

Abstract
Human preterm infants with necrotizing enterocolitis (NEC) have increased circulating and luminal levels of platelet-activating factor (PAF) and decreased serum PAF-acetylhydrolase (PAF-AH), the enzyme that inactivates PAF. Formula supplemented with recombinant PAF-AH decreases NEC in a neonatal rat model. We hypothesized that endogenous PAF-AH contributes to neonatal intestinal homeostasis, and therefore developed PAF-AH−/− mice using standard approaches to study the role of this enzyme in the neonatal NEC model. Following exposure to a well-established NEC model, intestinal tissues were evaluated for histology, pro-inflammatory cytokine mRNA synthesis, and death using standard techniques. We found that mortality rates were significantly lower in PAF-AH−/− pups compared to wild-type controls before 24 hours of life but surviving PAF-AH−/− animals were more susceptible to NEC development compared to wild-type controls. Increased NEC incidence was associated with prominent inflammation characterized by elevated intestinal mRNA expression of sPLA2, iNOS and CXCL1. In conclusion, the data support a protective role for endogenous PAF-AH in the development of NEC, and since preterm neonates have endogenous PAF-AH deficiency, this may place them at increased risk for disease.
INTRODUCTION
Neonatal necrotizing enterocolitis (NEC) is a common and devastating condition that afflicts premature newborn infants and is characterized by acute onset of intestinal inflammatory necrosis. The cause of NEC has not been elucidated, and mortality rates remain unacceptably high (1). Prematurity, formula feeding, intestinal ischemia, and bacterial colonization may contribute to the pathogenesis of NEC, as these stresses initiate pro-inflammatory signaling and intestinal injury in susceptible hosts. Platelet-activating factor (PAF, 1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine) is a potent pro-inflammatory mediator that contributes to the pathogenesis of NEC (2, 3). PAF initiates signaling following binding to a G-protein-coupled receptor [PAF receptor, PAFR (4)] and activation of downstream events that result in epithelial apoptosis, leukocyte activation, and the initiation of NEC in experimental systems (5–7). Studies in human infants with NEC revealed increased luminal and systemic accumulation of PAF and decreased expression of PAF acetylhydrolase [PAF-AH, also known as PLA2G7, LpPLA2 (8, 9)]. PAF-AH catalyzes the hydrolysis of the acetyl group esterifying the sn-2 position of PAF, thereby abolishing its biological activity (10). PAF-AHs exist in secreted (11) and intracellular (12) forms. The secreted form is found in biological fluids, including plasma (11) and breast milk (13), but is absent in infant formula. Human neonates are deficient in PAF-AH expression and enteral administration of the recombinant enzyme markedly reduced the incidence of NEC in a rat model of the disease (14). To assess the importance of endogenous PAF-AH activity in neonatal NEC, intestinal inflammatory responses, and survival, we developed genetically engineered mice that lack expression of the plasma form of this enzyme. We found that deletion of PAF-AH reduced early mortality associated with bacterial exposure and asphyxia, but that survivors had significantly higher incidence of NEC and increased expression of pro-inflammatory mediators compared to wild-type mice. These findings suggest that endogenous PAF-AH protects against NEC and that deficiency of this protein, a feature characteristic of preterm infants, increases the risk of developing this disease.
METHODS
Animal model of NEC
All animal protocols were reviewed and approved by the NorthShore University Health System Institutional Animal Care and Use Committee. To investigate the impact of PAF-AH in neonatal NEC, we utilized a previously described rodent model of the disease (15, 16). Neonatal mice were obtained from timed-pregnant animals via Cesarean section at post-conception days E20–21. Wild-type controls were C57BL/6J and the PAF-AH knockouts were backcrossed 8 times onto the C57BL/6J parental strain before initiating experiments. Neonatal pups were gavaged using a 1.9 F silicone catheter (PICC-NATER®, Utah Medical Products) containing Esbilac formula (PetAg, Inc.; 1.8 kcal/mL) every three hours and supplemented with 1×107 or 1×104 bacterial loads of Serratia marcescens and Streptococcus viridans in their first feeding after birth and on each succeeding day. Pups were exposed to a brief episode of asphyxia (60 seconds in 100% N2) followed by cold stress (4°C for 10 minutes) twice daily. Neonatal mice were monitored for approximately 24–48 hours, a time during which NEC is known to develop. The pups were euthanized when signs of distress were apparent, or at 24–48 hours, depending on the experimental design. We then isolated intestinal sections for histological analyses. Portions of the tissues were snap-frozen in liquid N2 and kept at −80°C for RNA extraction.
Histology
The intestine was removed and linearized in ice-cold normal saline solution. Duodenum, jejunum, ileum, and colon were embedded in OCT, sectioned, and stained with hematoxylin and eosin (H&E). A score was assigned by a blinded reviewer, using the NEC scoring system we published previously (17). Briefly, intact villi received a score of 0, sloughing of epithelial cells on villous tips was scored 1, mid-villous damage was scored 2, a score of 3 represented complete villous necrosis, and 4 indicated transmural necrosis. Scores greater than or equal to 2 were considered indicative of NEC.
