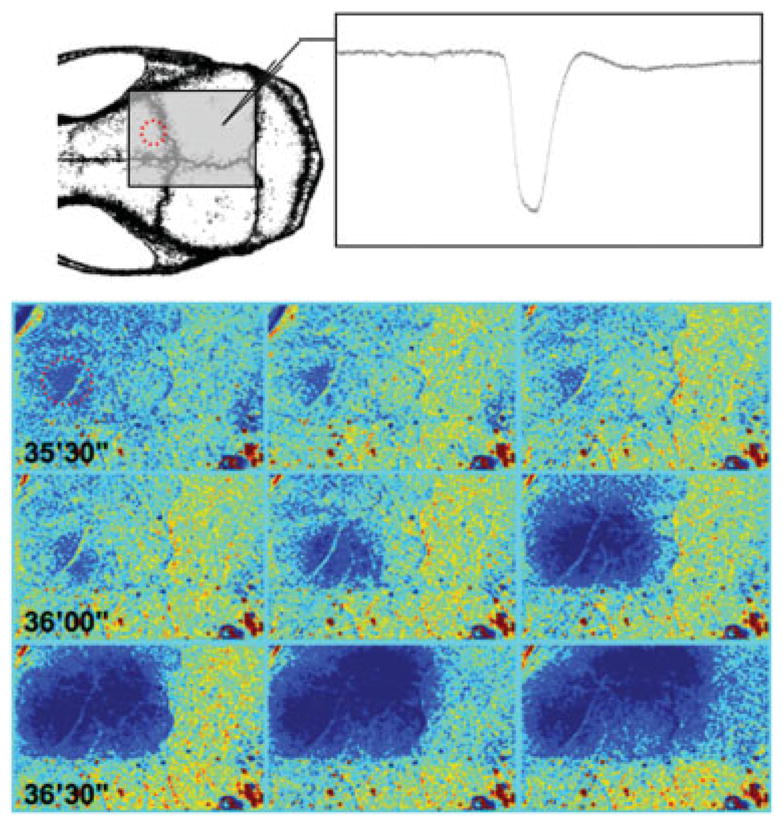
Cortical spreading depression (CSD) originates from a hypoperfused cortical focus. Representative time-lapse laser speckle images of relative cerebral blood flow changes taken approximately 35 minutes after cholesterol microembolization show the initiation of a CSD from a frontal cortical focus of hypoperfusion (red circle). A CSD was initiated from this ischemic focus after approximately 30 seconds (36′00‴), and propagated throughout the ipsilateral cortex (centrifugally spreading blue hypoperfusion wave), confirmed by concurrent electrophysiological recordings using a glass micropipette (inset). Images were acquired at 0.1Hz. Laser speckle imaging field and electrode positions are shown on the upper left drawing.
To test whether the occurrence of CSD was related to perturbations in blood flow, we integrated the magnitude of CBF deficit and its duration (i.e., area under CBF curve [AUC]) using the formula AUC = [100 - CBF] · t, where CBF is the average CBF (%) between the injection of microemboli and the onset of CSD or recovery of CBF to baseline without CSD, and t is the latency to CSD in minutes (Fig 3, inset). We found that mice that developed CSD had significantly higher AUC values, regardless of the type of microemboli. Indeed, a threshold of approximately 60% · min appeared to be a requisite for CSD occurrence in our study (see Fig 3A, horizontal shaded area).
FIGURE 3.

Ischemic burden and its relationship to cortical spreading depression (CSD) and infarct occurrence. Ischemic burden (area under the curve [AUC]) was calculated by measuring the area under the cerebral blood flow (CBF) curve between the injection of microemboli (blue circle) and the onset of CSD (red circle), as shown in the inset on the right. (A) The occurrence of CSD was associated with significantly higher AUC values than experiments without CSD; 60% · min (horizontal shaded area) was the AUC threshold for evoking CSD. (B) AUC values did not differ significantly between the animals with or without infarcts, regardless of the embolic particle. (C) The AUC values did not differ between the 3 types of microemboli. Individual data points as well as median (25–75% range) are given for each group. *p < 0.01 vs no CSD; Mann-Whitney rank sum test. MS = microsphere; Chol = = cholesteol.
