

Abstract
β-Site amyloid precursor protein cleaving enzyme 1 (BACE1) initiates amyloid-β (Aβ) generation that is central to the pathophysiology of Alzheimer’s disease (AD). Therefore, lowering Aβ levels by BACE1 manipulations represents a key therapeutic strategy, but it remains unclear whether partial inhibition of BACE1, as expected for AD treatments, can improve memory deficits. In this study, we used heterozygous BACE1 gene knockout (BACE1+/−) mice to evaluate the effects of partial BACE1 suppression on different types of synaptic and cognitive dysfunctions in Alzheimer’s transgenic mice (5XFAD model). We found that ~50% BACE1 reductions rescued deficits of 5XFAD mice not only in hippocampus-dependent memories as tested by contextual fear conditioning and spontaneous alternation Y-maze paradigms but also in cortex-dependent remote memory stabilization during 30 days after contextual conditioning. Furthermore, 5XFAD-associated impairments in long-term potentiation (a synaptic model of learning and memory) and declines in synaptic plasticity/learning-related BDNF-TrkB signaling pathways were prevented in BACE1+/−·5XFAD mice. Finally, these improvements were related with reduced levels of β-secretase-cleaved C-terminal fragment (C99), Aβ peptides and plaque burden in relevant brain regions of BACE1+/−·5XFAD mice. Therefore, our findings provide compelling evidence for beneficial effects of partially BACE1-inhibiting approaches on multiple forms of functional defects associated with AD.
Keywords: β-secretase, knockout, amyloid-β, fear conditioning, long-term potentiation, brain-derived neurotrophic factor
The processing of amyloid precursor protein (APP) by β-site APP-cleaving enzyme 1 (BACE1) is the first and rate-limiting step in releasing neurotoxic amyloid-β (Aβ) peptides and thus an excellent therapeutic target for the treatment of Alzheimer’s disease (AD). In spite of extensive medicinal chemistry efforts, much work still remains for the development of small-molecule BACE1-inhibiting drugs that can substantially lower cerebral Aβ levels after systemic administration (John 2006; Evin and Kenche 2007; Hills and Vacca 2007; Ghosh et al. 2008). Studies from our laboratory and others have demonstrated that BACE1 gene deletion (BACE1−/−) blocks Aβ production in brain, prevents pathological changes such as Aβ deposition, glial activation and neuron loss, and improves memory dysfunctions in mouse models of AD (Ohno et al. 2004, 2006, 2007; Laird et al. 2005; Ohno 2006, 2008). Although these results support the idea that BACE1 inhibition may be beneficial, efficacies of partial BACE1 suppression, as expected for future AD treatments with the appropriate dosage of suitable β-secretase inhibitors, remain unclear.
Consistent with the discovery of various BACE1 physiological substrates besides APP such as neuregulins, α-2,6-sialyltransferase and β-subunits of the voltage-gated sodium channel (Ohno 2006, 2008; Cole and Vassar 2007; Vassar et al. 2009), recent studies have revealed potential liabilities of complete BACE1 deletion including both cognitive and non-cognitive impairments, hippocampal synaptic dysfunctions, deregulated excitability of cortical neurons and hypomyelination (Ohno et al. 2004, 2006, 2007; Dominguez et al. 2005; Laird et al. 2005; Hu et al. 2006; Willem et al. 2006; Savonenko et al. 2008; Wang et al. 2008). Although the abnormalities associated with BACE1−/− genotype may raise some concern with respect to the safety of therapeutic BACE1 inhibition, it should be emphasized that these phenotypes are not observed in heterozygous BACE1 knockout (BACE1+/−) mice (Dominguez et al. 2005; Laird et al. 2005; Hu et al. 2006; Savonenko et al. 2008). Therefore, potential mechanism-based side effects might occur only in completely abolishing BACE1 activity but not in partially inhibiting it. Notably, recent data demonstrated that BACE1+/− deletion can reduce AD neuropathological changes such as Aβ plaques and neuritic dystrophy in PDAPP transgenic mice (McConlogue et al. 2007). However, it remains to be investigated whether BACE1+/− ablation suffices to improve cognitive and synaptic dysfunctions associated with AD.
We evaluated the effects of BACE1+/− deletion in transgenic mice that co-overexpress human APP and presenilin-1 (PS1) harboring five familial AD (FAD) mutations (5XFAD model) (Oakley et al. 2006; Ohno et al. 2006, 2007). 5XFAD mice start to develop visible amyloid deposition as early as ~2 months of age consistent with their dramatically accelerated Aβ42 production due to a combination of the multiple FAD mutations, thus representing an early-onset and aggressive amyloid mouse model (Oakley et al. 2006). At 4–6 months of age, 5XFAD mice not only show hippocampal synaptic and memory dysfunctions (Ohno et al. 2006; Kimura and Ohno 2009; Ohno 2009) but also exhibit remote memory destabilization caused by cortical Aβ accumulation (Kimura and Ohno 2009). For our study 5XFAD mice were crossed with BACE1+/− mice, and we applied a broad battery of behavioral and electrophysiological analyses combined with measurements of biochemical markers, plaque burden and Aβ levels to the 6-month-old mice and determined the degree of BACE1 inhibition that is required to achieve functional improvements. The findings provide a comprehensive experimental framework for the efficacies of partially BACE1-inhibiting and Aβ-lowering strategies for the treatment of AD.
Materials and methods
Animals
We used 5XFAD APP/PS1 doubly transgenic mice (Tg6799 line) that co-express and co-inherit FAD mutant forms of human APP (the Swedish mutation: K670N, M671L; the Florida mutation: I716V; the London mutation: V717I) and PS1 (M146L; L286V) transgenes under transcriptional control of the neuron-specific mouse Thy-1 promoter (Oakley et al. 2006; Ohno et al. 2006). Hemizygous 5XFAD transgenic mice (B6/SJL hybrid background) were crossbred to BACE1 homozygous knockout (BACE1−/−) mice (C57BL/6 background, The Jackson Laboratory, Bar Harbor, ME) (Cai et al. 2001; Laird et al. 2005) or C57BL/6 control mice. The resultant F1 heterozygous BACE1 knockout mice and hemizygous 5XFAD transgenic mice were further intercrossed, yielding animals with four different genotypes (wild-type, BACE1+/−, 5XFAD+/−, and BACE1+/−·5XFAD+/−) in the F2 progeny. All experiments were done with these mice at 6 months of age, blind with respect to their genotype, and were conducted with the approval of the Nathan Kline Institute Animal Care and Use Committee.
Immunoblot analysis
Hemibrain or hippocampal samples were taken from the mice under deep isoflurane anesthesia and were snap-frozen for biochemical assays. For Western blot analysis, each sample was homogenized in 5 volumes of modified RIPA buffer containing 150 mM NaCl, 50 mM Tris HCl (pH 8.0), 1 mM EDTA, 1% IGEPAL, 0.5% sodium deoxycholate, 0.1% SDS and protease inhibitor cocktail (Calbiochem, La Jolla, CA), and centrifuged at 10,000 g for 10 min to remove any insoluble material. Protein concentrations were determined by a BCA protein assay kit (Pierce, Rockford, IL), and 20–50 µg of protein was run on 12 or 4–12% NuPAGE gels (Invitrogen, Carlsbad, CA) and transferred to nitrocellulose membrane. After blocking, membranes were probed with anti-BACE1 (1:1,000, MAB5308, Millipore, Billerica, MA), an antibody that recognizes C-terminal epitope in APP (C1/6.1, kindly provided by Dr. Paul Mathews, Nathan Kline Institute) to detect full-length APP (1:1,000)/C-terminal fragments (1:500), an antibody specific for the soluble fragment cleaved n-terminus to the β-secretase cleavage site of APP (sAPPβ) (1:1,000, SIG-39138, Covance, Emeryville, CA), anti-brain-derived neurotrophic factor (BDNF) (1:1,000, ab72439, Abcam, Cambridge, MA), an antibody against phosphorylated tyrosine kinase receptor B (TrkB) (1:500, ab52191, Abcam) or anti-β-actin (1:15,000, AC-15, Sigma, St. Louis, MO), and were incubated with horseradish peroxidase-conjugated secondary IgG. Immunoblot signals were visualized by a Novex ECL chemiluminescence substrate reagent kit (Invitrogen) and were quantified by densitometric scanning and image analysis using Quantity One software (Bio-Rad Laboratories, Hercules, CA).