PAF-AH activity assays
Intestinal PAF-AH activity was assayed according to Narahara et al. and Miwa et al. (18, 19), with minor modifications. Intestinal lysates were diluted to a concentration of 0.5 mg/ml using 0.25 M sucrose, prior to the assays. The mixtures included 50 μl of diluted samples, 300 μl of 50 mM Tris-HCl (pH 7.5) containing 0.2% BSA, 25 nmol of cold PAF, and 1 μl of 1-O-hexadecyl-2-[3H-acetyl] PAF (0.1 mCi/ml, PerkinElmer Life and Analytical Sciences, Inc., Boston, MA) in fatty acid-free bovine serum albumin (0.2% final concentration). The volume was adjusted to 0.5 ml, and the reactions were terminated after 20 minutes with trichloroacetic acid (14%). One-tenth of the supernatant was added to 5 ml of scintillation fluid (PerkinElmer Life and Analytical Sciences) and the released [3H]-acetate was quantified by scintillation counting. Plasma PAF-AH activity was determined as previously described (20).
Quantitative RT-PCR
RNA samples were extracted from the ileum using RNeasy mini kits and Qiacube (Qiagen), following instructions provided by the manufacturer. RNA concentrations were measured by staining using SYBR Green II (Molecular Probes, Inc.). Blanks, standards, and samples were stained in DNAse and RNAse-free TE buffer (1:10,000-fold dilution). We determined the fluorescence intensity of RNA/SYBR Green II complexes and then calculated RNA concentrations based on a standard curve. Each PCR reaction (total volume = 10 μl) contained 0.5 μl of cDNA, 0.5 μl of GAPDH primers (final concentration = 0.5 μM), 0.5 μl of primer mix of gene of interest (final concentration = 0.5 μM), 5 μl of PerfeCTaTM qPCR SuperMix, UNG (Quanta BioSciences, Inc.), and 3.5 μl of DNAse-free, RNAse-free water. Reactions utilized cycling conditions that were optimized for these experiments. mRNA levels are expressed relative to those of GAPDH. The sequences of the primers utilized were previously described (16, 21).
Statistics
Parametric data are presented as mean ± SEM. Comparisons between two groups were carried out using Student’s t-test for unpaired data. Comparisons between three or more groups were performed using ANOVA and differences between groups were assessed using Tukey’s post hoc test. Analysis of two independent variables was performed with a two-way ANOVA test. Ordinate data to compare mortality rates and NEC incidence rates were analyzed using a Chi-square test. Unless otherwise stated, a P-value lower than 0.05 was considered significant,
RESULTS
Targeted disruption of the mouse PAF-AH gene
A 15.3 kb clone isolated from an adult 129/SvJgenomic DNA library spanned exons 2–7 of the mouse PAF-AH gene and was utilized to generate a targeting vector in which exons 3 and 4 were replaced by a self-excision cassette, ACN (22) (Figure 1A). The resulting DNA (PAF-AHACN) was cloned into a plasmid that contained two copies of the TK gene in pBluescript, to generate a targeting vector. Screening was performed by long-range PCR using a primer located 218 bp upstream of the region of homology (p1, Figure 1B) combined with control (p2) and diagnostic (p3) primers (Figure 1B). A positive ES clone was microinjected into 126 blastocysts at Xenogen, and a chimeric male (χ) was bred to C57BL/6J females. Genomic DNA samples from positive and negative ES cells, from a chimeric mouse (χ), and from positive and negative F1 progeny were analyzed in three independent PCR reactions (Figure 1C). The mRNA generated in PAF-AH−/− animals lacked exons 3 and 4, as expected (not shown). We determined the levels of PAF-AH activity in the plasma ofwild-type and PAF-AH−/− adult mice. The expression levels were consistent with our predictions based on genetic analyses (Figure 1D). PAF-AH-deficient mice were generated at the University of Utah under the direction of DMS.
Figure 1. Targeted disruption of the mouse PAF-AH gene.

(A). Mouse PAF-AH genomic locus and targeting strategy. E: EcoRI site; P, loxP site; S, SalI site; X: XhoI site. (B). Long-range PCR analysis used to detect correct targeting in ES cell clones. Each clone was evaluated using control PCR primers (p1 and p2) and diagnostic primers (p1 and p3). The bottom panel shows examples of positive (1c8) and negative (1c9, 1c10) ES clones. (C). Germline transmission of the targeted allele to the F1 generation was determined by PCR using a variety of primer combinations. The right panels show analyses of wild-type (−) and targeted (+) ES cell DNA, tail DNA from a chimeric mouse (χ), and DNA from F1+/+ and F1−/+ mice. (D). Levels of serum PAF-AH activity in wild-type (n=20) and PAF-AH deficient mice (n=14).
