

Abstract
Developing new techniques to induce β-cells to replicate is a major goal in diabetes research. Endogenous β-cells replicate in response to metabolic changes, such as obesity and pregnancy, which increase insulin requirement. Mouse genetic models promise to reveal the pathways responsible for compensatory β-cell replication. However, no simple, short-term, physiological replication stimulus exists to test mouse models for compensatory replication. Here, we present a new tool to induce β-cell replication in living mice. Four-day glucose infusion is well tolerated by mice as measured by hemodynamics, body weight, organ weight, food intake, and corticosterone level. Mild sustained hyperglycemia and hyperinsulinemia induce a robust and significant fivefold increase in β-cell replication. Glucose-induced β-cell replication is dose and time dependent. β-Cell mass, islet number, β-cell size, and β-cell death are not altered by glucose infusion over this time frame. Glucose infusion increases both the total protein abundance and nuclear localization of cyclin D2 in islets, which has not been previously reported. Thus, we have developed a new model to study the regulation of compensatory β-cell replication, and we describe important novel characteristics of mouse β-cell responses to glucose in the living pancreas.
Diabetes causes considerable morbidity, elevates health care costs, and shortens lifespan. For these reasons, finding effective prevention or treatment is a high priority. Although type 1 and type 2 diabetes have different etiologies, in both cases, the final pathway to abnormal glucose homeostasis involves loss of functional β-cells (1,2). Thus, a primary goal for diabetes research is to develop ways to increase the number of β-cells (3).
An important potential source of new β-cells is replication of existing β-cells or β-cell precursors, which supply additional β-cells in times of need (4–7). Evidence suggests that most adult mouse β-cells are derived from preexisting differentiated β-cells rather than stem cells (8). In fact, although adult β-cells normally replicate slowly (9), under certain conditions, such as pregnancy and insulin resistance, β-cells replicate more quickly (7,10–12). Notably, obese human beings have increased β-cell mass when compared with lean subjects, suggesting that the β-cell compensatory response to increased insulin requirement may be common between mice and humans (1,2). Several growth factors induce β-cell replication in living mice (13–21). Cell cycle regulatory proteins, in particular cyclin D2, cyclin D1, and cyclin-dependent kinase (cdk) 4, are critical for postnatal β-cell replication (22–25).
Advances in mouse genetics have brought rapid progress in understanding the fundamental properties of β-cell differentiation and function. To optimally use mouse genetics to discover the mechanisms involved in β-cell compensatory replication, mice lacking candidate pathway members should be evaluated with respect to response to a β-cell replication stimulus. The ideal stimulus should be short term and physiological, to avoid interfering effects of systemic adaptive mechanisms. It should be applied after development is complete to avoid unknown intrinsic alterations in β-cells. Existing methods of inducing β-cell replication in adult mice, including high-fat diet, pancreatectomy, and pregnancy, cause widespread changes in whole-body physiology and result in altered levels of known and unknown factors that may affect β-cells. To date, no simple, short-term, physiological intervention known to directly stimulate β-cell replication has been available to test mouse genetic models for β-cell compensatory replication. Thus, we set out to adapt multiple-day glucose infusion, a well-characterized tool in rats, for use in the study of β-cell homeostasis in mice.
Four-day glucose infusion induces β-cell replication in rats; β-cell size and mass also increase (4,26–29). We have adapted this glucose infusion model for use in mice, taking particular pains to maintain a physiological, nonstressed state to avoid the influence of unrelated systemic responses to treatment. We find that a mild sustained increase in circulating blood glucose and insulin results in a marked increase in β-cell replication. Temporal analysis of β-cell replication in response to glucose shows that in contrast to the rapid functional increase in insulin secretion, the compensatory replication response occurs several days later. In mice, β-cell mass, islet number, β-cell size, and β-cell death are unchanged by glucose infusion. Interestingly, we find that glucose infusion increases islet cyclin D2 protein abundance and nuclear localization, suggesting that cyclin D2 may be involved in postnatal glucose-induced compensatory β-cell expansion. With this work, we have developed the first model of sustained glucose infusion to study mechanisms of β-cell replication in living mice.
RESEARCH DESIGN AND METHODS
Animal care, surgery, and recovery
Animal handling was in accordance with approved institutional animal care and use committee protocols at the University of Pittsburgh. Detailed protocols regarding catheterization, tether system, housing, catheter maintenance, arterial blood sampling, and infusions are located in the online appendix (available at http://dx.doi.org/10.2337/db06-1513). Briefly, 8- to 12-week-old, 20- to 25-g C57bl/6J male mice with free access to food and water were kept on a 12-h light/dark cycle. For catheterization, mice were anesthetized with inhaled 2% isoflurane. Micro-renathane catheters (MRE-025; Braintree Scientific, Braintree, MA) were prepared by heating, pulling, cutting to appropriate diameter, shaping into a J form in hot oil, and sterilizing (ethylene oxide). Catheters were inserted in the left femoral artery and vein, sutured in place, stabilized with superglue (Henkel, Rocky Hill, CT), tunneled subcutaneously to the upper back by threading through a blunt needle, taped to a wire attached to posterior cervical muscles for stiffness (792500; A-M-Systems, Sequim, WA), and connected to a 360° dual channel swivel designed for mice (375/D/22QM; Instech, Plymouth Meeting, PA). Catheters were maintained by continuously flushing 7 μl/h saline containing 20 units/ml heparin (Baxter, Deerfield, IL) using a syringe pump with multisyringe adaptor (R99-EM; Razel Scientific Instruments, St. Albans, VT). Arterial catheters were monitored for patency daily and kept unclogged by manual flushes using a 1-cc syringe with 26-gauge needle when necessary.
Glucose infusions
Continuous hemodynamic measurements were recorded using a pressure transducer (Argon, Athens, TX) connected to the arterial catheter. Mice received infusions of 0.9% sodium chloride, 25% dextrose, or 50% dextrose, with 500 μg/ml bromodeoxyuridine (BrdU; Sigma) added when indicated, at a constant rate of 100 μl/h. For a 25-g mouse, this translates to 0, 1, or 2 mg · kg−1 · h−1 glucose, in comparison with rat glucose infusion studies in which rats received 2.8–4 mg · kg−1 · h−1 glucose (4,26–28,30,31). Two different infusion protocols were employed; for the initial experiment, mice received either saline or glucose, with BrdU, for 4 days. For the time-course experiment, mice received saline or glucose for 1, 2, 3, or 4 days, with BrdU added for the final 24 h only. Arterial blood was sampled twice daily: 100 μl whole blood was removed; 2 μl was used for blood glucose, the remainder was centrifuged, and erythrocytes were resuspended with 20 μl saline with 100 units/ml heparin and reinfused into the mouse. Plasma was stored at −80°C for measurement of insulin and corticosterone. Food intake was recorded daily. After infusion, mice were weighed and anesthetized and organs removed and weighed. Data from all mice receiving 4 days of glucose, including the initial experiment and the time-course experiment, were combined for metabolic studies.