ELISA of Aβ40 and Aβ42
Sandwich Aβ ELISAs were performed as described previously (Oakley et al. 2006; Ohno et al. 2006). Briefly, each hemibrain sample was extracted in 8X cold 5 M guanidine HCl plus 50 mM Tris HCl (pH 8.0) buffer, and centrifuged at 20,000 g for 1 h at 4°C to remove insoluble material. Final guanidine HCl concentrations were below 0.1 M. Protein concentrations were determined by a BCA kit (Pierce). To quantitate total levels of cerebral Aβ40 and Aβ42, supernatant fractions were analyzed by well-established human Aβ40 and Aβ42 ELISA kits (KHB3481 and KHB3441, Invitrogen), respectively, according to the protocol of the manufacturer. These ELISAs cannot discriminate among Aβ peptides with heterogeneous N-termini; however, they specifically detect Aβ(x-40)/Aβ(x-42) in the absence of cross-reactivity with p3 fragments. Optical densities at 450 nm of each well were read on a VersaMax tunable microplate reader (Molecular Devices, Sunnyvale, CA), and sample Aβ40 and Aβ42 concentrations were determined by comparison with the respective standard curves. Aβ40 and Aβ42 concentration values were normalized to total brain protein concentrations and expressed in nanograms per milligram of total protein.
Aβ immunohistochemistry
Mice were perfused with 4% paraformaldehyde in phosphate buffered saline (PBS) under deep isoflurane anesthesia. The brain was removed and sectioned coronally at 30 µm on a vibratome (VT1200, Leica Microsystems, Wetzlar, Germany), and successive sections were stored in PBS containing 0.01% sodium azide at 4°C. Three to four sections per mouse were stained by the avidin-biotin peroxidase complex method (Oakley et al. 2006; Ohno et al. 2007) for immunohistochemical analysis of amyloid deposition in the regions of interest. Each section was separated by ~90 µm and taken at levels of +1.2/+0.8 mm (for the anterior cingulate cortex) and −1.5/−1.9 mm (for the hippocampus) to bregma according to the mouse brain atlas of Franklin and Paxinos (2008). The sections were incubated overnight at 4°C with monoclonal anti-Aβ1–16 antibody (1:200; 6E10, Signet, Dedham, MA). The ABC kit (PK-2200, Vector Laboratories, Burlingame, CA) was utilized with 3,3’-diaminobenzidine tetrahydrochloride as a chromogen to visualize the reaction product. The sections were then mounted on charged slides, dehydrated in a series of alcohol, cleared in xylene, and covered with a coverslip. Light microscopy was conducted on an Axioskop 2 microscope equipped with an AxioCaM HRc digital camera (Zeiss, Munich, Germany) for capturing images. Semi-quantitative analysis was performed using AxioVision imaging software with the AutoMeasure module (Zeiss). Identified objects after thresholding were individually inspected to confirm the object as a plaque or not in a blinded manner. Percentage area occupied by Aβ plaques in the hippocampus and anterior cingulate cortex were assessed bilaterally, and the average of the individual measurements from each mouse was used to calculate group medians.
Fear conditioning
Contextual fear conditioning was tested as described previously (Ohno et al. 2001; Kimura and Ohno 2009). The experiments were performed using four standard conditioning chambers, each of which was housed in a soundproof isolation cubicle and equipped with a stainless-steel grid floor connected to a solid-state shock scrambler. Each scrambler was connected to an electronic constant-current shock source that was controlled via an interface connected to a Windows XP computer running FreezeFrame software (Coulbourn Instruments, Allentown, PA). A digital camera was mounted on the steel ceiling of each chamber, and video signals were sent to the same computer for analysis. During training, mice were placed in the conditioning chamber for 3 min and then received two unsignaled footshocks (1.0 mA, 2 s) at 1 min intervals. When mice were tested for 30-day remote memories, they received more intensive training with three shocks (1 min intervals) to ensure their initial hippocampal memory formation. After the last shock delivery, mice were left in the chamber for another 30 s. Hippocampus-dependent contextual fear memory formation and the subsequent remote memory stabilization were evaluated by scoring freezing behavior (the absence of all movement except for that needed for breathing) for 3 min when the mice were placed back into the same conditioning chamber 1 day and 30 days after training, respectively. To avoid the confounding effects of extinction due to repeated exposures to the chamber, separate groups of mice were tested at each retention delay. The automated FreezeFrame system (Coulbourn Instruments), which digitizes the video signal at 4 Hz and compares movement frame by frame, was used to score the amount of freezing.
Spontaneous alternation Y-maze test
Spontaneous alternation performance was tested using a symmetrical Y-maze, as described previously (Ohno et al. 2004, 2007). Each mouse was placed in the center of the Y-maze and was allowed to explore freely through the maze during an 8-min session. The sequence and total number of arms entered were recorded. Arm entry was considered to be complete when the hind paws of the mouse had been completely placed in the arm. Percentage alternation is the number of triads containing entries into all three arms divided by the maximum possible alternations (the total number of arms entered minus 2) X 100.
Electrophysiology
After brains were removed from mice following decapitation under deep anesthesia with isoflurane, transverse hippocampal slices (400 µm thick) were prepared using a vibratome (VT1200, Leica Microsystems, Wetzlar, Germany) and were maintained in an artificial cerebral spinal fluid (aCSF)-filled holding chamber at room temperature for at least 1 h. The aCSF contained (in mM) 124 NaCl, 3 KCl, 2.4 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3 and 10 D-glucose, and was equilibrated with 95% O2 and 5% CO2. Slices were individually transferred to the submerged glass-bottom recording chamber, which was constantly perfused with oxygenated aCSF (2 ml/min) at 30°C. Field excitatory postsynaptic potential (fEPSP) was recorded with a metallic (Pt/Ir) electrode (FHC, Bowdoin, ME) from the stratum radiatum layer of CA1 area, and the Schaffer collateral afferents were stimulated with 100-µs test pulses via a bipolar cluster electrode (FHC) (Ohno et al. 2001; Kimura and Ohno 2009). To evaluate basal synaptic transmission, we applied different stimulation strengths (20 µA to 100 µA in steps of 10 µA) and plotted the amplitudes of presynaptic fiber volleys versus the corresponding fEPSP slopes to compare the slope of input/output (I/O) curves of fEPSP. For long-term potentiation (LTP) experiments, the stimulus strength was set to elicit 40–50% of the maximum fEPSP amplitude and test pulses were delivered to Schaffer collaterals once every 30 seconds. LTP was induced by 3-theta-burst stimulation (3-TBS) protocols (each burst consisted of 4 pulses at 100 Hz with a 200-ms interburst interval). Before 3-TBS application, the responses were monitored for at least 20 min to ensure a stable baseline of fEPSP. To determine whether the magnitude of LTP differed significantly between groups, average responses during the last 5-min block of recordings (35–40 min after 3-TBS) were compared.
Statistical analysis
The significance of differences between the groups was determined by a one-way ANOVA and post-hoc Fisher’s PLSD tests were performed when appropriate. Data were presented as mean ± SEM and the level of significance was set for p value less than 0.05.
Results
Partial ablation of BACE1 reduces sAPPβ, C99 and Aβ in 5XFAD mice
To evaluate the effects of partial BACE1 deletion on APP processing in the 5XFAD model, we first measured levels of BACE1, full-length APP and its β-cleaved fragments in brains of BACE1+/−·5XFAD mice at 6 months of age compared with those of 5XFAD littermate control mice (Fig. 1). Immunoblot analysis of hemibrain homogenates (Fig. 1a) demonstrated that BACE1+/− genotype significantly reduced cerebral BACE1 expression (by ~50%) in 5XFAD mice in concordance with the reduction in gene copy number (F(1,13) = 6.71, p < 0.05) (Fig. 1b). Transgene-derived overexpression of human APP in 5XFAD mice was several folds relative to endogenous levels of APP detected in wild-type controls (Fig. 1a), while the full-length APP expression levels were not different between BACE1+/−·5XFAD and 5XFAD mice (F(1,8) = 0.27, p > 0.05) (Fig. 1c). On the other hand, levels of the secreted ectodomain of APP produced by β-secretase (sAPPβ) and the β-secretase-cleaved C-terminal fragment (C99) were dramatically elevated in 5XFAD mouse brains as opposed to wild-type control brains that showed only background levels of both immunoreactive bands (Fig. 1a). Notably, cerebral levels of sAPPβ (F(1,13) = 11.73, p < 0.05) and C99 (F(1,8) = 6.59, p < 0.05) were significantly reduced in BACE1+/−·5XFAD mice compared to 5XFAD mice (Fig. 1d and 1e, respectively).
Fig. 1.