Developmental control of PAF-AH expression in mice
Human infants express low levels of circulating PAF-AH activity and expression levels progressively reach adult values by 6 weeks of life (9). We measured serum PAF-AH activity in pups of various ages, and compared these values to those found in adult mouse serum samples. We found that neonatal and adult mice expressed comparable levels of PAF-AH activity in the plasma (Figure 2A). All mice expressed higher levels of PAF-AH activity compared to humans (Figure 2A), as previously reported (23).
Figure 2. PAF-AH activity in plasma and intestinal tissues of wild-type and PAF-AH−/− mice.

PAF-AH activity was determined as the rate of [3H] acetate released from 1-O-hexadecyl-[3H-acetyl]-PAF and expressed as nmol/min/mg. (A). Box-and-Whisker plot showing median serum PAF-AH activity in wild-type mice during development. The levels of PAF-AH activity in adult mouse and human serum samples are shown for comparison. * represents P < 0.01. (B). Gut PAF-AH activity in adult and neonatal wild-type (black bars) and PAF-AH−/− mice (white bars).
*p < 0.0001, age effect,
**p < 0.0001, genotype effect.
Expression of PAF-AH in intestinal tissues
We next determined the levels of PAF-AH activity in intestinal tissues of neonatal and adult mice (Figure 2B). We found that wild-type neonatal mice expressed higher intestinal PAF-AH activity compared to adult animals (Figure 2B, 2-way ANOVA, p<0.0001, Bonferroni post hoc test). PAF-AH−/− mice expressed significantly lower activity compared to wild-type animals, regardless of age (Figure 2B, 2-way ANOVA, p<0.0001, Bonferroni post hoc test). The most robust differences in expression were observed by comparing neonatal PAF-AH−/− vs. wild-type mice (Figure 2B). These results confirm that deletion of the plasma form of PAF-AH abolishes expression of serum and intestinal activities.
Deletion of PAF-AH increases pro-inflammatory gene expression
We next sought to characterize the consequences of PAF-AH deletion on the expression of several key pro-inflammatory mediators in healthy, mother-fed neonatal mice. We measured CXCL1 and iNOS mRNA expression because these mediators regulate key pathways in intestinal inflammation (24, 25) and repair (26). In addition, we evaluated PAFR mRNA levels because this receptor regulates PAF-induced signaling. We found that PAF-AH−/− pups expressed higher levels of CXCL1 compared to wild-type controls (Figure 3A). In addition, we observed that CXCL1 expression decreased with age in mice of both genotypes (Figure 3A, p<0.05; 2-way ANOVA). Deletion of PAF-AH also resulted in higher intestinal iNOS expression (Figure 3B, p<0.01, 2-way ANOVA). While the levels of iNOS remained constant in wild-type mice over the period studied, iNOS expression increased significantly by two days of life in PAF-AH−/− animals compared to wild-type controls (Figure 3B). In contrast, PAFR expression was similar between wild-type and knockout mice (Figure 3C).
Figure 3. Expression of CXCL1 (A), iNOS (B), and PAFR (C) mRNA in the ileum of wild-type and PAF-AH−/− mice during development.

mRNA levels were quantified by RT-PCR in the ileum of mother-fed wild-type (■) and PAF-AH−/− (□) animals at 0, 1, and 2 days after birth. For CXCL1, *p < 0.01, age effect and **p < 0.01, genotype effect. Two-day old pups displayed statistically higher iNOS levels compared to day 0 and 1 (*p<0.01). In contrast, we detected no genotype- or age-dependent differences in PAFR mRNA levels.
Survival of wild-type and PAF-AH−/− mice following bacterial challenge
To evaluate whether deletion of PAF-AH affected survival and/or NEC incidence, we utilized our model of NEC which includes asphyxia, formula feeding and bacterial supplementation (16). When we subjected C57BL/6J wild-type and PAF-AH knockout mice to our model of NEC, we observed high early mortality rates in wild-type cohorts and lower incidence of early death in the PAF-AH knockout strain (Figure 4). The etiology of early death was initially unclear, but further studies revealed the presence of Serratia marscesens and Streptococcal viridans in the bloodstream of a high percentage of animals. To reduce the risk for sepsis and early death associated with exogenous bacterial supplementation, we lowered the bacterial load to 1×104 organisms/animal/day. However, this alteration did not significantly decrease the death rate (Figure 4). Thus, deletion of PAF-AH decreased the incidence of early death following bacterial supplementation (Fisher’s exact test, p<0.001). These results suggest that accumulation of PAF and perhaps other PAF-AH substrates is protective against neonatal bacterial infection.
Figure 4. Mortality rates of wild-type and PAF-AH−/− mice before and after 24 hours of life.