Biochemical measurements
Blood glucose was measured using an Ascencia Elite XL glucometer (Bayer, Tarrytown, NY). Plasma insulin (Linco, St. Charles, MO) and corticosterone (MP Biomedicals, Solon, OH) were measured by radioimmunoassay.
Immunostaining
Tissues were fixed in Bouin’s solution (Sigma, St. Louis, MO) for 4 h at room temperature, dehydrated, embedded in paraffin, and sectioned (5 μm). For BrdU/insulin staining, sections were treated with 1 N HCl at 37°C for 60 min, blocked in PBS/5% rabbit serum/1% BSA, and incubated with rat anti-BrdU (1:250; Abcam, Cambridge, MA) and guinea pig anti-insulin (1:50; Invitrogen, Carlsbad, CA) followed by anti-rat (1:200; Alexa 594; Invitrogen) and anti–guinea pig (1:200; Alexa 488; Invitrogen) secondary antibodies. Insulin immunohistochemistry was performed as above, without the HCl step, except block contained goat serum, and detection was by Link/Label and DAB kits (BioGenex, San Ramon, CA) with hematoxylin counterstain. For E-cadherin immunofluorescence, sections were boiled in 10 mmol/l citrate, pH 6.0, for 20 min, blocked in 50 mmol/l Tris/5% rabbit serum/1% BSA/10 mmol/l CaCl2, and stained for E-cadherin (1:200; Calbiochem; San Diego, CA), insulin, and Hoescht 33342 (Invitrogen) to detect nuclei. For transferase-mediated dUTP nick-end labeling (TUNEL), the Pro-mega fluorescence Dead End kit (Promega, Madison, WI) was used following the manufacturer’s instructions; sections were then stained for insulin and Hoescht as above. Cyclin D2 immunohistochemistry was performed as for insulin immunohistochemistry, using citrate unmasking as per E-cadherin staining and cyclin D2 primary antibody (1:50; Neomarkers Ab-4; Lab Vision, Fremont, CA).
Histological analyses
For BrdU, islets were photographed at ×400 and assigned blinded filenames, and the number of β-cells and BrdU (+) β-cells were manually counted. At least 1,500 β-cells were counted per mouse, from two sections separated by at least 50 μm. To measure β-cell mass, two to three insulin-stained pancreas sections from each mouse, separated by at least 50 μm, were scanned in entirety (Nikon CoolScan), red and blue hues were separated, and the images were converted to gray scale and thresholded using Adobe Photoshop (Adobe, San Jose, CA). Pancreatic and islet areas were quantified using Image J (National Institutes of Health, Bethesda, MD). Islet number per section was quantified in two ways: 1) digitally, Image J–generated islet areas were tallied and 2) manually, all islet diameters were estimated using an intraocular calibrated grid. β-Cell size was measured in two ways: 1) ×400 images of insulin-stained islets were converted to gray scale and thresholded, and the insulin-positive area was digitally measured using image J; insulin area was divided by the number of β-cells (manually counted) to obtain an average β-cell area. At least 1,500 β-cells were measured per mouse. 2) E-cadherin–stained islets were photographed at ×1,000; cells staining for both E-cadherin and insulin with a nucleus in the plane of section were manually traced using Adobe Photoshop, and cell area was measured using the histogram function. Five islets were imaged per mouse, with 20 cells measured per islet. Microscopy was performed using an Olympus Provis upright scope; imaging on a Confocal Olympus Fluoview 1000 (inverted) confirmed that BrdU-positive nuclei were associated with insulin-positive cells.
Islet isolation, Western blots, and real-time PCR
After 4-day infusion of saline or 50% glucose, mice were weighed and killed, and the pancreatic duct was injected with 1.7 ml/cc Collagenase P (Sigma). Islets were isolated as described (16), handpicked, washed once in PBS, and frozen at −80°C. For Western blots, islets were lysed in 125 mmol/l Tris, pH 6.8, 2% SDS, 1 mmol/l dithiothreitol, 20 μg/ml p-amidinophenyl methanesulfonyl fluoride hydrochloride, and protease-inhibitor cocktail; sonicated; centrifuged; and loaded onto SDS-PAGE precast gels (Bio-Rad). Antibodies used include cyclin D1 and cyclin D2 (Neomarkers, Fremont, CA) and cyclin D3 and cdk4 (Abcam). For real-time PCR, RNA was extracted using the RNeasy micro kit (Qiagen, Valencia, CA) and reverse transcribed (1 μg total RNA, 1 μl random primer [50 μmol/l; Applied Biosystems], 1× reverse transcriptase buffer, and 10 units Moloney murine leukemia virus reverse transcriptase [Promega]) in a total volume of 20 μl. The RNA and primer were heated to 72°C and slowly cooled before reverse transcription at 42°C for 1 h. The room temperature reaction was then diluted to 100 μl with RNase-free water. For real-time PCR analysis, 2.5% of the total room temperature reaction was used as input for PCR using SYBR Green Master Mix (Applied Biosystems) on an ABI 7300 Real Time PCR System. Primers were as follows: cyclin D1, gcgtaccctgacaccaatct and ca caacttctcggcagtcaa; cyclin D2, gctatggagctgctgtgct and ccaagaaacggtccaggtaa; cyclin D3, ggaagctatggaccagcaag and tttgcacgcactggaagtag; cdk4, tatgaacccgt ggctgaaat and ccttgatgtcccgatcagtt; cdk6, gcctatgggaaggtgttcaa and gggctctg gaactttatcca; actin, agccatgtacgtagccatcc and ctctcagctgtggtggtgaa.
Statistical analysis
All data are presented as means ± SE. Comparisons between two means were determined by unpaired Student’s two-tailed t test. For comparison of multiple means or multiple measurements over time, ANOVA was performed; SDs between means were determined using Tukey’s multiple comparisons analysis. P < 0.05 was considered significant.