Effects of heterozygous BACE1 deletion on APP processing and Aβ levels in brains of 5XFAD mice at 6 months of age. (a) Immunoblot analysis of protein extracts from hemibrain homogenates of wild-type mice and 5XFAD mice with BACE1+/+ or BACE1+/− genotype. (b–e) Intensities of immunoreactive bands on blots for BACE1 (b), full-length APP (c), sAPPβ (d) and C99 (e) were quantified by phosphorimaging and expressed as percentage of 5XFAD levels (n = 5–8 mice per group). Note that ~50% elimination of BACE1 expression results in ~51% and ~55% reductions in sAPPβ and C99, respectively (* p < 0.05 versus 5XFAD), while APP overexpression levels are not changed in BACE1+/−·5XFAD mice compared to 5XFAD mice. (f–g) Levels of total Aβ40 (f) and Aβ42 (g) were quantified by sandwich ELISAs of guanidine extracts of hemibrain samples and expressed in nanograms per milligram of total protein (n = 5–7 mice per group). Note that excessive levels of Aβ40 and Aβ42 are significantly reduced in BACE1+/−·5XFAD mice as compared with 5XFAD mice (by ~66% and ~57%, respectively; * p < 0.05). All data are presented as mean ± SEM.
Furthermore, we performed sandwich ELISAs to measure cerebral Aβ levels (Fig. 1f, g). Consistent with our previous results (Oakley et al. 2006; Ohno et al. 2006), 5XFAD mice exhibited much higher levels of Aβ42 than Aβ40. Importantly, BACE1+/− ablation significantly and almost equivalently lowered excessive levels of Aβ40 and Aβ42 in 5XFAD mouse brains (Aβ40, F(1,10) = 8.46, p < 0.05; Aβ42, F(1,10) = 18.69, p < 0.05) (Fig. 1f and 1g, respectively). Taken collectively, biochemical analyses demonstrated that partial BACE1 reduction due to heterozygous gene deletion significantly suppressed the β-cleavage of APP in 5XFAD mouse brains, resulting in reduced levels of β-products such as sAPPβ, C99, Aβ40 and Aβ42 by ~51%, ~55%, ~66% and ~57%, respectively (Fig. 1d–g).
Partial ablation of BACE1 rescues recent memory deficits in 5XFAD mice
We examined whether BACE1+/− deletion ameliorated deficits in hippocampus-dependent recent memory observed in the 5XFAD mouse model at 6 months of age (Fig. 2). We first analyzed memory with a contextual fear conditioning paradigm, in which mice learn to associate a distinct context (CS: conditioned stimuli) with aversive footshocks (US: unconditioned stimuli) (Fanselow 2000). Wild-type mice exhibited a robust conditioned fear response as assessed by freezing (the absence of all but respiratory movements) when placed back into the conditioning chamber 1 day after training with two CS-US parings (Fig. 2a). The levels of contextual freezing were significantly different between the four groups of mice when analyzed by a one-way ANOVA (F(3,99) = 3.12, p < 0.05). Notably, post-hoc Fisher’s PLSD test revealed that 5XFAD mice at 6 months of age exhibited significantly lower freezing compared to wild-type controls (p < 0.05), whereas BACE1+/−·5XFAD bigenic mice showed significantly higher levels of freezing than did 5XFAD mice (p < 0.05) and were equivalent to wild-type littermate controls. Meanwhile, BACE1+/− mice were normal in contextual fear conditioning.
Fig. 2.

Effects of heterozygous BACE1 deletion on deficits in hippocampus-dependent memory in 5XFAD mice at 6 months of age. (a) Mice were trained with two CS-US pairings for contextual fear conditioning. 5XFAD mice show significantly lower levels of contextual freezing than wild-type mice when tested 1 day after training (* p < 0.05). Note that BACE1+/−·5XFAD are rescued completely back to wild-type control levels of contextual memory (# p < 0.05 versus 5XFAD). n = 20–38 mice per group. (b) Spatial working memory of mice was tested by spontaneous alternation performance in the Y-maze. Note that 5XFAD mice perform poorly (only slightly above 50% chance levels) compared to wild-type controls (* p < 0.05), while BACE1+/−·5XFAD are improved completely back to wild-type levels of alternation performance (# p < 0.05 versus 5XFAD). n = 11–21 mice per group. (c) Total number of arm entries reflecting exploratory activities of mice in the Y-maze does not differ between the four groups. n = 11–21 mice per group. All data are presented as mean ± SEM.
We also tested the mice with another hippocampus-dependent learning task, spontaneous alternation performance in the Y-maze that represents a measure of spatial working memory (Lalonde 2002). A one-way ANOVA similarly revealed significant differences in percent alternation between the four groups tested (F(3,63) = 9.98, p < 0.05) (Fig. 2b). 5XFAD mice at 6 months of age showed dramatically reduced levels of spontaneous alternation performance in the Y-maze as compared with wild-type controls (p < 0.05). Importantly, the spatial working memory deficits were also rescued to wild-type levels in 5XFAD mice with BACE1+/− genotype, which showed significantly higher spontaneous alternation than did 5XFAD mice (p < 0.05). In contrast to BACE1−/− mice that exhibited poor spontaneous alternation in the Y-maze in our previous studies (Ohno et al. 2004; Ohno et al. 2007), BACE1+/− mice were normal in this spatial working memory performance. Furthermore, the total number of arm entries in the alternation Y-maze test was not significantly different between the four groups of mice tested (F(3,63) = 0.62, p > 0.05) (Fig. 2c). Therefore, the levels of exploratory activity were not affected in these mice including BACE1+/− mice, which was also in contrast with our previous observation that BACE1−/− mice were hyperactive during Y-maze test (Ohno et al. 2004; Ohno et al. 2007). Therefore, BACE1+/− gene deletion was efficacious in preventing memory deficits assessed in 5XFAD mice by two different hippocampus-dependent learning tasks.
Partial ablation of BACE1 rescues remote memory deficits in 5XFAD mice
It is established that memories depend initially on the hippocampus, while they become increasingly independent of this structure and memory traces are gradually stabilized and eventually transformed into remote memories in cortical networks (McClelland et al. 1995; Dudai 2004; Frankland and Bontempi 2005; Squire and Bayley 2007). Our previous study revealed that remote memory stabilization during 30 days after contextual fear conditioning significantly declined in 5XFAD mice consistent with their massive Aβ accumulation in the cerebral cortex (Kimura and Ohno 2009). Therefore, we next evaluated the remote memory function by re-exposing separate groups of mice to the conditioning chamber 30 days after training (Fig. 3). To this end, we applied stronger training protocols with three CS-US pairings, since the reduced levels of initial memory formation following milder training (i.e., two shocks; Fig. 2a) would preclude specific analyses of the subsequent remote memory stabilization processes in 5XFAD mice at 6 months of age. 5XFAD mice exhibited robust contextual freezing that was comparable to wild-type controls 1 day after training with three footshocks (F(1,35) = 0.22, p > 0.05) (Fig. 3a), indicating that they successfully overcame the impairment of initial hippocampus-dependent contextual learning with more intensive training. A one-way ANOVA revealed significant differences in freezing levels between the four groups that were tested for remote contextual fear memory 30 days after three CS-US pairings (F(3,54) = 3.90, p < 0.05) (Fig. 3b). Remarkably, the comparisons with post-hoc Fisher’s PLSD test indicated that 5XFAD mice exhibited significantly lower freezing compared to wild-type control mice (p < 0.05), while the deficient remote memory stabilization during 30 days after training was completely restored to wild-type control levels in BACE1+/−·5XFAD bigenic mice (p < 0.05). Meanwhile, BACE1+/− mice were normal in remote contextual memory tested 30 days after conditioning. Altogether, the results indicated that partial reduction of BACE1 due to its heterozygous knockout improved deficits of 5XFAD mice not only in initial hippocampal memory formation but also in the subsequent prolonged processing of remote memory stabilization.
Fig. 3.

Effects of heterozygous BACE1 deletion on deficits in remote memory stabilization in 5XFAD mice at 6 months of age. (a) To ensure initial hippocampus-dependent memory formation in 5XFAD mice, they were trained by more intensive protocols with three CS-US pairings for contextual fear conditioning. 5XFAD mice show levels of contextual freezing equivalent to those of wild-type mice when tested 1 day after the stronger training. n = 15–22 mice per group. (b) 5XFAD mice exhibit significantly lower levels of freezing than wild-type controls when tested 30 days after training with three CS-US pairings (* p < 0.05). Note that BACE1+/−·5XFAD are rescued completely back to wild-type levels of 30-day remote contextual memory (# p < 0.05 versus 5XFAD). n = 12–17 mice per group. All data are presented as mean ± SEM.