Microembolic CSDs Develop Without Requisite Brain Infarcts
Despite extensive histopathological evaluation of approximately 4,000 sections from 42 brains, ischemic infarcts were detected only in 12 of 28 brains after microsphere or cholesterol crystal injection (Table 2); lesions were identified by the presence of shrunken neurons with eosinophilic cytoplasm and pyknotic and karyorrhexic nuclei (Fig 4). The neuropil was slightly vacuolated, and most infarcts had little or no gliosis, consistent with early subacute infarction. No evidence of inflammatory cell infiltration was observed, except in a single animal in the microsphere group. Infarcts were typically between 100 and 200μm but varied in size from 100 to 1,200μm (see Table 2). The majority of microinfarcts were found in the cortex (see Table 2) exclusively within ipsilateral middle cerebral artery (MCA) and bilateral ACA territories. No infarcts were found in the brainstem or cerebellum. The presence of microinfarcts in this model could not be predicted by the ischemic burden (see Fig 3B) or CSD occurrence.
TABLE 2.
Occurrence and Characteristics of Microembolic Infarction
| Microemboli | Diameter (μm) | Volume (μl) | Particles | Time of Evaluation | No. | Occurrence | Infarcts/Animal | Infarct Diammeter (μm) | Infarct Distribution (No. infarcts/No. mice) | White Matter | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cortex | Striatum | Hippocampus | ||||||||||
| Saline | — | 10 | — | 3 days [2–3] | 8 | 1 | 1 | 200 | 1/1 | — | — | — |
| Air | — | 0.5 | — | 2 days [2–3] | 6 | 0 | — | — | — | — | — | — |
| Microspheres | 10 | 1 | 20,000 | 3 days [3–3] | 16 | 5 | 1 [1–3] | 400 [200–800] | 5/3 | — | 1/1 | 1/1 |
| Cholesterol | <70 | 15 | 4,000 | 4 days[3–12] | 12 | 7 | 4 [2–16] | 225 [100–1200] | 33/6 | 2/1 | 2/2 | — |
Data are presented as median [25–75% range].
FIGURE 4.
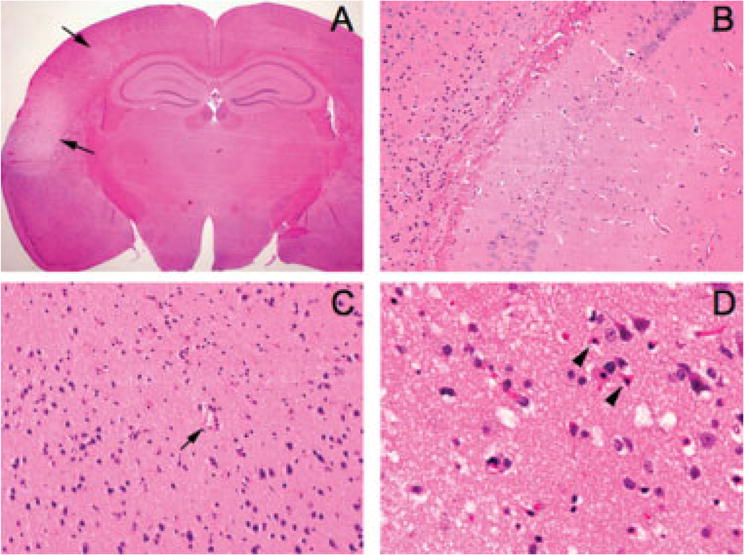
Histopathology of microembolic infarcts. (A) A representative coronal section showing 2 cortical infarcts (arrows) 72 hours after injection of microspheres (10μm diameter, 20,000 particles; original magnification, ×12.5). (B) Although the majority of infarcts were in the ipsilateral cortex, the hippocampal formation also showed microinfarcts in a few brains (original magnification, ×200). (C) Most cortical infarcts were centered around a small blood vessel (arrow) (original magnification, ×200). (D) High-power image of a representative cortical microinfarct demonstrating the characteristic shrunken, eosinophilic neurons with pyknotic nuclei (arrowheads) adjacent to intact neurons at the infarct edge (original magnification, ×400).