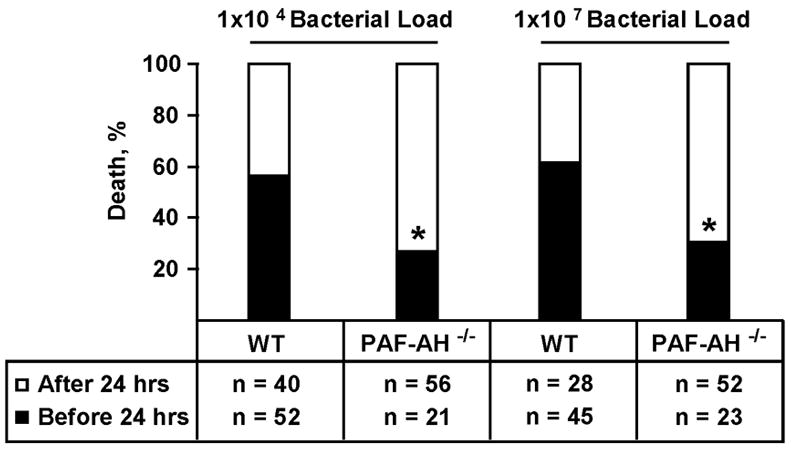
Pups retrieved from timed-pregnant wild-type and PAF-AH−/− mice by Cesarean section between E20–E21 were subjected to our NEC protocol using a 1×104 or 1×107 bacterial load of Serratia marcescens and Streptococcus viridans in their first feeding after birth and on each succeeding day. Mortality incidence was significantly higher in wild-type compared to PAF-AH−/− mice before 24 hours of life (Fisher’s exact test, * p<0.001).
Susceptibility of wild-type and PAF-AH−/− mice to NEC
We next compared the incidence of NEC in wild-type and PAF-AH−/− animals that survived after 24 hours. In these studies, we utilized a bacterial load of 1×104 organisms/animal/day and found higher incidence of NEC in PAF-AH−/− animals (43%, 19 NEC-positive vs 25 NEC-negative animals) compared to wild-type pups (20%, 6 NEC positive vs 24 NEC-negative animals, Chi-square test, p<0.05) (Figure 5). Similar to our previous studies, the severity of histologic injury was quite variable in both wild-type and PAF-AH−/− animals, and anatomic location was most prominent in ileum and proximal colon.
Figure 5. NEC incidence in wild-type and PAF-AH−/− mice after 24 hours of life.

Intestinal samples were scored on a scale of 0–4 to evaluate NEC severity. Animals that survived longer than 24 hours with a score ≥2 were considered to have NEC. Data were analyzed using a Chi squared test. The incidence of NEC in PAF-AH−/− pups (43%) was significantly higher than that observed in wild-type controls (20%, *p<0.05).
Pro-inflammatory gene expression in cold- and asphyxia-stressed wild-type and PAF-AH−/−mice
We found that PAF-AH−/− pups expressed significantly higher levels of CXCL1 and iNOS compared to wild-type animals (Figure 6, A–B, p<0.05; unpaired t test with Welch’s correction). These differences were observed regardless of whether the animals died before or after 24 hours of life (Figure 6, A–B) and surpassed the increases in expression observed between healthy, mother-fed, wild-type and PAF-AH−/− mice (Figure 3). We detected no differences in ileal PAFR mRNA levels in wild-type versus PAF-AH−/− pups subjected to our NEC model (Figure 6C), suggesting that the capacity for PAFR-mediated signaling was not affected by deletion of PAF-AH. To investigate whether the potential for PAF production was differentially affected in wild-type and PAF-AH−/− pups, we assessed the levels of ileal sPLA2 in both cohorts. We found that PAF-AH deficient mice subjected to our NEC model expressed elevated levels of sPLA2 mRNA compared to equally treated wild-type animals, and that the extent of up-regulation varied according to survival. PAF-AH−/− animals that perished before 24 hours displayed 3.7-fold higher levels of sPLA2 mRNA compared to wild-type controls (Figure 6D). PAF-AH−/− pups that survived longer than 24 hours displayed 8-fold higher ileal sPLA2 compared to wild-type pups (p<0.05; unpaired t test with Welch’s correction, Figure 6D). Increased expression of one of the rate-limiting enzymes in the PAF biosynthetic pathway suggests the possibility that a positive feedback loop leading to additional PAF synthesis is operative in PAF-AH−/− pups.
Figure 6. Expression of CXCL1 (A), iNOS (B), PAFR (C), and sPLA2 (D) mRNA in mice subjected to a model of NEC.

mRNA levels were quantified by RT-PCR in the ileum of wild-type and PAF-AH−/− mice subjected to our model of NEC using 1×104 bacterial load of Serratia marcescens and Streptococcus viridans. PAF-AH−/− expressed higher levels of mRNA for CXCL1 (*p<0.05; t-test), iNOS (*p<0.05; t-test) and sPLA2 (*p<0.05; t-test). In contrast, we detected no genotype-or time-of-death-dependent differences in PAFR expression.