Partial ablation of BACE1 alleviates amyloid pathology in 5XFAD mice
To investigate the relationship between changes in memory performances and levels of regional Aβ accumulation, we further conducted Aβ immunostaining (Fig. 4). The hippocampus is important for mediating contextual memory formation and spatial working memory in the Y-maze (Fanselow 2000; Lalonde 2002), while the anterior cingulate cortex is critically involved in contextual remote memory stabilization (Frankland and Bontempi 2005). Therefore, we focused on these two brain structures to analyze the effects of BACE1+/− deletion on amyloid plaque load with relevance to the recent and remote memory improvements in BACE1+/−·5XFAD mice, as indicated in Figs. 2 and 3. Immunostaining with the human APP/Aβ-specific monoclonal antibody 6E10 showed that brains of BACE1+/−·5XFAD mice at 6 months of age including the hippocampus (Fig. 4c) and anterior cingulate cortex (Fig. 4f) stain less intensely than the respective areas of 5XFAD littermate control mice (Fig. 4b, e). This result most likely indicates reduced levels of Aβ deposition as human APP levels remain unchanged (Fig. 1a, c) and Aβ ELISA values significantly decrease (Fig. 1f, g) in BACE1+/−·5XFAD mouse brains. Although stereology-based method may need to be applied to provide fully quantitative results, our semi-quantitative analysis of the percentage area covered by 6E10 immunoreactivity clearly revealed that plaque burden is significantly reduced in the hippocampus (F(1,12) = 21.43, p < 0.05) (by ~78%, Fig. 4g) and anterior cingulate cortex (F(1,13) = 8.68, p < 0.05) (by ~44%, Fig. 4h) of BACE1+/−·5XFAD mice compared with the respective brain regions of 5XFAD mice with normal levels of BACE1.
Fig. 4.
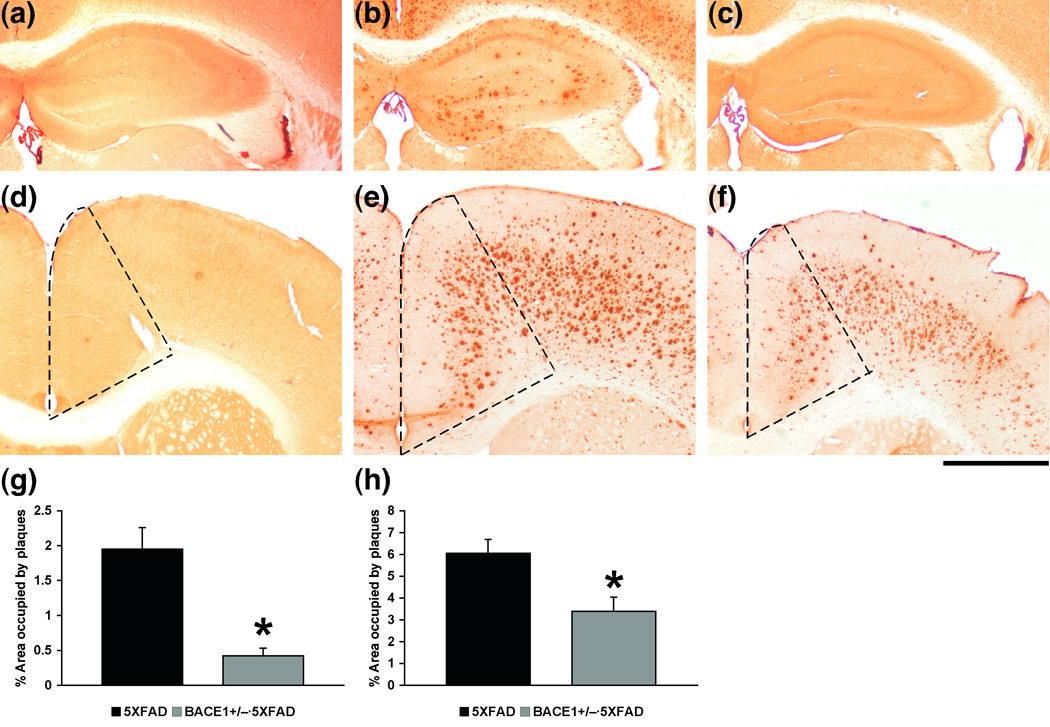
Effects of heterozygous BACE1 deletion on amyloid deposition in the hippocampus and anterior cingulate cortex of 5XFAD mice at 6 months of age. (a–f) Brain sections from wild-type control (a, d), 5XFAD (b, e) and BACE1+/−·5XFAD (c, f) mice were immunostained with the 6E10 anti-APP/Aβ antibody. Shown are representative photomicrographs of the hippocampus (a–c) and anterior cingulate cortex (d–f) (areas enclosed by the dashed line). Scale bar = 1 mm. (g–h) Percentage area covered by amyloid plaque in the hippocampus (g) and anterior cingulate cortex (h) was measured for quantification. Note that BACE1+/−·5XFAD mice show significantly lower levels of Aβ plaque burden in both brain regions compared to those of 5XFAD mice (* p < 0.05). n = 7 mice per group. All data are presented as mean ± SEM.
Partial ablation of BACE1 rescues LTP deficits in 5XFAD mice
Concomitant with the occurrence of memory deficits, 5XFAD mice at 6 months of age show hippocampal synaptic dysfunctions, as assessed by reductions in basal transmission and LTP (a measure of synaptic plasticity representing a cellular basis of learning and memory) at Schaffer collateral-CA1 pathways (Kimura and Ohno 2009). We examined whether BACE1+/− deletion rescued these synaptic failures observed in hippocampal slices from 6-month-old 5XFAD mice (Fig. 5). First, input/output (I/O) responses relating presynaptic fiber volley amplitude to the initial slope of the corresponding fEPSP were similarly affected in 5XFAD and BACE1+/−·5XFAD bigenic mice, as compared with those of their wild-type littermate controls (Fig. 5a). A one-way ANOVA and post-hoc Fisher’s PLSD test revealed that baseline synaptic transmission as assessed by the average slope of I/O curves was significantly reduced in 5XFAD mice (F(2,27) = 6.64, p < 0.05), and that basal transmission remained significantly impaired at hippocampal CA1 synapses in BACE1+/−·5XFAD mice (p < 0.05) that were indistinguishable from 5XFAD mice with BACE1+/+ genotype (Fig. 5b).
Fig. 5.

Effects of heterozygous BACE1 deletion on hippocampal synaptic dysfunctions in 5XFAD mice at 6 months of age. (a) The amplitudes of presynaptic fiber volleys are plotted against the slopes of corresponding fEPSPs in response to different stimulation strengths (20–100 µA) to compare input/output (I/O) curves at Schaffer collateral-CA1 synapses. (b) The summary bar graph shows that basal synaptic transmission as measured by the average slope of I/O curves is significantly impaired in both 5XFAD and BACE1+/−·5XFAD mice (* p < 0.05 versus wild-type controls). n = 8–14 mice per group. (c) LTP was induced by 3-TBS protocols at Schaffer collateral-CA1 synapses. Each point indicates the fEPSP slope normalized to the average baseline response before 3-TBS is applied at time 0. (d) The summary bar graph shows that LTP as measured by the average normalized fEPSP slopes during 35–40 min after 3-TBS is significantly impaired in 5XFAD mice (* p < 0.05 versus wild-type controls), while BACE1+/−·5XFAD mice are rescued almost completely back to wild-type control levels of LTP (# p < 0.05 versus 5XFAD). n = 8–12 mice per group. Traces are the average of fEPSPs recorded during baseline and 35–40 min after 3-TBS. Calibration: 0.5 mV, 10 ms. All data are presented as mean ± SEM.
We next examined whether BACE1+/− mutation improved synaptic plasticity at hippocampal CA1 synapses of 5XFAD mice (Fig. 5c). Consistent with our previous results (Kimura and Ohno 2009), LTP induced by 3-TBS protocols was significantly reduced in 5XFAD mice at 6 months of age. Importantly, while the earlier or induction phase of LTP remained impaired in BACE1+/−·5XFAD mice, the later or maintenance phase of LTP was rescued at CA1 synapses of these bigenic mice. A one-way ANOVA and post-hoc Fisher’s PLSD test, which compared the average magnitude of LTP during 35–40 min after induction between the three groups, revealed that LTP was significantly reduced in 5XFAD mice compared to wild-type controls, while the LTP deficit was almost completely restored to wild-type control levels in 5XFAD mice with BACE1+/− genotype (F(2,25) = 3.62, p < 0.05) (Fig. 5d). Together, the results indicated that deficient LTP, but not impaired basal transmission, at hippocampal CA1 synapses of 5XAFD mice was rescued by BACE1+/− deletion.