MRI lesions were not found in 10 of 11 animals after microsphere embolization (10μm diameter; 20,000 particles) or in 4 control mice. CSD was evoked in 4 of these 11 mice, and microinfarctions (200μm [25–75% range, 100–2,000μm]) were found in 5 by histological analysis. MRI imaging identified a hyperintense lesion (2,000μm) of subcortical infarction in 1 animal, which was subsequently confirmed by histopathology.
Discussion
Microemboli injected into the carotid artery triggered hypoperfusion and CSD in rodent brain. CSD occurrence depended on the duration and degree of occlusion as well as on the size and composition of the emboli. Larger particles (>20μm microspheres or cholesterol crystals) caused more lesions, an observation of potential importance to cryptogenic stroke in humans. We observed a close correspondence between the ischemic burden (ie, magnitude × duration of CBF reduction) and the appearance of CSD; when the CBF response was large and/or long-lasting, CSD was evoked. Air microinjection in the mouse, although larger than the volume injected during a bubble test for PFO in humans, was the most reliable CSD trigger, and this may be related clinically to the infrequent occurrence of microbubble-induced attacks of migraine with aura in humans.23 Air and polystyrene beads tended to cause short-lived but significant global flow reductions within the entire ipsilateral carotid territory, whereas cholesterol crystals triggered small foci of prolonged hypoperfusion most often within the ipsilateral MCA territory. The prolonged period of blood flow reduction was most likely caused by cholesterol crystals (< 70μm) that did not traverse small vessels to reach the lung, as was observed with small fluorescent microspheres (unpublished observation). The irregular shape (tapered, spiculated, and spikey) probably contributed to partial blood flow obstruction, in contrast to the rounded microsphere particles (10μm) or air (0.5μl), which caused more severe but transient obstruction either by mechanical blockage or via reflexive vasoconstriction. The blood flow response to cholesterol crystals, which may also be due to intravascular coagulation and release of vasoactive substances,24 indicates that only small areas of hypoperfused brain were required to evoke CSD following microembolization.
The findings reported herein indicate that CSDs triggered by microemboli do not necessarily cause tissue injury as they do in other ischemic conditions,25 nor develop as a consequence of sustained tissue damage.10 However, we did detect microinfarcts in 13 of 42 brains, distributed within the ipsilateral hemisphere (cortex > hippocampus > striatum), but not brainstem or cerebellum. Had we injected more biologically relevant blood components such as biodegradable fibrin particles or platelets instead of nondegradable microspheres or cholesterol crystals, a lower rate of microinfarction might have been anticipated. Because platelets have been implicated in migraine aura attacks,26 and aggregates may restrict local blood flow and release vasoactive molecules, strategies to reduce platelet responsiveness may prove useful, especially in patients with arteriovenous or cardiac shunts, and those migraineurs at greatest risk for small vascular-like brain lesions.