DISCUSSION
In this study, we demonstrate that neonatal mice deficient in endogenous PAF-AH are more susceptible to NEC in response to formula feeding, bacterial colonization, and asphyxia/cold stress, compared to wild-type pups. Since neonatal human infants have very low endogenous PAF-AH activity, this new model provides significant insight into the pathophysiology of neonatal NEC. PAF-AH−/− mice exposed to the NEC protocol expressed significantly higher sPLA2, CXCL1, and iNOS compared to wild-type animals. These findings suggest that PAF-AH deficiency increases sPLA2 and subsequent PAF accumulation, resulting in robust up-regulation of pro-inflammatory gene expression, intestinal inflammation, and NEC.
Several studies have investigated the importance of PAF and PAF-AH in the pathogenesis of NEC. In animal models, intra-aortic injection of PAF into adult rats caused ischemic intestinal necrosis similar to that observed in patients with NEC (27); pretreatment with dexamethasone or medroxyprogesterone significantly increased plasma PAF-AH activity and prevented the gross and histologic features of NEC (28). These observations support a role for endogenous PAF and PAF-AH activity as opposite players in the control of acute intestinal necrosis. We previously developed a neonatal rat model of NEC that incorporates key features of the human disease, including asphyxia, formula feeding, and bacterial colonization (14). In this model, enteral administration of recombinant PAF-AH significantly reduced the incidence of NEC (14). Our observation that neonatal rodents express high levels of endogenous plasma and intestinal PAF-AH activity (this study) while neonatal humans are deficient (9), led us to hypothesize that neonatal mice deficient in the endogenous enzyme might be a better model of human NEC. The present study demonstrates that deficiency of intestinal and circulating PAF-AH markedly increases the severity of inflammation and the incidence of NEC, and due to adequate PAF-degrading capacity, wild-type control mice may not be a reliable model for human, neonatal disease.
We previously demonstrated a significant role for PAF-induced intestinal injury in experimental NEC (2, 5). The mechanism whereby deletion of PAF-AH increases NEC severity is likely to involve exacerbated PAF signaling. PAF homeostasis is defined at the biosynthetic and catabolic levels (4). PAF biosynthesis through the remodeling pathway requires participation of specific PLA2 isoforms and recently described lyso-PAF acetyltransferases (29, 30). PAF-AH activities down-regulate PAF signaling in several biological compartments. The observation that human neonates are deficient in circulating PAF-AH activity, combined with the presence of active enzyme in breast milk, led to the hypothesis that milk PAF-AH may confer protection against NEC development in preterm newborns (13). Recent work by Chen and co-workers showed that inhibition of PAF-AH induces PAF accumulation equivalent to that observed following agonist stimulation in quiescent neutrophils and monocytes (31). These combined observations point to increased PAF signaling as a feature associated with PAF-AH deletion. The functional consequences associated with exacerbated PAF signaling in the intestine remain to be completely characterized, but previous work demonstrated that PAF up-regulates expression of sPLA2 in this organ (32). In this study we found that deletion of PAF-AH was associated with higher levels of ileal sPLA2 mRNA in pups challenged with our NEC protocol. Thus, a feed forward mechanism of increased sPLA2 and subsequent PAF production may account for enhanced NEC severity in PAF-AH−/− neonatal mice.
Increased inflammation is a feature of neonatal NEC in humans and in animal models of the disease (33–35). The pro-inflammatory functions of PAF suggest key roles in the final common pathway leading to NEC (2). We found that deficiency of PAF-AH was associated with increased levels of iNOS and CXCL1 (IL-8 in humans), two mediators whose expression has been shown to contribute to intestinal inflammation (36, 37). Interestingly, the expression of these pro-inflammatory genes was elevated in un-stressed, PAF-AH deficient animals. However, stress from bacterial exposure, formula feeding, and asphyxia further increased CXCL1 and iNOS mRNA levels in PAF-AH−/− pups compared to wild-type animals. These observations underscore that endogenous PAF-AH has important anti-inflammatory functions in neonatal animals.
Our studies revealed interesting and unexpected findings related to the role of endogenous PAF-AH in the intestine. We found that early mortality was lower (20–30%) in PAF-AH knockout compared to wild-type mice, regardless of bacterial load. Our ability to observe these differences was likely facilitated by the unexpected finding that the C57BL/6J background conferred neonatal mice with exquisite sensitivity to bacterial exposure. Blood culture results from these animals confirmed that most pups died from septic shock. The observation that PAF-AH−/− animals survived significantly longer than wild-type pups when subjected to identical stress suggests that increased local and systemic PAF and/or other PAF-AH substrates may be protective. PAF has been shown to be required for the ability of neutrophils to kill bacteria (38), and this property may attenuate early bacterial translocation, thus limiting events that lead to septic shock in newborns. Our combined results are consistent with a model whereby suboptimal PAF levels predispose an organism to infection, while excessive production of the mediator contributes to inflammation and bowel necrosis.