Partial ablation of BACE1 rescues BDNF-TrkB signaling deficits in 5XFAD mice
A growing body of work indicates that BDNF levels decline in brains of AD patients and APP transgenic mice (Peng et al. 2005; Hu and Russek 2008; Peng et al. 2009; Zuccato and Cattaneo 2009). Furthermore, it is known that BDNF and its receptor TrkB are critically involved in the late phase of LTP rather than its induction mechanisms (Pang and Lu 2004; Minichiello 2009). Therefore, we investigated BDNF-TrkB signaling pathways to address potential mechanisms underlying the rescue of LTP maintenance in BACE1+/−·5XFAD mice (Fig. 6). Western blot analysis of hippocampal homogenate samples (Fig. 6a) showed that BDNF levels were significantly reduced in 5XFAD mice at 6 months of age compared with wild-type controls (by ~35%), while BDNF was restored to wild-type levels in 5XFAD mice with BACE1+/− genotype (F(2,13) = 4.47, p < 0.05) (Fig. 6b). In accord, levels of phosphorylated TrkB, an activated form of the BDNF receptor, were also reduced in the hippocampus of 5XFAD mice (by ~67%), whereas this reduction was significantly attenuated in that of BACE1+/−·5XFAD mice (F(2,14) = 5.87, p < 0.05) (Fig. 6c). Therefore, BACE1+/− deletion improved deficiencies in hippocampal BDNF-TrkB signaling in 5XFAD mice in line with the rescue from their CA1 LTP impairments.
Fig. 6.

Effects of heterozygous BACE1 deletion on impaired hippocampal BDNF-TrkB signaling in 5XFAD mice at 6 months of age. (a) Immunoblot analysis of protein extracts from hippocampal homogenates of wild-type mice and 5XFAD mice with BACE1+/+ or BACE1+/− genotype. (b–c) Intensities of immunoreactive bands on blots for BDNF (b) and phosphorylated TrkB (p-TrkB) (c) were quantified by phosphorimaging and expressed as percentage of wild-type levels (n = 5–7 mice per group). Note that both BDNF and p-TrkB levels are significantly reduced in the hippocampus of 5XFAD mice (* p < 0.05 versus wild-type), while these BDNF-TrkB signaling defects are increased back to wild-type control levels in BACE1+/−·5XFAD mice (# p < 0.05 versus 5XFAD). All data are presented as mean ± SEM.
Discussion
Despite considerable progress in understanding of the genetic and molecular basis of AD amyloidogenesis, much work remains for the validation and practical application of Aβ-lowering strategies including the secretase inhibition to AD therapies (Aisen 2005). Not only the fact that BACE1 initiates Aβ production in the amyloid cascade but also recent findings that BACE1 expression and activities are elevated in sporadic AD brains favor the notion that this enzyme represents one of the prime therapeutic targets (Cole and Vassar 2007; Zacchetti et al. 2007; Ohno 2008). Previous studies with BACE1 null knockout mice have provided a critical step in evaluating efficacies of BACE1-inhibiting approaches for the treatment of AD and memory deficits (Ohno et al. 2004, 2006, 2007; Laird et al. 2005; Ohno 2006, 2008). However, even if suitable inhibitors are developed, it will be almost impossible to completely suppress BACE1 enzymatic activity in vivo. The impacts of partial BACE1 suppression, which would be more relevant to therapies in a clinical setting, have been examined using APP transgenic mice with heterozygous BACE1 ablation (Luo et al. 2001; Laird et al. 2005; McConlogue et al. 2007). These studies showed that while BACE1+/− deletion causes only a slight decrease (~12%) of soluble Aβ in younger APP mice at pre-pathological stages, it results in dramatic reductions (~30–90%) in Aβ levels and plaque load in older APP mice at moderate pathological stages. We report here the first demonstration that partial suppression of β-secretase due to BACE1+/− mutation is effective in preventing synaptic and mnemonic dysfunctions in a mouse model of AD.
In this study, ~50% reductions in BACE1 expression with heterozygous gene deletion significantly affected the β-cleavage of APP in 5XFAD mice, resulting in ~51% and ~55% decreases in sAPPβ and C99 levels, respectively. Consequently, BACE1+/− deletion caused ~66% and ~57% reductions in brain levels of Aβ40 and Aβ42, respectively. This partial suppression of BACE1 sufficed to rescue deficits of 5XFAD mice in hippocampus-dependent contextual memory tested 1 day after fear conditioning and spatial working memory in the spontaneous alternation Y-maze paradigm. Furthermore, amyloid deposition found in the hippocampus of 5XFAD mice was also suppressed (by ~78%) in that of BACE1+/−·5XFAD mice. It should be noted that Aβ42 is more hydrophobic and more prone to assemble into neurotoxic oligomers and aggregates than Aβ40 (McGowan et al. 2005; Haass and Selkoe 2007) and that Aβ42 is highly overproduced due to a combination of the multiple FAD mutations in 5XFAD mice (Oakley et al. 2006; Ohno et al. 2006). Therefore, it is likely that reductions in Aβ42, which is the species of particular importance in neuronal dysfunctions and early plaque formation, more critically contribute to the benefits observed in BACE1+/−·5XFAD mice, although two major forms of the Aβ peptide are almost equivalently diminished. Anyway, our results support the idea that levels of Aβ (especially, more pathogenic Aβ42) determine its effects on cognitive function and their ~50% reduction is widely accepted as it is likely efficacious in improving memories.
Our data from 5XFAD mice with BACE1+/− gene deletion is in agreement with recent findings that ~60% knock-down of hippocampal BACE1 with lentiviral-vectored siRNAs not only reduces C99 and total Aβ levels by ~60% and ~40%, respectively, but also significantly ameliorates spatial memory deficits of APP751Swe·Lon transgenic mice in a Morris water maze (Singer et al. 2005). Furthermore, chronic immunization of APPSwe Tg2576 mice with BACE1 ectodomain produces ~35% reductions in Aβ40 and Aβ42 levels presumably by interfering with the β-cleavage of APP in endocytic pathways and results in better hippocampus-dependent memory performance in the water maze (Chang et al. 2007). Therefore, different lines of evidence clearly demonstrate that partial suppression of BACE1 activities can impact the β-cleavage of APP to reduce cerebral Aβ levels and is effective in improving hippocampal cognitive declines in APP transgenic mouse models of AD. However, the therapeutic implications of these findings may need to be interpreted with some caution, since a recent report indicated that BACE1+/− gene deletion did not affect cerebral levels of endogenous Aβ in wild-type non-transgenic mice (Nishitomi et al. 2006). It is possible that the Swedish mutant form of APP, which is overexpressed in transgenic models including 5XFAD mice and represents the more efficient substrate for BACE1 than wild-type APP, may render the β-cleavage more sensitive to Aβ reductions by partial BACE1 inhibition. In this regard, BACE1+/− ablation significantly reduces Aβ levels and amyloid burden in PDAPP mice, of which APP has the V717F London mutation but is wild-type at the β-secretase cleavage site (McConlogue et al. 2007). Therefore, the Swedish mutation is not prerequisite; however, we cannot completely rule out the possibility that highly overexpressed levels of the substrate APP may lead to the augmented efficacy of partial BACE1 suppression in reducing cerebral Aβ concentrations in transgenic AD models. Given no clear evidence that APP levels are elevated in sporadic AD cases (Brouwers et al. 2006; Theuns et al. 2006), it will be important to re-evaluate whether partial suppression of the β-secretase enzyme activity can gain benefits in wild-type as well as APP transgenic mice using suitable BACE1 inhibitors when they become available.