RESULTS
Four-day glucose infusion is well tolerated in mice
After 4-day infusion of saline or glucose (2 mg · kg−1 · h−1 glucose; Fig. 1A), mice appeared healthy and unstressed. Presurgical body weight was not significantly different from postinfusion weight (saline, presurgical 24.77 ± 0.50 g, postinfusion 23.49 ± 0.44 g; 25% glucose, presurgical 24.57 ± 0.36 g, postinfusion 23.46 ± 0.4 g; and 50% glucose, presurgical 24.52 ± 0.61 g, postinfusion 23.52 ± 0.48 g; Fig. 1B), nor was postinfusion weight of glucose-infused mice different from saline-infused mice. To indirectly monitor for systemic salt- and water-balance derangements, we measured organ weight at death of the heart, lungs, and brain (heart, saline 114 ± 3 mg, 25% glucose 107 ± 2 mg, and 50% glucose 111 ± 4 mg; lungs, saline 151 ± 4 mg, 25% glucose 142 ± 4 mg, and 50% glucose 144 ± 3 mg; and brain, saline 459 ± 5 mg, 25% glucose 456 ± 5 mg, and 50% glucose 462 ± 5 mg); no significant differences were observed, implying that the volume of saline or glucose infused is well tolerated over the 4-day treatment (Fig. 1C). To further assess mouse health, continuous hemodynamic monitoring was recorded. Mean arterial blood pressure and heart rate were not different between saline- and 50% glucose–infused mice (Fig. 1D). To assess animal stress more directly, we measured morning plasma corticosterone levels in a subset of saline- and 50% glucose–infused mice. Corticosterone levels confirm that neither saline-infused nor glucose-infused mice experience significant stress (Fig. 1E).
FIG. 1.

Four-day glucose infusion is well tolerated in mice. A: Experiment timeline. Glucose infusion does not alter body weight (B), organ weight (C), or mean arterial blood pressure (BP) and heart rate (HR) (D). E: Plasma corticosterone is not elevated; gray area indicates normal range for mice (49). Data are means ± SE.
Glucose infusion causes mild sustained hyperglycemia and hyperinsulinemia
When 25% or 50% glucose was infused at 100 μl/h, nonfasting blood glucose increased within 8 h and remained elevated for the 4-day infusion (average blood glucose, saline 108.3 ± 1.7 mg/dl, 25% glucose 120.7 ± 2.5 mg/dl, and 50% glucose 136.2 ± 2.2 mg/dl; P < 0.001 for all three comparisons; Fig. 2A). For a subset of mice, the blood glucose was checked at 1, 2, and 4 h and was not significantly higher than the 8-h value (data not shown). In an attempt to increase blood glucose further, a separate group of mice received saline or 50% glucose at a higher rate (200 μl/h, 4 mg glucose · kg−1 · h−1 for 25-g mouse). Three of three saline mice showed no ill effects (mean blood glucose 98.2 ± 4.4 mg/dl), but three of four glucose mice died in the 3rd or 4th infusion day (mean blood glucose 169 ± 12.6; data not shown). Consequently, we used the 100 μl/h rate for all subsequent experiments. For mice receiving infusions at 100 μl/h, plasma insulin levels increased within 8 h and remained elevated (Fig. 2B; average insulin, saline 1.2 ± 0.07 ng/ml, 25% glucose 1.7 ± 0.13 ng/ml, and 50% glucose 2.7 ± 0.17 ng/ml; P < 0.001 for 50% glucose vs. saline and 50% vs. 25% glucose; P < 0.05 for 25% glucose vs. saline). We measured daily food consumption to determine the impact of glucose infusion on total caloric intake. Mice consumed little chow in the first postoperative day but increased to a normal caloric intake by the 3rd day of recovery (Fig. 2C and D). Of note, mice receiving either 25% or 50% glucose reduced their oral intake of chow by approximately the amount of calories contained in the infusion, to maintain a normal daily total caloric intake (Fig. 2C and D). In sum, this glucose infusion protocol induced mild, sustained increases in circulating glucose and insulin without causing major metabolic perturbations.
FIG. 2.

Glucose infusion induces mild metabolic alterations within the physiological range. Twenty-five and 50% glucose (Glu) significantly increase blood glucose (A) and plasma insulin (B). Sources of calories in 25% glucose-infused (C) and 50% glucose-infused (D) mice include infusate (dotted line) and chow (gray circles). Glucose-infused mice reduce chow intake, so total caloric intake (black diamonds) is similar to saline mice (white squares; saline data are repeated in C and D for comparison). Data are means ± SE; *P < 0.001, ‡P < 0.05 for one-way ANOVA using Tukey’s post-test comparison with saline.
Glucose infusion increases β-cell replication in a dose-dependent fashion
Pancreas sections stained for BrdU and insulin showed a marked increase in the number of BrdU-positive β-cells in glucose-infused mice compared with saline-infused mice (Fig. 3A). Confocal microscopy confirmed that BrdU-positive nuclei belonged to insulin-positive cells (Fig. 3A, inset). Quantification revealed a robust and significant increase in β-cell replication in mice receiving 50% glucose when compared with saline (Fig. 3B; saline 0.8 ± 0.2%, 50% glucose 4.5 ± 0.9%; P < 0.01 for 50% glucose vs. saline). Twenty-five–percent glucose infusion induced a strong trend toward increased β-cell replication (1.9 ± 0.5%, P = 0.07 for 25% glucose vs. saline, P < 0.05 for 25% glucose vs. 50% glucose), suggesting that the increase is dose dependent. To determine whether replication in response to glucose varied with islet size, we stratified islets by number of β-cells in islet cross section (Fig. 3C). Interestingly, in saline-infused mice, very small islets (<20 β-cells seen in cross section) had lower rates of replication than larger islets, although this trend did not reach significance (P = 0.085 for <20 vs. >200). Infusion of 50% glucose increased β-cell replication in all islet sizes, although only midsize islets reached significance (Fig. 3C). The 25% glucose group trended toward a similar pattern, although no islet size reached significance compared with saline-infused islets.
FIG. 3.

Glucose infusion increases β-cell replication in a dose-dependent fashion. A: Representative islets stained for BrdU and insulin show glucose-induced replication; intestine is a positive control for BrdU exposure and staining. Inset: Confocal microscopy confirms BrdU-positive nuclei belong to insulin-positive cells. B: 50% glucose increases β-cell replication (vs. 25% glucose or saline); 25% glucose also shows a strong trend (vs. saline). C: Glucose (Glu) induces more replication in midsize islets than small islets. *P < 0.05 vs. saline; #P < 0.05 vs. “<20” islets. Scale bars = 20 μm; mean ± SE. Sal, saline.