In conclusion, we have demonstrated that genetically deficient PAF-AH mice are protected from early mortality in response to bacterial exposure, formula feeding, and asphyxia, but develop significantly more NEC after 24 hours compared to wild-type controls. The incidence of NEC correlates with increased intestinal pro-inflammatory gene expression in PAF-AH knockout animals. These novel findings identify two different roles for PAF-AH in the regulation of key responses during neonatal development. Our results highlight the potential utility of exogenous PAF-AH supplementation for the prevention of NEC in premature infants.
Acknowledgments
We are indebted to Liping Chen for key contributions to the development of PAF-AH-deficient mice before her untimely death. We are indebted to Katrina A. Lund, Ethan C. Reichert, and Alison A. Gardner (University of Utah) for excellent technical assistance. We also thank Drs. Michaeline Bunting, Stephen M. Prescott, Kenneth W. Spitzer, Kirk Thomas, and Matthew K. Topham for valuable advice and discussions.
Financial support: This work was supported by NIH grants HD37581 and AI058128 [to M.S.C. and T.J.] and HL35828 [to D.M.S.].
Abbreviations
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- NEC
necrotizing enterocolitis
- PAF(R)
platelet-activating factor (receptor)
- PAF-AH
PAF- acetylhydrolase
- PLA2
phospholipase A2
Footnotes
Publisher's Disclaimer: Pediatric Research Articles Ahead of Print contains articles in unedited manuscript form that have been peer-reviewed and accepted for publication. As a service to our readers, we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting and review of the resulting proof before it is published in its final definitive form. Please note that during the production process errors may be discovered, which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Henry MC, Moss RL. Necrotizing enterocolitis. Annu Rev Med. 2009;60:111–124. doi: 10.1146/annurev.med.60.050207.092824. [DOI] [PubMed] [Google Scholar]
- 2.Caplan MS, MacKendrick W. Inflammatory mediators and intestinal injury. Clin Perinatol. 1994;21:235–246. [PubMed] [Google Scholar]
- 3.Caplan MS, Hedlund E, Adler L, Hsueh W. Role of asphyxia and feeding in a neonatal rat model of necrotizing enterocolitis. Pediatr Pathol. 1994;14:1017–1028. doi: 10.3109/15513819409037698. [DOI] [PubMed] [Google Scholar]
- 4.Prescott SM, Zimmerman GA, Stafforini DM, McIntyre TM. Platelet-activating factor and related lipid mediators. Annu Rev Biochem. 2000;69:419–445. doi: 10.1146/annurev.biochem.69.1.419. [DOI] [PubMed] [Google Scholar]
- 5.Caplan MS, Kelly A, Hsueh W. Endotoxin and hypoxia-induced intestinal necrosis in rats: the role of platelet activating factor. Pediatr Res. 1992;31:428–434. doi: 10.1203/00006450-199205000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Hsueh W, Caplan MS, Sun X, Tan X, MacKendrick W, Gonzalez-Crussi F. Platelet-activating factor, tumor necrosis factor, hypoxia and necrotizing enterocolitis. Acta Paediatr Suppl. 1994;396:11–17. doi: 10.1111/j.1651-2227.1994.tb13234.x. [DOI] [PubMed] [Google Scholar]
- 7.Sun X, Caplan MS, Hsueh W. Tumour necrosis factor and endotoxin synergistically activate intestinal phospholipase A2 in mice. Role of endogenous platelet activating factor and effect of exogenous platelet activating factor. Gut. 1994;35:215–219. doi: 10.1136/gut.35.2.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caplan MS, Hsueh W, Sun XM, Gidding SS, Hageman JR. Circulating plasma platelet activating factor in persistent pulmonary hypertension of the newborn. Am Rev Respir Dis. 1990;142:1258–1262. doi: 10.1164/ajrccm/142.6_Pt_1.1258. [DOI] [PubMed] [Google Scholar]
- 9.Caplan M, Hsueh W, Kelly A, Donovan M. Serum PAF acetylhydrolase increases during neonatal maturation. Prostaglandins. 1990;39:705–714. doi: 10.1016/0090-6980(90)90030-y. [DOI] [PubMed] [Google Scholar]
- 10.Stafforini DM. Biology of platelet-activating factor acetylhydrolase (PAF-AH, lipoprotein associated phospholipase A2) Cardiovasc Drugs Ther. 2009;23:73–83. doi: 10.1007/s10557-008-6133-8. [DOI] [PubMed] [Google Scholar]