Previous studies have analyzed APP mice for the acquisition of learning or memories shortly (~24 h) after training and thus mainly evaluated their defects in hippocampal encoding of information (Ashe 2001; Kobayashi and Chen 2005; Ohno 2006; Eriksen and Janus 2007). Behavioral experiments combined with brain imaging and inactivation techniques have revealed the temporary role of the hippocampus in the formation and maintenance of memories, and provided convergent evidence for the time shift from hippocampus-dependent recent memories (~1 day post-training) to hippocampus-independent (but cortex-dependent) remote memories (~30 days post-training) in a variety of learning tasks including contextual fear conditioning (Bontempi et al. 1999; Frankland et al. 2004; Maviel et al. 2004; Frankland and Bontempi 2005; Squire and Bayley 2007). Our results clearly demonstrated that BACE1+/− deletion not only rescued hippocampal recent memory deficits tested 1 day after contextual fear conditioning but also improved the subsequent cortical remote memory stabilization during 30 days after training in 5XFAD mice. Consistent with this, Aβ burden was also significantly reduced (by ~44%) in the anterior cingulate cortex of BACE1+/−·5XFAD mice, a key cortical subregion that is responsible for contextual remote memory stabilization (Frankland et al. 2004; Frankland and Bontempi 2005). Interestingly, recent evidence suggests that functional connectivity between the hippocampus and cortical areas including the medial prefrontal and anterior cingulate cortices (Wang et al. 2006) as well as neocortical synaptic plasticity like LTP (Battaglia et al. 2007) is disrupted in AD patients and APP transgenic mice. Therefore, it is likely that memory traces cannot be successfully transferred or stabilized as remote memories into cortical neuron networks in APP mice, which should add to cognitive declines due to their reduced levels of initial encoding within hippocampal circuits. Collectively, the results presented here suggest that partial inhibition of BACE1 can benefit the multiple memory systems by ameliorating failures in hippocampal, cortical, and their interactive mnemonic processing in 5XFAD mice as a consequence of Aβ reductions in both brain structures.
Electrophysiological studies with hippocampal slices from BACE1 mutant mice demonstrated that BACE1 deficiency does not affect basal synaptic transmission or plasticity such as LTP and long-term depression (LTD) at Schaffer collateral-CA1 pathways (Laird et al. 2005). However, LTP at mossy fiber-CA3 synapses, which mainly depends on increases in presynaptic release in contrast with CA1 LTP, is almost completely abolished in BACE1−/− mice (Wang et al. 2008). This is consistent with the observation that BACE1 is highly localized to nerve terminals including the giant boutons of mossy fiber pathways in the hippocampal CA3 region (Laird et al. 2005; Zhao et al. 2007). Therefore, BACE1 may play a role in regulating presynaptic function in hippocampal circuits under physiological conditions. To determine synaptic mechanisms by which partial BACE1 reduction may improve memory loss associated with AD, we investigated the effects of BACE1+/− deletion on synaptic deficits in 5XFAD mice. As reported previously (Kimura and Ohno 2009), 5XFAD mice at 6 months of age exhibited deficits in both basal transmission and LTP at Schaffer collateral-CA1 synapses in conjunction with the onset of hippocampal memory failure. Importantly, we demonstrated that only LTP deficits were restored to normal consistent with memory improvements and basal synaptic transmission remained significantly reduced in BACE1+/−·5XFAD mice. Although both types of synaptic dysfunctions are thought to represent neurophysiological consequences of Aβ accumulation in a series of AD mouse models (Selkoe 2002; Rowan et al. 2003), our results indicate that deficient LTP rather than reduced levels of baseline transmission at CA1 synapses is more closely related with hippocampus-dependent memory dysfunctions in 5XFAD mice. Conceivably, the restored LTP may, at least in part, account for the beneficial effects of partial reductions of BACE1, and consequently of Aβ, on cognitive dysfunction in 5XFAD mice.
What mechanisms may underlie the improvement of LTP and memory in BACE1+/−·5XFAD mice? While many plasticity-related signaling molecules are affected in APP transgenic mouse brains (Dickey et al. 2003; Dickey et al. 2004), evidence is accumulating that a decline in cerebral BDNF levels may be associated with the pathogenesis of AD (Peng et al. 2005; Hu and Russek 2008; Zuccato and Cattaneo 2009). In fact, BDNF reductions are also reported to occur in APP mice (Peng et al. 2009) and experimental strategies aimed at elevating central BDNF levels are effective in exerting neuroprotective effects against Aβ toxicity in diverse models of AD (Arancibia et al. 2008; Tapia-Arancibia et al. 2008; Nagahara et al. 2009). However, the relationship between deficient BDNF signaling and impaired synaptic/cognitive functions in AD remains elusive. We clearly showed that reductions in BDNF found in the hippocampus of 5XFAD mice (by ~35%) were completely abolished in that of BACE1+/−·5XFAD mice concomitant with their rescue from deficits in CA1 LTP, in particular, the maintenance phase of LTP. Importantly, heterozygous and homozygous knockouts of BDNF similarly impair LTP maintenance (rather than its induction processes) at CA1 synapses and cause spatial long-term memory deficits in the water maze (Korte et al. 1995; Patterson et al. 1996; Pang and Lu 2004; Pang et al. 2004), suggesting that a critical level of BDNF may be required for normal hippocampal functions. Furthermore, genetic reductions of TrkB receptors also specifically deteriorate LTP maintenance and water maze learning (Minichiello et al. 1999; Minichiello 2009), indicating that BDNF contributes to synaptic plasticity and memory function through TrkB. We also demonstrated that levels of phosphorylated TrkB, an activated form of BDNF receptors involved in modulating learning and LTP (Rattiner et al. 2004; Liu et al. 2008; Minichiello 2009), were reduced in the hippocampus of 5XFAD mice (by ~67%), while this reduction was significantly alleviated by BACE1+/− deletion. Taken collectively, our results support the idea that the ability of partial BACE1 inhibition to ameliorate deficient LTP and memory may be, at least in part, mediated by the restoration of impaired BDNF-TrkB signaling pathways in 5XFAD mice, although further study is needed to fully address the underlying mechanisms.
While 5XFAD-associated functional deficits were rescued by genetic reductions of BACE1, transgene expression levels were not affected by BACE1+/− or BACE1−/− deletion in 5XFAD mice in this study as well as in our previous experiments (Ohno et al. 2006, 2007). Therefore, it is unlikely that merely overexpressing FAD mutant forms of APP/PS1 may be the cause of synaptic or cognitive dysfunctions in 5XFAD mice. On the other hand, it is important to note that BACE1+/− deletion partially suppresses build-up of the β-secretase-cleaved C-terminal fragment C99 (by ~55%) in 5XFAD mouse brains. Transgenic overexpression or central administration of the potentially amyloidogenic C99 fragments has been reported to impair LTP and memory in a broad battery of behavioral assays (Nalbantoglu et al. 1997; Song et al. 1998; Berger-Sweeney et al. 1999; Choi et al. 2001; Lee et al. 2006). Taken together, although it is very likely that partial BACE1 inhibition benefits synaptic plasticity and memories through central lowering of Aβ peptides in APP transgenic mice, we cannot exclude the possibility that reduced levels of neurotoxic C99 may contribute to the functional improvements observed in BACE1+/−·5XFAD mice.
BACE1+/− mice were normal in contextual fear memory (including 30-day remote memory) and spatial working memory in the Y-maze. These results are consistent with the observation that partial BACE1 suppression caused by lentivirus-delivered siRNA does not affect normal learning and memory performances of non-transgenic control mice in the water maze (Singer et al. 2005). Notably, BACE1−/− mice, but not BACE1+/− mice, exhibit hippocampus-dependent memory deficits as tested by the auditory trace fear conditioning, water maze, and spontaneous alternation Y-maze paradigms (Ohno et al. 2004, 2006, 2007; Laird et al. 2005; Savonenko et al. 2008). Furthermore, BACE1−/− mice also show a broad range of non-cognitive abnormalities such as increased exploratory activities in the open-field and Y-maze tests, deceased swim speed during water maze training, lower levels of anxiety in the elevated plus-maze and impaired prepulse inhibition, whereas BACE1+/− mice completely lack these phenotypes (Ohno et al. 2004, 2006, 2007; Laird et al. 2005; Savonenko et al. 2008). Therefore, to be therapeutically effective, it would be crucial to partially inhibit β-secretase activities and reduce excess Aβ peptides to levels that are required to specifically ameliorate AD-related memory deficits but obviate potential adverse effects due to complete or too much suppression of BACE1.