Despite the presence of microinfarcts in 30% of mice, we did not observe sustained behavioral deficits, nor were lesions visible on high–field-strength T2-weighted MRI in 10 of 11 animals. This discrepancy between positive histology, but negative brain imaging and clinical outcome in most animals was probably due to the small size and isolated lesions caused by the injected microemboli. Considering the >0.5–1mm in-plane resolution of routine magnetic resonance images, it would not be too surprising if many human brain lesions similar in size to those reported herein escape detection. We posit that some forms of migraine aura reside on a hypoperfusion continuum depending, in part, on particle size and the intensity and duration of transient microvascular insult. Perhaps these relationships explain the high comorbidity between stroke, migraine auras, and PFO.27
A higher prevalence of PFO is reported in migraineurs.28–30 Anzola et al found that 48% of patients with migraine and aura had a PFO, as did 23% of migraineurs without aura and 20% of control subjects.31 A recent meta-analysis of 134 publications confirmed this association.29 PFO enables venous blood to traverse through an incomplete atrial septum, the closure of which normally takes place shortly after birth. As a result, blood flows from right to left atria, and can thereby bypass the lung filtering mechanism for particulate and vasoactive substances. Large and small emboli originating within the venous system may, hence, reach the intracranial arterial circulation to cause either stroke or migraine aura, depending on size and location.32
However, clinical trials testing the impact of PFO closure produced mixed results.30,33 In retrospective studies, success rates ranged from 5 to 90%34–37; the grade of evidence was low. The only prospective, randomized controlled trial30 failed to achieve its desired endpoint of migraine headache cure, in our view an unlikely endpoint considering the refractory nature of these patients, the continued exposure to other triggers for migraine attacks, and an underlying genetically susceptible brain.
Migraine aura may also pose a risk for silent brain lesions. Kruit et al demonstrated small (approximately 7mm) lesions located in cerebellar microwatershed regions that they posited to be microemboli.38,39 Although a higher incidence of cortical lesions might have been expected from our data, this was not found by Kruit and colleagues. Perhaps the studied patient population may not have harbored PFOs as the only cause of their microinfarcts, or the intensity of the transient hypoperfusion was sufficient to trigger CSDs but not cortical microinfarcts. Also, the imaging techniques used in the Kruit study may not have been sufficiently sensitive to detect small microinfarcts (<200μm), as we found in our MRI study.
Migraine auras commonly develop in a number of vasculopathies, suggesting that mechanisms intrinsic to blood vessels are important to migraine pathogenesis in addition to embolization. These include focal vessel narrowing,40 decreased endothelium dependent relaxation,41 thrombocytosis, or coagulopathy.42 Because the majority of grey matter can generate CSD and is clinically silent, the same pathophysiologic mechanisms may be implicated in a subset of migraineurs without aura, and especially those who respond to drugs suppressing CSD susceptibility43 and those who experience migraine headaches both with and without aura.44,45 Our findings support the notion that in a subset of migraineurs, transient hypoxic ischemic events serve as a CSD generator and possibly a triggering mechanism for aura without requisite tissue injury.
Acknowledgments
This project was supported by the National Institute of Neurological Disorders and Stroke (grant NS061505, C.A.; grant NS35611, M.A.M.), Massachusetts; General Hospital Department of Anesthesia, and NIH National Research Service (grant GM07592, A.N.).
We thank Joseph H. Rapp, Rudy Davis, and Turgay Dalkara for insightful advice.