- 11.Stafforini DM, McIntyre TM, Carter ME, Prescott SM. Human plasma platelet-activating factor acetylhydrolase. Association with lipoprotein particles and role in the degradation of platelet-activating factor. J Biol Chem. 1987;262:4215–4222. [PubMed] [Google Scholar]
- 12.Arai H. Platelet-activating factor acetylhydrolase. Prostaglandins Other Lipid Mediat. 2002;68–69:83–94. doi: 10.1016/s0090-6980(02)00023-0. [DOI] [PubMed] [Google Scholar]
- 13.Moya FR, Eguchi H, Zhao B, Furukawa M, Sfeir J, Osorio M, Ogawa Y, Johnston JM. Platelet-activating factor acetylhydrolase in term and preterm human milk: a preliminary report. J Pediatr Gastroenterol Nutr. 1994;19:236–239. doi: 10.1097/00005176-199408000-00015. [DOI] [PubMed] [Google Scholar]
- 14.Caplan MS, Lickerman M, Adler L, Dietsch GN, Yu A. The role of recombinant platelet-activating factor acetylhydrolase in a neonatal rat model of necrotizing enterocolitis. Pediatr Res. 1997;42:779–783. doi: 10.1203/00006450-199712000-00010. [DOI] [PubMed] [Google Scholar]
- 15.Caplan MS, Jilling T. The role of polyunsaturated fatty acid supplementation in intestinal inflammation and neonatal necrotizing enterocolitis. Lipids. 2001;36:1053–1057. doi: 10.1007/s11745-001-0816-3. [DOI] [PubMed] [Google Scholar]
- 16.Jilling T, Simon D, Lu J, Meng FJ, Li D, Schy R, Thomson RB, Soliman A, Arditi M, Caplan MS. The roles of bacteria and TLR4 in rat and murine models of necrotizing enterocolitis. J Immunol. 2006;177:3273–3282. doi: 10.4049/jimmunol.177.5.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jilling T, Lu J, Jackson M, Caplan MS. Intestinal epithelial apoptosis initiates gross bowel necrosis in an experimental rat model of neonatal necrotizing enterocolitis. Pediatr Res. 2004;55:622–629. doi: 10.1203/01.PDR.0000113463.70435.74. [DOI] [PubMed] [Google Scholar]
- 18.Narahara H, Frenkel RA, Johnston JM. Secretion of platelet-activating factor acetylhydrolase following phorbol ester-stimulated differentiation of HL-60 cells. Arch Biochem Biophys. 1993;301:275–281. doi: 10.1006/abbi.1993.1144. [DOI] [PubMed] [Google Scholar]
- 19.Miwa M, Miyake T, Yamanaka T, Sugatani J, Suzuki Y, Sakata S, Araki Y, Matsumoto M. Characterization of serum platelet-activating factor (PAF) acetylhydrolase. Correlation between deficiency of serum PAF acetylhydrolase and respiratory symptoms in asthmatic children. J Clin Invest. 1988;82:1983–1991. doi: 10.1172/JCI113818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stafforini DM, McIntyre TM, Prescott SM. Platelet-activating factor acetylhydrolase from human plasma. Methods Enzymol. 1990;187:344–357. doi: 10.1016/0076-6879(90)87041-z. [DOI] [PubMed] [Google Scholar]
- 21.Caplan MS, Russell T, Xiao Y, Amer M, Kaup S, Jilling T. Effect of polyunsaturated fatty acid (PUFA) supplementation on intestinal inflammation and necrotizing enterocolitis (NEC) in a neonatal rat model. Pediatr Res. 2001;49:647–652. doi: 10.1203/00006450-200105000-00007. [DOI] [PubMed] [Google Scholar]
- 22.Bunting M, Bernstein KE, Greer JM, Capecchi MR, Thomas KR. Targeting genes for self-excision in the germ line. Genes Dev. 1999;13:1524–1528. doi: 10.1101/gad.13.12.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gardner AA, Reichert EC, Topham MK, Stafforini DM. Identification of a domain that mediates association of platelet-activating factor acetylhydrolase with high density lipoprotein. J Biol Chem. 2008;283:17099–17106. doi: 10.1074/jbc.M802394200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heuvelin E, Lebreton C, Grangette C, Pot B, Cerf-Bensussan N, Heyman M. Mechanisms involved in alleviation of intestinal inflammation by bifidobacterium breve soluble factors. PLoS One. 2009;4:e5184. doi: 10.1371/journal.pone.0005184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singer II, Kawka DW, Scott S, Weidner JR, Mumford RA, Riehl TE, Stenson WF. Expression of inducible nitric oxide synthase and nitrotyrosine in colonic epithelium in inflammatory bowel disease. Gastroenterology. 1996;111:871–885. doi: 10.1016/s0016-5085(96)70055-0. [DOI] [PubMed] [Google Scholar]