In conclusion, the results presented here demonstrate that BACE1+/− deletion can rescue multiple forms of synaptic and memory failure in 5XFAD mice, representing a “proof-of-concept” for therapeutic efficacies of partially β-secretase-inhibiting and Aβ-reducing approaches for the treatment of AD. We evaluated the effects of BACE1+/− ablation using 6-month-old 5XFAD mice, in which moderate levels of AD-related pathological changes develop and synaptic or cognitive dysfunctions have just started to occur (Oakley et al. 2006; Kimura and Ohno 2009; Ohno 2009). This would probably enable us to sensitively detect any benefit that may result from modest lowering of neurotoxic Aβ in BACE1+/−·5XFAD mice. Importantly, if BACE1 activities are only partially blocked, APP transgenic mice slowly but continuously accumulate Aβ peptides in their brains. Consequently, the efficacy of BACE1+/− deletion in reducing cerebral Aβ and plaque burden is likely to become smaller and eventually disappear in APP mice as disease progresses into the severer pathological stages (Laird et al. 2005; McConlogue et al. 2007). In this regard, further investigation is needed to determine whether synaptic and cognitive improvements due to partial BACE1 suppression may remain unchanged or diminish in 5XFAD mice with BACE1+/− genotype at advanced ages, especially correlative to alterations of their Aβ levels in relevant brain regions.
Acknowledgements
This work was supported by National Institutes of Health grant R01 MH067251 (M.O.) and Alzheimer’s Association grant IIRG-08-91231 (M.O.).
References
- Aisen PS. The development of anti-amyloid therapy for Alzheimer's disease: from secretase modulators to polymerisation inhibitors. CNS Drugs. 2005;19:989–996. doi: 10.2165/00023210-200519120-00002. [DOI] [PubMed] [Google Scholar]
- Arancibia S, Silhol M, Mouliere F, Meffre J, Hollinger I, Maurice T, Tapia-Arancibia L. Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol Dis. 2008;31:316–326. doi: 10.1016/j.nbd.2008.05.012. [DOI] [PubMed] [Google Scholar]
- Ashe KH. Learning and memory in transgenic mice modeling Alzheimer's disease. Learn Mem. 2001;8:301–308. doi: 10.1101/lm.43701. [DOI] [PubMed] [Google Scholar]
- Battaglia F, Wang HY, Ghilardi MF, Gashi E, Quartarone A, Friedman E, Nixon RA. Cortical plasticity in Alzheimer's disease in humans and rodents. Biol Psychiatry. 2007;62:1405–1412. doi: 10.1016/j.biopsych.2007.02.027. [DOI] [PubMed] [Google Scholar]
- Berger-Sweeney J, McPhie DL, Arters JA, Greenan J, Oster-Granite ML, Neve RL. Impairments in learning and memory accompanied by neurodegeneration in mice transgenic for the carboxyl-terminus of the amyloid precursor protein. Brain Res Mol Brain Res. 1999;66:150–162. doi: 10.1016/s0169-328x(99)00014-5. [DOI] [PubMed] [Google Scholar]
- Bontempi B, Laurent-Demir C, Destrade C, Jaffard R. Time-dependent reorganization of brain circuitry underlying long-term memory storage. Nature. 1999;400:671–675. doi: 10.1038/23270. [DOI] [PubMed] [Google Scholar]
- Brouwers N, Sleegers K, Engelborghs S, Bogaerts V, Serneels S, Kamali K, Corsmit E, De Leenheir E, Martin JJ, De Deyn PP, Van Broeckhoven C, Theuns J. Genetic risk and transcriptional variability of amyloid precursor protein in Alzheimer's disease. Brain. 2006;129:2984–2991. doi: 10.1093/brain/awl212. [DOI] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nat Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- Chang WP, Downs D, Huang XP, Da H, Fung KM, Tang J. Amyloid-β reduction by memapsin 2 (β-secretase) immunization. FASEB J. 2007;21:3184–3196. doi: 10.1096/fj.06-7993com. [DOI] [PubMed] [Google Scholar]
- Choi SH, Park CH, Koo JW, Seo JH, Kim HS, Jeong SJ, Lee JH, Kim SS, Suh YH. Memory impairment and cholinergic dysfunction by centrally administered Aβ and carboxyl-terminal fragment of Alzheimer's APP in mice. FASEB J. 2001;15:1816–1818. doi: 10.1096/fj.00-0859fje. [DOI] [PubMed] [Google Scholar]
- Cole SL, Vassar R. The Alzheimer's disease β-secretase enzyme, BACE1. Mol Neurodegener. 2007;2:22. doi: 10.1186/1750-1326-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Loring JF, Montgomery J, Gordon MN, Eastman PS, Morgan D. Selectively reduced expression of synaptic plasticity-related genes in amyloid precursor protein + presenilin-1 transgenic mice. J Neurosci. 2003;23:5219–5226. doi: 10.1523/JNEUROSCI.23-12-05219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Gordon MN, Mason JE, Wilson NJ, Diamond DM, Guzowski JF, Morgan D. Amyloid suppresses induction of genes critical for memory consolidation in APP + PS1 transgenic mice. J Neurochem. 2004;88:434–442. doi: 10.1111/j.1471-4159.2004.02185.x. [DOI] [PubMed] [Google Scholar]
- Dominguez D, Tournoy J, Hartmann D, Huth T, Cryns K, Deforce S, Serneels L, Camacho IE, Marjaux E, Craessaerts K, Roebroek AJ, Schwake M, D'Hooge R, Bach P, Kalinke U, Moechars D, Alzheimer C, Reiss K, Saftig P, De Strooper B. Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J Biol Chem. 2005;280:30797–30806. doi: 10.1074/jbc.M505249200. [DOI] [PubMed] [Google Scholar]
- Dudai Y. The neurobiology of consolidations, or, how stable is the engram? Annu Rev Psychol. 2004;55:51–86. doi: 10.1146/annurev.psych.55.090902.142050. [DOI] [PubMed] [Google Scholar]
- Eriksen JL, Janus CG. Plaques, tangles, and memory loss in mouse models of neurodegeneration. Behav Genet. 2007;37:79–100. doi: 10.1007/s10519-006-9118-z. [DOI] [PubMed] [Google Scholar]
- Evin G, Kenche VB. BACE inhibitors as potential therapeutics for Alzheimer's disease. Recent Pat CNS Drug Discov. 2007;2:188–199. doi: 10.2174/157488907782411783. [DOI] [PubMed] [Google Scholar]
- Fanselow MS. Contextual fear, gestalt memories, and the hippocampus. Behav Brain Res. 2000;110:73–81. doi: 10.1016/s0166-4328(99)00186-2. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Bontempi B. The organization of recent and remote memories. Nat Rev Neurosci. 2005;6:119–130. doi: 10.1038/nrn1607. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Bontempi B, Talton LE, Kaczmarek L, Silva AJ. The involvement of the anterior cingulate cortex in remote contextual fear memory. Science. 2004;304:881–883. doi: 10.1126/science.1094804. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. New York: Academic Press; 2008. [Google Scholar]
- Ghosh AK, Kumaragurubaran N, Hong L, Koelsh G, Tang J. Memapsin 2 (beta-secretase) inhibitors: drug development. Curr Alzheimer Res. 2008;5:121–131. doi: 10.2174/156720508783954730. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hills ID, Vacca JP. Progress toward a practical BACE-1 inhibitor. Curr Opin Drug Discov Devel. 2007;10:383–391. [PubMed] [Google Scholar]
- Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9:1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- Hu Y, Russek SJ. BDNF and the diseased nervous system: a delicate balance between adaptive and pathological processes of gene regulation. J Neurochem. 2008;105:1–17. doi: 10.1111/j.1471-4159.2008.05237.x. [DOI] [PubMed] [Google Scholar]
- John V. Human β-secretase (BACE) and BACE inhibitors: progress report. Curr Top Med Chem. 2006;6:569–578. doi: 10.2174/156802606776743084. [DOI] [PubMed] [Google Scholar]
- Kimura R, Ohno M. Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol Dis. 2009;33:229–235. doi: 10.1016/j.nbd.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi DT, Chen KS. Behavioral phenotypes of amyloid-based genetically modified mouse models of Alzheimer's disease. Genes Brain Behav. 2005;4:173–196. doi: 10.1111/j.1601-183X.2005.00124.x. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC. BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci. 2005;25:11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde R. The neurobiological basis of spontaneous alternation. Neurosci Biobehav Rev. 2002;26:91–104. doi: 10.1016/s0149-7634(01)00041-0. [DOI] [PubMed] [Google Scholar]