References
- 1.Lipton RB, Stewart WF, Diamond S, et al. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache. 2001;41:646–657. doi: 10.1046/j.1526-4610.2001.041007646.x. [DOI] [PubMed] [Google Scholar]
- 2.Henrich JB, Horwitz RI. A controlled study of ischemic stroke risk in migraine patients. J Clin Epidemiol. 1989;42:773–780. doi: 10.1016/0895-4356(89)90075-9. [DOI] [PubMed] [Google Scholar]
- 3.Etminan M, Takkouche B, Isorna FC, Samii A. Risk of ischaemic stroke in people with migraine: systematic review and meta-analysis of observational studies. BMJ. 2005;330:63. doi: 10.1136/bmj.38302.504063.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moskowitz MA. The neurobiology of vascular head pain. Ann Neurol. 1984;16:157–168. doi: 10.1002/ana.410160202. [DOI] [PubMed] [Google Scholar]
- 5.Leao AAP. Spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944;7:359–390. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- 6.Hadjikhani N, Sanchez Del Rio M, Wu O, et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001;98:4687–4692. doi: 10.1073/pnas.071582498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andersen AR, Friberg L, Olsen TS, Olesen J. Delayed hyperemia following hypoperfusion in classic migraine. Single photon emission computed tomographic demonstration. Arch Neurol. 1988;45:154–159. doi: 10.1001/archneur.1988.00520260040017. [DOI] [PubMed] [Google Scholar]
- 8.Woods RP, Iacoboni M, Mazziotta JC. Brief report: bilateral spreading cerebral hypoperfusion during spontaneous migraine headache. N Engl J Med. 1994;331:1689–1692. doi: 10.1056/NEJM199412223312505. [DOI] [PubMed] [Google Scholar]
- 9.Matsushima K, Schmidt-Kastner R, Hogan MJ, Hakim AM. Cortical spreading depression activates trophic factor expression in neurons and astrocytes and protects against subsequent focal brain ischemia. Brain Res. 1998;807:47–60. doi: 10.1016/s0006-8993(98)00716-1. [DOI] [PubMed] [Google Scholar]
- 10.Dohmen C, Sakowitz OW, Fabricius M, et al. Spreading depolarizations occur in human ischemic stroke with high incidence. Ann Neurol. 2008;63:720–728. doi: 10.1002/ana.21390. [DOI] [PubMed] [Google Scholar]
- 11.Dreier JP, Woitzik J, Fabricius M, et al. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain. 2006;129(pt 12):3224–3237. doi: 10.1093/brain/awl297. [DOI] [PubMed] [Google Scholar]
- 12.Strong AJ, Fabricius M, Boutelle MG, et al. Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke. 2002;33:2738–2743. doi: 10.1161/01.str.0000043073.69602.09. [DOI] [PubMed] [Google Scholar]
- 13.Wolff H. Headache and Other Head Pain. New York, NY: Oxford University Press; 1963. [Google Scholar]
- 14.Van Harreveld A, Stamm J. Vascular concomitants of spreading cortical depression. J Neurophysiol. 1952;15:487–496. doi: 10.1152/jn.1952.15.6.487. [DOI] [PubMed] [Google Scholar]
- 15.Olesen J, Friberg L, Olsen TS, et al. Ischaemia-induced (symptomatic) migraine attacks may be more frequent than migraine-induced ischaemic insults. Brain. 1993;116(pt 1):187–202. doi: 10.1093/brain/116.1.187. [DOI] [PubMed] [Google Scholar]
- 16.Del Sette M, Angeli S, Leandri M, et al. Migraine with aura and right-to-left shunt on transcranial Doppler: a case-control study. Cerebrovasc Dis. 1998;8:327–330. doi: 10.1159/000015875. [DOI] [PubMed] [Google Scholar]
- 17.Post MC, Letteboer TG, Mager JJ, et al. A pulmonary right-to-left shunt in patients with hereditary hemorrhagic telangiectasia is associated with an increased prevalence of migraine. Chest. 2005;128:2485–2489. doi: 10.1378/chest.128.4.2485. [DOI] [PubMed] [Google Scholar]