β-Cell mass and islet number are unchanged by 4-day glucose infusion
Sections stained for insulin did not show obvious differences in islet size or number between saline- and glucose-infused mice (Fig. 4A). Similar to reports of glucose-infused rats (4,26,29,30), pancreas weight was significantly reduced in glucose-infused mice (saline 0.24 ± 0.01 g, 50% glucose 0.20 ± 0.01 g; P = 0.0009; Fig. 4B). Histomorphometric analysis revealed that percent β-cell area per pancreatic area (saline 0.45 ± 0.04, 50% glucose 0.52 ± 0.6; P = 0.37; Fig. 4C) and β-cell mass (saline 1.05 ± 0.07 mg, 50% glucose 1.02 ± 0.1 mg; P = 0.76; Fig. 4D) did not change in glucose- versus saline-infused mice. We evaluated the number and size distribution of islets using two different methods. Whether measured digitally (Fig. 4E) or by manual islet measurement using a calibrated intraocular grid (Fig. 4F), the distribution of islet sizes was unaltered. As expected, the number of very small islets (<25 μm diameter) was greater using the more sensitive manual counting technique. We observed no significant differences in singlets, doublets, or insulin-positive ductal cells in saline- vs. glucose-infused mice by manual counting (Fig. 4F and G).
FIG. 4.

β-Cell mass and islet number are unchanged after 4-day glucose infusion. A: Representative low-power images of pancreas sections stained for insulin and hematoxylin. B: Pancreas weight is reduced after glucose infusion. Percent β-cell area per pancreas area (C) and β-cell mass (D) are not increased in glucose-infused mice. Islet number is unchanged in glucose (Glu)-infused mice, whether counted digitally (E) or manually (F–G) using a calibrated intraocular grid. G: The number of singlets, doublets, and insulin-positive ductal cells is unchanged in glucose-infused mice. Scale bars = 1 mm. Data are means ± SE.
β-Cell size and death are unchanged by glucose infusion
Glucose is known to activate nutrient signaling pathways that increase cell size (32,33), and glucose infusion in rats increases β-cell size (4,29). Measurement of β-cell size digitally (saline 127 ± 12 μm2, 50% glucose 128 ± 8 μm2; P = 0.22; Fig. 5A and B) or by manually tracing cell borders (saline 89.6 ± 4.8 μm2, 50% glucose 90.5 ± 3.9 μm2; P = 0.88; Fig. 5C and D) showed no evidence of increased β-cell size in glucose-infused mice. Thus, mild hyperglycemia for 4 days in mice does not induce β-cell hypertrophy.
FIG. 5.
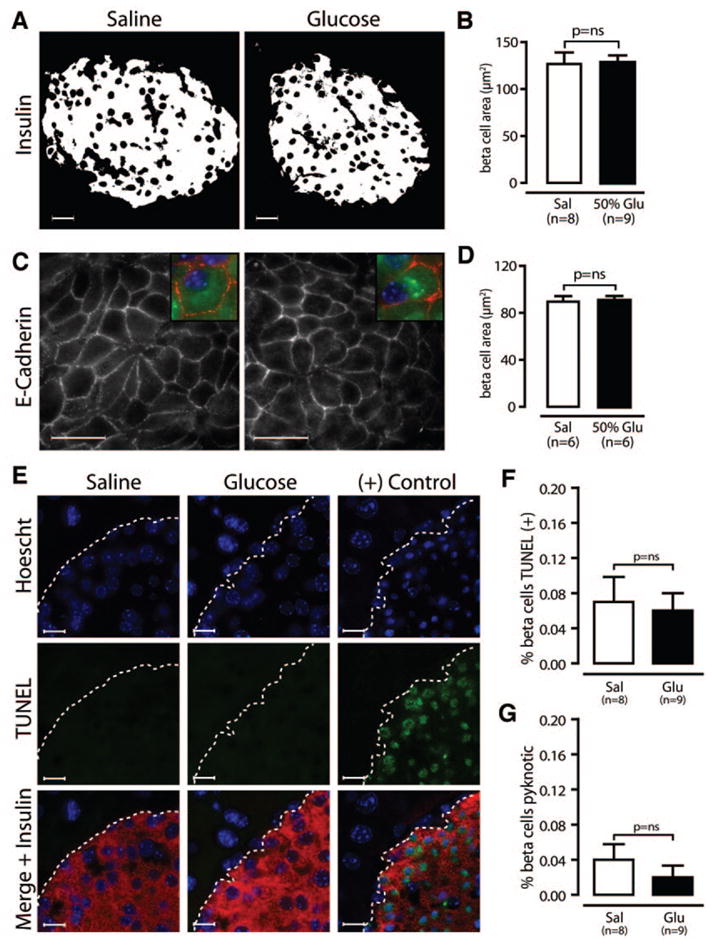
β-Cell size and death are unchanged by glucose infusion. A and B: Integrated insulin area per β-cell is unchanged in glucose (Glu)-infused mice. C and D: Using sections stained for E-cadherin (red), insulin (green), and Hoechst (blue), measurement of β-cell area by tracing cell borders confirms that β-cell size is unchanged. E–G: Using sections stained for TUNEL (green), insulin (red), and Hoescht (blue), we find the rates of TUNEL (F) and pyknosis (G) are low in both groups. The positive control is a mouse treated with streptozotocin. Scale bars in A and C = 20 μm and in E = 10 μm. Data are means ± SE. Sal, saline.
Because glucose can either promote (34) or prevent (30,35,36) β-cell death, we stained pancreas sections with insulin, with TUNEL to detect fragmented DNA, and with Hoechst to detect pyknotic nuclei. Islets from saline- and glucose-infused mice showed very low levels of β-cell death (Fig. 5E). Quantification of TUNEL (saline 0.07 ± 0.03%, 50% glucose 0.06 ± 0.02%; P = 0.83; Fig. 5F) or pyknotic nuclei (saline 0.04 ± 0.02%, 50% glucose 0.02 ± 0.01%; P = 0.85; Fig. 5G) showed no difference in β-cell death rates. Thus, glucose infusion neither caused β-cell death nor protected against it, with the caveat that we might not detect a reduction from the already low baseline death rate.