- Lee KW, Im JY, Song JS, Lee SH, Lee HJ, Ha HY, Koh JY, Gwag BJ, Yang SD, Paik SG, Han PL. Progressive neuronal loss and behavioral impairments of transgenic C57BL/6 inbred mice expressing the carboxy terminus of amyloid precursor protein. Neurobiol Dis. 2006;22:10–24. doi: 10.1016/j.nbd.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Liu YF, Chen HI, Yu L, Kuo YM, Wu FS, Chuang JI, Liao PC, Jen CJ. Upregulation of hippocampal TrkB and synaptotagmin is involved in treadmill exercise-enhanced aversive memory in mice. Neurobiol Learn Mem. 2008;90:81–89. doi: 10.1016/j.nlm.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R. Mice deficient in BACE1, the Alzheimer's β-secretase, have normal phenotype and abolished β-amyloid generation. Nat Neurosci. 2001;4:231–232. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- Maviel T, Durkin TP, Menzaghi F, Bontempi B. Sites of neocortical reorganization critical for remote spatial memory. Science. 2004;305:96–99. doi: 10.1126/science.1098180. [DOI] [PubMed] [Google Scholar]
- McClelland JL, McNaughton BL, O'Reilly RC. Why there are complementary learning systems in the hippocampus and neocortex: insights from the successes and failures of connectionist models of learning and memory. Psychol Rev. 1995;102:419–457. doi: 10.1037/0033-295X.102.3.419. [DOI] [PubMed] [Google Scholar]
- McConlogue L, Buttini M, Anderson JP, Brigham EF, Chen KS, Freedman SB, Games D, Johnson-Wood K, Lee M, Zeller M, Liu W, Motter R, Sinha S. Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP transgenic mice. J Biol Chem. 2007;282:26326–26334. doi: 10.1074/jbc.M611687200. [DOI] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T. Aβ42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci. 2009;10:850–860. doi: 10.1038/nrn2738. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kuhn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbantoglu J, Tirado-Santiago G, Lahsaini A, Poirier J, Goncalves O, Verge G, Momoli F, Welner SA, Massicotte G, Julien JP, Shapiro ML. Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature. 1997;387:500–505. doi: 10.1038/387500a0. [DOI] [PubMed] [Google Scholar]
- Nishitomi K, Sakaguchi G, Horikoshi Y, Gray AJ, Maeda M, Hirata-Fukae C, Becker AG, Hosono M, Sakaguchi I, Minami SS, Nakajima Y, Li HF, Takeyama C, Kihara T, Ota A, Wong PC, Aisen PS, Kato A, Kinoshita N, Matsuoka Y. BACE1 inhibition reduces endogenous Abeta and alters APP processing in wild-type mice. J Neurochem. 2006;99:1555–1563. doi: 10.1111/j.1471-4159.2006.04178.x. [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M. Genetic and pharmacological basis for therapeutic inhibition of β- and β-secretases in mouse models of Alzheimer's memory deficits. Rev Neurosci. 2006;17:429–454. doi: 10.1515/revneuro.2006.17.4.429. [DOI] [PubMed] [Google Scholar]
- Ohno M. β-Secretase as a prime therapeutic target for Alzheimer’s disease: a perspective from mouse model studies. In: Araki W, editor. Recent Advances in the Biology of Secretases, Key Proteases in Alzheimer’s Disease. Kerala: Research Signpost; 2008. pp. 1–25. [Google Scholar]
- Ohno M. Failures to reconsolidate memory in a mouse model of Alzheimer's disease. Neurobiol Learn Mem. 2009;92:455–459. doi: 10.1016/j.nlm.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Frankland PW, Chen AP, Costa RM, Silva AJ. Inducible, pharmacogenetic approaches to the study of learning and memory. Nat Neurosci. 2001;4:1238–1243. doi: 10.1038/nn771. [DOI] [PubMed] [Google Scholar]
- Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron. 2004;41:27–33. doi: 10.1016/s0896-6273(03)00810-9. [DOI] [PubMed] [Google Scholar]
- Ohno M, Chang L, Tseng W, Oakley H, Citron M, Klein WL, Vassar R, Disterhoft JF. Temporal memory deficits in Alzheimer's mouse models: rescue by genetic deletion of BACE1. Eur J Neurosci. 2006;23:251–260. doi: 10.1111/j.1460-9568.2005.04551.x. [DOI] [PubMed] [Google Scholar]
- Ohno M, Cole SL, Yasvoina M, Zhao J, Citron M, Berry R, Disterhoft JF, Vassar R. BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiol Dis. 2007;26:134–145. doi: 10.1016/j.nbd.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang PT, Lu B. Regulation of late-phase LTP and long-term memory in normal and aging hippocampus: role of secreted proteins tPA and BDNF. Ageing Res Rev. 2004;3:407–430. doi: 10.1016/j.arr.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306:487–491. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Peng S, Wuu J, Mufson EJ, Fahnestock M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer's disease. J Neurochem. 2005;93:1412–1421. doi: 10.1111/j.1471-4159.2005.03135.x. [DOI] [PubMed] [Google Scholar]
- Peng S, Garzon DJ, Marchese M, Klein W, Ginsberg SD, Francis BM, Mount HT, Mufson EJ, Salehi A, Fahnestock M. Decreased brain-derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer's disease. J Neurosci. 2009;29:9321–9329. doi: 10.1523/JNEUROSCI.4736-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattiner LM, Davis M, French CT, Ressler KJ. Brain-derived neurotrophic factor and tyrosine kinase receptor B involvement in amygdala-dependent fear conditioning. J Neurosci. 2004;24:4796–4806. doi: 10.1523/JNEUROSCI.5654-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan M, Klyubin I, Cullen W, Anwy lR. Synaptic plasticity in animal models of early Alzheimer's disease. Philos Trans R Soc Lond B Biol Sci. 2003;358:821–828. doi: 10.1098/rstb.2002.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savonenko AV, Melnikova T, Laird FM, Stewart KA, Price DL, Wong PC. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc Natl Acad Sci U S A. 2008;105:5585–5590. doi: 10.1073/pnas.0710373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Singer O, Marr RA, Rockenstein E, Crews L, Coufal NG, Gage FH, Verma IM, Masliah E. Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat Neurosci. 2005;8:1343–1349. doi: 10.1038/nn1531. [DOI] [PubMed] [Google Scholar]
- Song DK, Won MH, Jung JS, Lee JC, Kang TC, Suh HW, Huh SO, Paek SH, Kim YH, Kim SH, Suh YH. Behavioral and neuropathologic changes induced by central injection of carboxyl-terminal fragment of β-amyloid precursor protein in mice. J Neurochem. 1998;71:875–878. doi: 10.1046/j.1471-4159.1998.71020875.x. [DOI] [PubMed] [Google Scholar]
- Squire LR, Bayley PJ. The neuroscience of remote memory. Curr Opin Neurobiol. 2007;17:185–196. doi: 10.1016/j.conb.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia-Arancibia L, Aliaga E, Silhol M, Arancibia S. New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res Rev. 2008;59:201–220. doi: 10.1016/j.brainresrev.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Theuns J, Brouwers N, Engelborghs S, Sleegers K, Bogaerts V, Corsmit E, De Pooter T, van Duijn CM, De Deyn PP, Van Broeckhoven C. Promoter mutations that increase amyloid precursor-protein expression are associated with Alzheimer disease. Am J Hum Genet. 2006;78:936–946. doi: 10.1086/504044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Kovacs DM, Yan R, Wong PC. The β-secretase enzyme BACE in health and Alzheimer's disease: regulation, cell biology, function, and therapeutic potential. J Neurosci. 2009;29:12787–12794. doi: 10.1523/JNEUROSCI.3657-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Song L, Laird F, Wong PC, Lee HK. BACE1 knock-outs display deficits in activity-dependent potentiation of synaptic transmission at mossy fiber to CA3 synapses in the hippocampus. J Neurosci. 2008;28:8677–8681. doi: 10.1523/JNEUROSCI.2440-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zang Y, He Y, Liang M, Zhang X, Tian L, Wu T, Jiang T, Li K. Changes in hippocampal connectivity in the early stages of Alzheimer's disease: evidence from resting state fMRI. Neuroimage. 2006;31:496–504. doi: 10.1016/j.neuroimage.2005.12.033. [DOI] [PubMed] [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, Destrooper B, Saftig P, Birchmeier C, Haass C. Control of peripheral nerve myelination by the β-secretase BACE1. Science. 2006;314:664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- Zacchetti D, Chieregatti E, Bettegazzi B, Mihailovich M, Sousa VL, Grohovaz F, Meldolesi J. BACE1 Expression and Activity: Relevance in Alzheimer's Disease. Neurodegener Dis. 2007;4:117–126. doi: 10.1159/000101836. [DOI] [PubMed] [Google Scholar]
- Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O'Connor T, Logan S, Maus E, Citron M, Berry R, Binder L, Vassar R. β-Site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J Neurosci. 2007;27:3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol. 2009;5:311–322. doi: 10.1038/nrneurol.2009.54. [DOI] [PubMed] [Google Scholar]