- 18.Rigatelli G. Migraine and patent foramen ovale: connecting flight or one-way ticket? Expert Rev Neurother. 2008;8:1331–1337. doi: 10.1586/14737175.8.9.1331. [DOI] [PubMed] [Google Scholar]
- 19.Nozari A, Dilekoz E, Sukhotinsky I, et al. Microembolization triggers cortical spreading depression both with and without microinfarcts: implications for a migraine-stroke continuum. Stroke. 2009;40:280. [Google Scholar]
- 20.Ayata C, Dunn AK, Gursoy-Ozdemir Y, Huang Z, Boas DA, Moskowitz MA. Laser speckle flowmetry for the study of cerebrovascular physiology in normal and ischemic mouse cortex. J Cereb Blood Flow Metab. 2004;24:744–755. doi: 10.1097/01.WCB.0000122745.72175.D5. [DOI] [PubMed] [Google Scholar]
- 21.Eikermann-Haerter K, Dilekoz E, Kudo C, et al. Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J Clin Invest. 2009;119:99–109. doi: 10.1172/JCI36059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weiss L, Harlos JP. The validity of negative necropsy reports for metastases in solid organs. J Pathol. 1986;148:203–206. doi: 10.1002/path.1711480302. [DOI] [PubMed] [Google Scholar]
- 23.Dinia L, Roccatagliata L, Bonzano L, et al. Diffusion MRI during migraine with aura attack associated with diagnostic micro-bubbles injection in subjects with large PFO. Headache. 2007;47:1455–1456. doi: 10.1111/j.1526-4610.2007.00948.x. [DOI] [PubMed] [Google Scholar]
- 24.Steiner TJ, Rail DL, Rose FC. Cholesterol crystal embolization in rat brain: a model for atheroembolic cerebral infarction. Stroke. 1980;11:184–189. doi: 10.1161/01.str.11.2.184. [DOI] [PubMed] [Google Scholar]
- 25.Busch E, Gyngell ML, Eis M, Hoehn-Berlage M, Hossmann KA. Potassium-induced cortical spreading depressions during focal cerebral ischemia in rats: contribution to lesion growth assessed by diffusion-weighted NMR and biochemical imaging. J Cereb Blood Flow Metab. 1996;16:1090–1099. doi: 10.1097/00004647-199611000-00002. [DOI] [PubMed] [Google Scholar]
- 26.Hanington E, Jones RJ, Amess JA, Wachowicz B. Migraine: a platelet disorder. Lancet. 1981;2:720–723. doi: 10.1016/s0140-6736(81)91049-7. [DOI] [PubMed] [Google Scholar]
- 27.Lamy C, Giannesini C, Zuber M, et al. Clinical and imaging findings in cryptogenic stroke patients with and without patent foramen ovale: the PFO-ASA Study. Atrial Septal Aneurysm Stroke. 2002;33:706–711. doi: 10.1161/hs0302.104543. [DOI] [PubMed] [Google Scholar]
- 28.Lechat P, Mas JL, Lascault G, et al. Prevalence of patent foramen ovale in patients with stroke. N Engl J Med. 1988;318:1148–1152. doi: 10.1056/NEJM198805053181802. [DOI] [PubMed] [Google Scholar]
- 29.Schwedt TJ, Demaerschalk BM, Dodick DW. Patent foramen ovale and migraine: a quantitative systematic review. Cephalalgia. 2008;28:531–540. doi: 10.1111/j.1468-2982.2008.01554.x. [DOI] [PubMed] [Google Scholar]
- 30.Dowson A, Mullen MJ, Peatfield R, et al. Migraine Intervention With STARFlex Technology (MIST) trial: a prospective, multi-center, double-blind, sham-controlled trial to evaluate the effectiveness of patent foramen ovale closure with STARFlex septal repair implant to resolve refractory migraine headache. Circulation. 2008;117:1397–1404. doi: 10.1161/CIRCULATIONAHA.107.727271. [DOI] [PubMed] [Google Scholar]
- 31.Anzola GP, Magoni M, Guindani M, et al. Potential source of cerebral embolism in migraine with aura: a transcranial Doppler study. Neurology. 1999;52:1622–1625. doi: 10.1212/wnl.52.8.1622. [DOI] [PubMed] [Google Scholar]