Glucose-induced β-cell replication increases late in infusion
We initially included BrdU during the entire 4-day infusion to maximize sensitivity and avoid introducing temporal bias; however, this approach does not give information about when β-cell replication occurs. To answer this question, we devised a new experimental protocol, designed to measure replication on day 1, 2, 3, or 4 (Fig. 6A). β-Cell replication increased steadily in glucose-infused mice during days 2, 3, and 4 of infusion, reaching significance on the 4th day (saline 0.27 ± 0.08%, glucose 1.35 ± 0.046%; P = 0.047; Fig. 6B). Thus, we find that although the metabolic effects of glucose infusion, including hyperglycemia, hyperinsulinemia, and decreased food intake, begin immediately after the infusion starts, β-cells are induced to replicate only after a delay of several days.
FIG. 6.

Glucose-induced replication increases over 4-day infusion. A: Mice received 1, 2, 3, or 4 days of saline or 50% glucose, with BrdU added only for the final 24 h. B: Glucose (Glu)-induced replication increases during days 2, 3, and 4. Data are means ± SE.
Glucose infusion induces cyclin D2 expression and nuclear localization
To investigate the mechanism by which glucose induces β-cell replication in vivo, we analyzed regulatory components of the G1/S transition of the cell cycle. D-type cyclins and cdk4, the predominant associated cdk in mouse β-cells (37), are activated by extracellular growth signals and are known to be required for the normal postnatal expansion of β-cell mass and replication (22–25). We assessed total protein levels of cyclin D1, cyclin D2, cyclin D3, and cdk4 by immunoblot of islets from saline- or glucose-infused mice, and we found that cyclin D2 was significantly increased in glucose-infused islets (cyclin D2, actin ratio 0.57 ± 0.08 vs. 1.58 ± 0.42; P = 0.038; Fig. 7A). Notably, the intracellular distribution of cyclin D2 was also affected by glucose infusion. Pancreas sections from glucose-infused mice showed greatly enhanced nuclear cyclin D2 staining in islets when compared with islets from saline-infused mice (Fig. 7B). Finally, we evaluated mRNA levels of D-type cyclins, cdk4, and cdk6 in islets isolated from saline- or glucose-infused mice, and we found that glucose infusion did not alter any of these genes at the RNA level (Fig. 7C), as reported for cdk4 in rats (38). Thus, glucose infusion increases both the total abundance and nuclear localization of cyclin D2 protein in islets without altering cyclin D2 mRNA quantity, suggesting that the regulation occurs at the protein level.
FIG. 7.
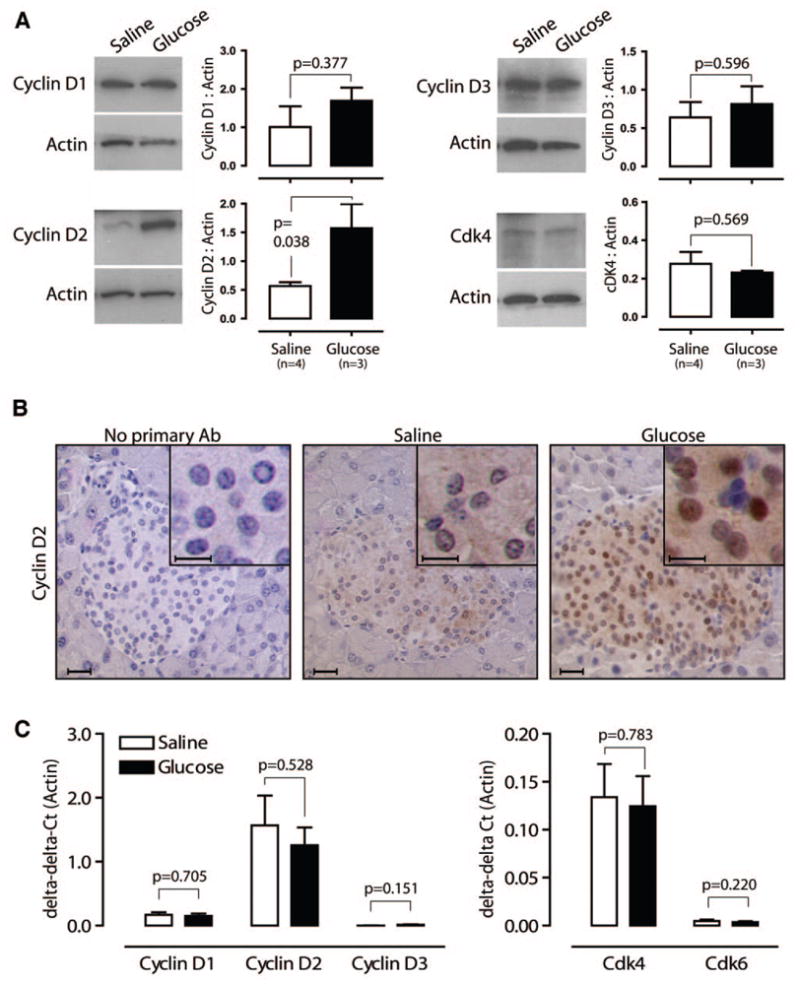
Glucose infusion increases cyclin D2 abundance and nuclear localization. A: Islets from mice infused with saline or glucose for 4 days show that cyclin D2 protein abundance significantly increases; cyclin D1, cyclin D3, and cdk4 are unchanged. B: Immunohistochemistry for cyclin D2 reveals pronounced nuclear localization in islets from glucose-infused mice. C: Real-time PCR demonstrates no difference in mRNA abundance of D-type cyclins and cdks 4 and 6 in islets from saline- or glucose-infused mice. Scale bars = 20 μm (10 μm in insets). Data are means ± SE.
DISCUSSION
With these studies, we have developed a new tool to study mouse β-cell replication in vivo. We achieved mild, sustained hyperglycemia and hyperinsulinemia over 4 days, without adverse effects as measured by caloric intake, blood pressure, heart rate, body weight, organ weights, and corticosterone level. β-Cell replication increased significantly over the 4-day infusion in a dose-dependent relationship with glucose concentration. Other parameters of β-cell expansion, such as β-cell size and islet number, were unchanged, suggesting that mild hyperglycemia uniquely stimulates replication over this time frame. Additionally, cyclin D2 is upregulated and shifts to a nuclear location in glucose-infused islets. These findings provide further support for a primary role for replication in β-cell homeostasis in mice, as previously proposed (8,22).