- 26.Bove PF, Wesley UV, Greul AK, Hristova M, Dostmann WR, van der Vliet A. Nitric oxide promotes airway epithelial wound repair through enhanced activation of MMP-9. Am J Respir Cell Mol Biol. 2007;36:138–146. doi: 10.1165/rcmb.2006-0253SM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzalez-Crussi F, Hsueh W. Experimental model of ischemic bowel necrosis. The role of platelet-activating factor and endotoxin. Am J Pathol. 1983;112:127–135. [PMC free article] [PubMed] [Google Scholar]
- 28.Furukawa M, Lee EL, Johnston JM. Platelet-activating factor-induced ischemic bowel necrosis: the effect of platelet-activating factor acetylhydrolase. Pediatr Res. 1993;34:237–241. doi: 10.1203/00006450-199308000-00027. [DOI] [PubMed] [Google Scholar]
- 29.Shimizu T. Lipid mediators in health and disease: enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annu Rev Pharmacol Toxicol. 2009;49:123–150. doi: 10.1146/annurev.pharmtox.011008.145616. [DOI] [PubMed] [Google Scholar]
- 30.Shindou H, Hishikawa D, Nakanishi H, Harayama T, Ishii S, Taguchi R, Shimizu T. A single enzyme catalyzes both platelet-activating factor production and membrane biogenesis of inflammatory cells. Cloning and characterization of acetyl-CoA:LYSO-PAF acetyltransferase. J Biol Chem. 2007;282:6532–6539. doi: 10.1074/jbc.M609641200. [DOI] [PubMed] [Google Scholar]
- 31.Chen J, Yang L, Foulks JM, Weyrich AS, Marathe GK, McIntyre TM. Intracellular PAF catabolism by PAF acetylhydrolase counteracts continual PAF synthesis. J Lipid Res. 2007;48:2365–2376. doi: 10.1194/jlr.M700325-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Tan XD, Wang H, Gonzalez-Crussi FX, Chang H, Gonzalez-Crussi F, Hsueh W. Platelet activating factor and endotoxin increase the enzyme activity and gene expression of type II phospholipase A2 in the rat intestine. Role of polymorphonuclear leukocytes. J Immunol. 1996;156:2985–2990. [PubMed] [Google Scholar]
- 33.Caplan MS, Hedlund E, Hill N, MacKendrick W. The role of endogenous nitric oxide and platelet-activating factor in hypoxia-induced intestinal injury in rats. Gastroenterology. 1994;106:346–352. doi: 10.1016/0016-5085(94)90591-6. [DOI] [PubMed] [Google Scholar]
- 34.De Plaen IG, Liu SX, Tian R, Neequaye I, May MJ, Han XB, Hsueh W, Jilling T, Lu J, Caplan MS. Inhibition of nuclear factor-kappaB ameliorates bowel injury and prolongs survival in a neonatal rat model of necrotizing enterocolitis. Pediatr Res. 2007;61:716–721. doi: 10.1203/pdr.0b013e3180534219. [DOI] [PubMed] [Google Scholar]
- 35.Caplan MS, Sun XM, Hseuh W, Hageman JR. Role of platelet activating factor and tumor necrosis factor-alpha in neonatal necrotizing enterocolitis. J Pediatr. 1990;116:960–964. doi: 10.1016/s0022-3476(05)80661-4. [DOI] [PubMed] [Google Scholar]
- 36.Nadler EP, Dickinson E, Knisely A, Zhang XR, Boyle P, Beer-Stolz D, Watkins SC, Ford HR. Expression of inducible nitric oxide synthase and interleukin-12 in experimental necrotizing enterocolitis. J Surg Res. 2000;92:71–77. doi: 10.1006/jsre.2000.5877. [DOI] [PubMed] [Google Scholar]
- 37.Nadler EP, Stanford A, Zhang XR, Schall LC, Alber SM, Watkins SC, Ford HR. Intestinal cytokine gene expression in infants with acute necrotizing enterocolitis: interleukin-11 mRNA expression inversely correlates with extent of disease. J Pediatr Surg. 2001;36:1122–1129. doi: 10.1053/jpsu.2001.25726. [DOI] [PubMed] [Google Scholar]
- 38.Rubin BB, Downey GP, Koh A, Degousee N, Ghomashchi F, Nallan L, Stefanski E, Harkin DW, Sun C, Smart BP, Lindsay TF, Cherepanov V, Vachon E, Kelvin D, Sadilek M, Brown GE, Yaffe MB, Plumb J, Grinstein S, Glogauer M, Gelb MH. Cytosolic phospholipase A2-alpha is necessary for platelet-activating factor biosynthesis, efficient neutrophil-mediated bacterial killing, and the innate immune response to pulmonary infection: cPLA2-alpha does not regulate neutrophil NADPH oxidase activity. J Biol Chem. 2005;280:7519–7529. doi: 10.1074/jbc.M407438200. [DOI] [PMC free article] [PubMed] [Google Scholar]