- 32.Steiner MM, Di Tullio MR, Rundek T, et al. Patent foramen ovale size and embolic brain imaging findings among patients with ischemic stroke. Stroke. 1998;29:944–948. doi: 10.1161/01.str.29.5.944. [DOI] [PubMed] [Google Scholar]
- 33.Dubiel M, Bruch L, Schmehl I, et al. Migraine headache relief after percutaneous transcatheter closure of interatrial communications. J Interv Cardiol. 2008;21:32–37. doi: 10.1111/j.1540-8183.2007.00316.x. [DOI] [PubMed] [Google Scholar]
- 34.Post MC, Thijs V, Herroelen L, Budts WI. Closure of a patent foramen ovale is associated with a decrease in prevalence of migraine. Neurology. 2004;62:1439–1440. doi: 10.1212/01.wnl.0000120756.25236.37. [DOI] [PubMed] [Google Scholar]
- 35.Schwerzmann M, Wiher S, Nedeltchev K, et al. Percutaneous closure of patent foramen ovale reduces the frequency of migraine attacks. Neurology. 2004;62:1399–1401. doi: 10.1212/01.wnl.0000120677.64217.a9. [DOI] [PubMed] [Google Scholar]
- 36.Shammas NW, Dippel EJ, Harb G, et al. Interatrial septal defect closure for prevention of cerebrovascular accidents: impact on recurrence and frequency of migraine headaches. J Invasive Cardiol. 2007;19:257–260. [PubMed] [Google Scholar]
- 37.Wilmshurst PT, Nightingale S, Walsh KP, Morrison WL. Effect on migraine of closure of cardiac right-to-left shunts to prevent recurrence of decompression illness or stroke or for haemodynamic reasons. Lancet. 2000;356:1648–1651. doi: 10.1016/s0140-6736(00)03160-3. [DOI] [PubMed] [Google Scholar]
- 38.Kruit MC, van Buchem MA, Hofman PAM, et al. Migraine as a risk factor for subclinical brain lesions. JAMA. 2004;291:427–434. doi: 10.1001/jama.291.4.427. [DOI] [PubMed] [Google Scholar]
- 39.Kruit MC, Launer LJ, Ferrari MD, van Buchem MA. Infarcts in the posterior circulation territory in migraine. The population-based MRI CAMERA study. Brain. 2005;128(pt 9):2068–2077. doi: 10.1093/brain/awh542. [DOI] [PubMed] [Google Scholar]
- 40.Friberg L, Olsen TS, Roland PE, Lassen NA. Focal ischaemia caused by instability of cerebrovascular tone during attacks of hemiplegic migraine. A regional cerebral blood flow study. Brain. 1987;110(pt 4):917–934. doi: 10.1093/brain/110.4.917. [DOI] [PubMed] [Google Scholar]
- 41.Olesen J, Thomsen LL, Iversen H. Nitric oxide is a key molecule in migraine and other vascular headaches. Trends Pharmacol Sci. 1994;15:149–153. doi: 10.1016/0165-6147(94)90075-2. [DOI] [PubMed] [Google Scholar]
- 42.Michiels JJ, Berneman Z, Schroyens W, et al. Platelet-mediated erythromelalgic, cerebral, ocular and coronary microvascular ischemic and thrombotic manifestations in patients with essential thrombocythemia and polycythemia vera: a distinct aspirin-responsive and coumadin-resistant arterial thrombophilia. Platelets. 2006;17:528–544. doi: 10.1080/09537100600758677. [DOI] [PubMed] [Google Scholar]
- 43.Ayata C, Jin H, Kudo C, et al. Suppression of cortical spreading depression in migraine prophylaxis. Ann Neurol. 2006;59:652–661. doi: 10.1002/ana.20778. [DOI] [PubMed] [Google Scholar]
- 44.Diener HC, Tfelt-Hansen P, Dahlof C, et al. Topiramate in migraine prophylaxis—results from a placebo-controlled trial with propranolol as an active control. J Neurol. 2004;251:943–950. doi: 10.1007/s00415-004-0464-6. [DOI] [PubMed] [Google Scholar]
- 45.Jensen R, Brinck T, Olesen J. Sodium valproate has a prophylactic effect in migraine without aura: a triple-blind, placebo-controlled crossover study. Neurology. 1994;44:647–651. doi: 10.1212/wnl.44.4.647. [DOI] [PubMed] [Google Scholar]