Our data confirm that glucose induces β-cell replication in situ in the mouse pancreas. Given the increasing use of mice as a model to study diabetes, confirming this basic tenet of islet biology in mice is important. Recent work suggests that glucose metabolism is required for compensatory β-cell replication in the setting of obesity and insulin resistance (39). Our finding that replication is induced by a small sustained increase in blood glucose supports a role for glucose in the normal regulation of β-cell homeostasis. In vitro studies show that glucose can directly alter β-cell replication without involving other organs (21,40–45). Whether glucose acts directly on mouse β-cells in vivo to stimulate replication cannot be inferred from the current studies.
It is possible that glucose-stimulated secreted insulin may act locally in the islet as a β-cell mitogen. The intraislet insulin concentration is likely to be sufficient to exert mitogenic effects. Insulin receptor–deficient β-cells in vitro show reduced replication (46). Mice lacking the insulin receptor in β-cells are reported to have decreased islet size (47), and additional elimination of one allele of the insulin-like growth factor receptor causes reduction in β-cell replication (48). Thus, glucose may act through biological intermediates, such as insulin. This model, applied to appropriate genetic models, will be ideal for studying this possibility further.
We find that cyclin D2 is upregulated and nuclear in islets from glucose-infused mice. The regulation of cyclin D2 appears to be posttranslational, which is consistent with previous work in β-cells (14). In pancreatic β-cells, cyclin D2 is more highly expressed during development and early postnatal life than during adulthood (22), suggesting the intriguing possibility that β-cell growth stimuli may shift differentiated adult β-cells toward a more developmental phenotype. Interestingly, many more islet cells show nuclear cyclin D2 than BrdU staining in glucose-infused mice. This may imply that nuclear cyclin D2 is not sufficient to induce cycling or that more β-cells are poised to replicate than the number detected during 4 days of infusion. The observation that replication occurs mostly on the 4th day of infusion supports the latter hypothesis. Evaluation of mice lacking cyclin D2 will determine whether this cell cycle regulator is required for glucose-induced β-cell replication in adult mice.
Our results are different from prior rat glucose infusion studies, primarily with respect to β-cell size and mass. β-Cell mass is determined by replication, hypertrophy, cell death, and neogenesis. It is not surprising that an absolute increase in replication of 3.8% did not translate into increased β-cell mass. This count includes all cells that have been through S phase during the 4-day infusion, including two daughter cells for each division, yielding a 1.9% increase in the total number of β-cells, which would not produce a detectable increase in β-cell mass. In our model, β-cell size, death, and suggested markers of neo-genesis are not altered, consistent with the unchanged β-cell mass. Rat glucose infusion protocols induced short-term, severe hyperglycemia and hyperinsulinemia (4,26–30), but our glucose and insulin profiles are in more physiological ranges and sustained. Perhaps β-cell replication is activated by mild hyperglycemia, but β-cell hypertrophy occurs in response to higher levels of glucose. Although we attempted to achieve higher circulating glucose levels by increasing the glucose load, we were unable to sustain the level of hyperglycemia seen in rats because of excess death.
In summary, we have developed a new model to induce mouse β-cells to replicate in vivo. This model can be applied to mice genetically modified to over- or under-express candidate genes in β-cell replication, including signaling pathway members, transcription factors, and cell cycle regulators. Our system will allow co-infusion of small molecule pathway activators and inhibitors for a more sophisticated analysis of signaling. Further molecular analysis of islets from glucose-infused mice may identify which glucose-induced pathways are instrumental in the replication response. Thus, this model opens the door for a variety of new experiments that will advance our understanding of β-cell biology and potentially direct efforts toward useful clinical targets as well.
Supplementary Material
Acknowledgments
L.C.A. has received National Institutes of Health Grant DK-076562. A.G.O. has received National Institutes of Health Grant DK067351. D.K.S. has received American Diabetes Association Grant ADA 7-04-RA-106. S.K.K. is supported by grants from the Juvenile Diabetes Research Foundation, the Snyder Foundation, and the National Institutes of Health.
We are grateful to Drs. Andrew Stewart, Rupangi Vasavada, Nathalie Fiaschi-Taesch, and Irene Cozar-Castellano for their thoughtful advice; to Dr. Simon Watkins for use of his outstanding imaging facility; and to Lia Romano, Jordan Pascoe, Taylor Rosa, Darinka Sipula, and Jennifer Roccisana for expert technical assistance.
- BrdU
bromodeoxyuridine
- cdk
cyclin-dependent kinase
- TUNEL
transferase-mediated dUTP nick-end labeling
Footnotes
Additional information for this article can be found in an online appendix at http://dx.doi.org/10.2337/db06-1513.
References
- 1.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-Cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 2.Yoon KH, Ko SH, Cho JH, Lee JM, Ahn YB, Song KH, Yoo SJ, Kang MI, Cha BY, Lee KW, Son HY, Kang SK, Kim HS, Lee IK, Bonner-Weir S. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab. 2003;88:2300–2308. doi: 10.1210/jc.2002-020735. [DOI] [PubMed] [Google Scholar]
- 3.Bonner-Weir S, Weir GC. New sources of pancreatic beta-cells. Nat Biotechnol. 2005;23:857–861. doi: 10.1038/nbt1115. [DOI] [PubMed] [Google Scholar]
- 4.Bonner-Weir S, Deery D, Leahy JL, Weir GC. Compensatory growth of pancreatic β-cells in adult rats after short-term glucose infusion. Diabetes. 1989;38:49–53. doi: 10.2337/diab.38.1.49. [DOI] [PubMed] [Google Scholar]
- 5.Lee HC, Bonner-Weir S, Weir GC, Leahy JL. Compensatory adaption to partial pancreatectomy in the rat. Endocrinology. 1989;124:1571–1575. doi: 10.1210/endo-124-3-1571. [DOI] [PubMed] [Google Scholar]
- 6.Jetton TL, Liu YQ, Trotman WE, Nevin PW, Sun XJ, Leahy JL. Enhanced expression of insulin receptor substrate-2 and activation of protein kinase B/Akt in regenerating pancreatic duct epithelium of 60%-partial pancreatectomy rats. Diabetologia. 2001;44:2056–2065. doi: 10.1007/s001250100011. [DOI] [PubMed] [Google Scholar]
- 7.Sorenson RL, Brelje TC. Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res. 1997;29:301–307. doi: 10.1055/s-2007-979040. [DOI] [PubMed] [Google Scholar]
- 8.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 9.Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of β-cells in aged adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- 10.Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, Zhang Y, Bernal D, Pons S, Shulman GI, Bonner-Weir S, White MF. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391:900–904. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- 11.Kido Y, Burks DJ, Withers D, Bruning JC, Kahn CR, White MF, Accili D. Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J Clin Invest. 2000;105:199–205. doi: 10.1172/JCI7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bruning JC, Winnay J, Bonner-Weir S, Taylor SI, Accili D, Kahn CR. Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell. 1997;88:561–572. doi: 10.1016/s0092-8674(00)81896-6. [DOI] [PubMed] [Google Scholar]
- 13.Buteau J, Spatz ML, Accili D. Transcription factor FoxO1 mediates glucagon-like peptide-1 effects on pancreatic β-cell mass. Diabetes. 2006;55:1190–1196. doi: 10.2337/db05-0825. [DOI] [PubMed] [Google Scholar]
- 14.Fatrai S, Elghazi L, Balcazar N, Cras-Meneur C, Krits I, Kiyokawa H, Bernal-Mizrachi E. Akt induces β-cell proliferation by regulating cyclin D1, cyclin D2, and p21 levels and cyclin-dependent kinase-4 activity. Diabetes. 2006;55:318–325. doi: 10.2337/diabetes.55.02.06.db05-0757. [DOI] [PubMed] [Google Scholar]
- 15.Drucker DJ. The biology of incretin hormones. Cell Metab. 2006;3:153–165. doi: 10.1016/j.cmet.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Garcia-Ocana A, Vasavada RC, Cebrian A, Reddy V, Takane KK, Lopez-Talavera JC, Stewart AF. Transgenic overexpression of hepatocyte growth factor in the β-cell markedly improves islet function and islet transplant outcomes in mice. Diabetes. 2001;50:2752–2762. doi: 10.2337/diabetes.50.12.2752. [DOI] [PubMed] [Google Scholar]
- 17.Vasavada RC, Garcia-Ocana A, Zawalich WS, Sorenson RL, Dann P, Syed M, Ogren L, Talamantes F, Stewart AF. Targeted expression of placental lactogen in the beta cells of transgenic mice results in beta cell proliferation, islet mass augmentation, and hypoglycemia. J Biol Chem. 2000;275:15399–15406. doi: 10.1074/jbc.275.20.15399. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Ocana A, Takane KK, Syed MA, Philbrick WM, Vasavada RC, Stewart AF. Hepatocyte growth factor overexpression in the islet of transgenic mice increases beta cell proliferation, enhances islet mass, and induces mild hypoglycemia. J Biol Chem. 2000;275:1226–1232. doi: 10.1074/jbc.275.2.1226. [DOI] [PubMed] [Google Scholar]
- 19.Vasavada RC, Cavaliere C, D’Ercole AJ, Dann P, Burtis WJ, Madlener AL, Zawalich K, Zawalich W, Philbrick W, Stewart AF. Overexpression of parathyroid hormone-related protein in the pancreatic islets of transgenic mice causes islet hyperplasia, hyperinsulinemia, and hypoglycemia. J Biol Chem. 1996;271:1200–1208. doi: 10.1074/jbc.271.2.1200. [DOI] [PubMed] [Google Scholar]
- 20.Buteau J, Foisy S, Joly E, Prentki M. Glucagon-like peptide 1 induces pancreatic β-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes. 2003;52:124–132. doi: 10.2337/diabetes.52.1.124. [DOI] [PubMed] [Google Scholar]
- 21.Liu JL, Coschigano KT, Robertson K, Lipsett M, Guo Y, Kopchick JJ, Kumar U, Liu YL. Disruption of growth hormone receptor gene causes diminished pancreatic islet size and increased insulin sensitivity in mice. Am J Physiol Endocrinol Metab. 2004;287:E405–E413. doi: 10.1152/ajpendo.00423.2003. [DOI] [PubMed] [Google Scholar]
- 22.Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest. 2004;114:963–968. doi: 10.1172/JCI22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, Sicinski P, White MF. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol. 2005;25:3752–3762. doi: 10.1128/MCB.25.9.3752-3762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, Barbacid M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- 25.Tsutsui T, Hesabi B, Moons DS, Pandolfi PP, Hansel KS, Koff A, Kiyokawa H. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol Cell Biol. 1999;19:7011–7019. doi: 10.1128/mcb.19.10.7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bernard C, Thibault C, Berthault MF, Magnan C, Saulnier C, Portha B, Pralong WF, Penicaud L, Ktorza A. Pancreatic β-cell regeneration after 48-h glucose infusion in mildly diabetic rats is not correlated with functional improvement. Diabetes. 1998;47:1058–1065. doi: 10.2337/diabetes.47.7.1058. [DOI] [PubMed] [Google Scholar]
- 27.Paris M, Bernard-Kargar C, Berthault MF, Bouwens L, Ktorza A. Specific and combined effects of insulin and glucose on functional pancreatic beta-cell mass in vivo in adult rats. Endocrinology. 2003;144:2717–2727. doi: 10.1210/en.2002-221112. [DOI] [PubMed] [Google Scholar]
- 28.Steil GM, Trivedi N, Jonas JC, Hasenkamp WM, Sharma A, Bonner-Weir S, Weir GC. Adaptation of beta-cell mass to substrate oversupply: enhanced function with normal gene expression. Am J Physiol Endocrinol Metab. 2001;280:E788–E796. doi: 10.1152/ajpendo.2001.280.5.E788. [DOI] [PubMed] [Google Scholar]
- 29.Topp BG, McArthur MD, Finegood DT. Metabolic adaptations to chronic glucose infusion in rats. Diabetologia. 2004;47:1602–1610. doi: 10.1007/s00125-004-1493-5. [DOI] [PubMed] [Google Scholar]
- 30.Bernard C, Berthault MF, Saulnier C, Ktorza A. Neogenesis vs. apoptosis as main components of pancreatic beta cell ass changes in glucose-infused normal and mildly diabetic adult rats. FASEB J. 1999;13:1195–1205. doi: 10.1096/fasebj.13.10.1195. [DOI] [PubMed] [Google Scholar]
- 31.Lipsett M, Finegood DT. β-Cell neogenesis during prolonged hyperglycemia in rats. Diabetes. 2002;51:1834–1841. doi: 10.2337/diabetes.51.6.1834. [DOI] [PubMed] [Google Scholar]
- 32.Briaud I, Lingohr MK, Dickson LM, Wrede CE, Rhodes CJ. Differential activation mechanisms of Erk-1/2 and p70(S6K) by glucose in pancreatic β-cells. Diabetes. 2003;52:974–983. doi: 10.2337/diabetes.52.4.974. [DOI] [PubMed] [Google Scholar]
- 33.Briaud I, Dickson LM, Lingohr MK, McCuaig JF, Lawrence JC, Rhodes CJ. Insulin receptor substrate-2 proteasomal degradation mediated by a mammalian target of rapamycin (mTOR)-induced negative feedback down-regulates protein kinase B-mediated signaling pathway in beta-cells. J Biol Chem. 2005;280:2282–2293. doi: 10.1074/jbc.M412179200. [DOI] [PubMed] [Google Scholar]
- 34.Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA, Donath MY. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest. 2002;110:851–860. doi: 10.1172/JCI15318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srinivasan S, Bernal-Mizrachi E, Ohsugi M, Permutt MA. Glucose promotes pancreatic islet beta-cell survival through a PI 3-kinase/Akt-signaling pathway. Am J Physiol Endocrinol Metab. 2002;283:E784–E793. doi: 10.1152/ajpendo.00177.2002. [DOI] [PubMed] [Google Scholar]
- 36.Hoorens A, Van de Casteele M, Kloppel G, Pipeleers D. Glucose promotes survival of rat pancreatic beta cells by activating synthesis of proteins which suppress a constitutive apoptotic program. J Clin Invest. 1996;98:1568–1574. doi: 10.1172/JCI118950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cozar-Castellano I, Weinstock M, Haught M, Velazquez-Garcia S, Sipula D, Stewart AF. Evaluation of β-cell replication in mice transgenic for hepatocyte growth factor and placental lactogen: comprehensive characterization of the G1/S regulatory proteins reveals unique involvement of p21cip. Diabetes. 2006;55:70–77. [PubMed] [Google Scholar]
- 38.Jonas JC, Laybutt DR, Steil GM, Trivedi N, Pertusa JG, Van de Casteele M, Weir GC, Henquin JC. High glucose stimulates early response gene c-Myc expression in rat pancreatic beta cells. J Biol Chem. 2001;276:35375–35381. doi: 10.1074/jbc.M105020200. [DOI] [PubMed] [Google Scholar]
- 39.Terauchi Y, Takamoto I, Kubota N, Matsui J, Suzuki R, Komeda K, Hara A, Toyoda Y, Miwa I, Aizawa S, Tsutsumi S, Tsubamoto Y, Hashimoto S, Eto K, Nakamura A, Noda M, Tobe K, Aburatani H, Nagai R, Kadowaki T. Glucokinase and IRS-2 are required for compensatory beta cell hyperplasia in response to high-fat diet-induced insulin resistance. J Clin Invest. 2007;117:246–257. doi: 10.1172/JCI17645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swenne I. The role of glucose in the in vitro regulation of cell cycle kinetics and proliferation of fetal pancreatic B-cells. Diabetes. 1982;31:754–760. doi: 10.2337/diab.31.9.754. [DOI] [PubMed] [Google Scholar]
- 41.De Vroede MA, In’ t Veld PA, Pipeleers DG. Deoxyribonucleic acid synthesis in cultured adult rat pancreatic B cells. Endocrinology. 1990;127:1510–1516. doi: 10.1210/endo-127-3-1510. [DOI] [PubMed] [Google Scholar]
- 42.Kaung HC. Effect of glucose on beta cell proliferation and population size in organ culture of foetal and neonatal rat pancreases. J Embryol Exp Morphol. 1983;75:303–312. [PubMed] [Google Scholar]
- 43.Gahr S, Merger M, Bollheimer LC, Hammerschmied CG, Scholmerich J, Hugl SR. Hepatocyte growth factor stimulates proliferation of pancreatic beta-cells particularly in the presence of subphysiological glucose concentrations. J Mol Endocrinol. 2002;28:99–110. doi: 10.1677/jme.0.0280099. [DOI] [PubMed] [Google Scholar]
- 44.Hugl SR, White MF, Rhodes CJ. Insulin-like growth factor I (IGF-I)-stimulated pancreatic beta-cell growth is glucose-dependent: synergistic activation of insulin receptor substrate-mediated signal transduction pathways by glucose and IGF-I in INS-1 cells. J Biol Chem. 1998;273:17771–17779. doi: 10.1074/jbc.273.28.17771. [DOI] [PubMed] [Google Scholar]
- 45.Cousin SP, Hugl SR, Myers MG, Jr, White MF, Reifel-Miller A, Rhodes CJ. Stimulation of pancreatic beta-cell proliferation by growth hormone is glucose-dependent: signal transduction via janus kinase 2 (JAK2)/signal transducer and activator of transcription 5 (STAT5) with no crosstalk to insulin receptor substrate-mediated mitogenic signalling. Biochem J. 1999;344:649–658. [PMC free article] [PubMed] [Google Scholar]
- 46.Ohsugi M, Cras-Meneur C, Zhou Y, Bernal-Mizrachi E, Johnson JD, Luciani DS, Polonsky KS, Permutt MA. Reduced expression of the insulin receptor in mouse insulinoma (MIN6) cells reveals multiple roles of insulin signaling in gene expression, proliferation, insulin content, and secretion. J Biol Chem. 2005;280:4992–5003. doi: 10.1074/jbc.M411727200. [DOI] [PubMed] [Google Scholar]
- 47.Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999;96:329–339. doi: 10.1016/s0092-8674(00)80546-2. [DOI] [PubMed] [Google Scholar]
- 48.Ueki K, Okada T, Hu J, Liew CW, Assmann A, Dahlgren GM, Peters JL, Shackman JG, Zhang M, Artner I, Satin LS, Stein R, Holzenberger M, Kennedy RT, Kahn CR, Kulkarni RN. Total insulin and IGF-I resistance in pancreatic beta cells causes overt diabetes. Nat Genet. 2006;38:583–588. doi: 10.1038/ng1787. [DOI] [PubMed] [Google Scholar]
- 49.Shimizu K, Amagaya S, Ogihara Y. Analysis of corticosterone in the serum of mice and rats using high-performance liquid chromatography. J Chromatogr. 1983;272:170–175. doi: 10.1016/s0378-4347(00)86114-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.